

Abstract
Objective
To investigate whether the FcγRIIIa-66R/H/L polymorphism influences net effective receptor function and to assess if the FCGR3A combined genotypes formed by FcγRIIIa-66R/H/L and FcγRIIIa-176F/V as well as copy number variation (CNV) confer risk for development of SLE and lupus nephritis.
Methods
FcγRIIIa variants, expressed on A20 IIA1.6 cells, were used in flow cytometry-based human IgG binding assays. FCGR3A SNP and CNV genotypes were determined by Pyrosequencing methodology in a cohort of 1728 SLE patients and 2404 healthy controls.
Results
The FcγRIIIa-66L/H/R (rs10127939) polymorphism influences ligand binding capacity in the context of the FcγRIIIa-176V (rs396991) allele. The low binding FcγRIIIa-176F allele was associated with SLE nephritis (p = 0.0609) in African Americans but not in European Americans (p > 0.10). Nephritis among African American SLE subjects was associated with FcγRIIIa low binding haplotypes containing the 66R/H/L and 176F variants (p = 0.03) and with low binding genotype combinations (p = 0.002). No association was observed in European American SLE patients. The distribution of FCGR3A CNV was not significantly different between controls and SLE patients with or without nephritis.
Conclusion
FcγRIIIa-66R/H/L influences ligand binding. The low binding haplotypes formed by 66R/H/L and 176F confer enhanced risk for lupus nephritis in African Americans. FCGR3A CNVs are not associated with SLE or SLE nephritis in either African Americans or European Americans.
Introduction
The contributions of genetic variants of Fcγ receptor genes to autoimmune diseases have attracted substantial interest given their implications for disease mechanisms. However, array-based genome wide association studies have been limited to probing variants in the centromeric, non-duplicated region of the classical low affinity cluster, which contains FCGR2A. Genome wide significance of association with disease phenotype for the coding variant affecting IgG binding in the extracellular domain of FcγRIIa (rs1801274; H131R) has been demonstrated in multiple conditions, including systemic lupus, rheumatoid arthritis and Kawasaki disease [1–4]. In order to avoid limitations of array-based genotyping for duplicated regions, specific candidate-gene genotyping strategies have been developed for other members of the cluster, and autoimmune disease associations have been shown for FCGR2B, FCGR3A and FCGR3B with some population and inter-study variations [5–25]. For example, the lower affinity FcγRIIIa phenylalanine allele (176F) encoded by SNP rs396991 has been associated with systemic lupus erythematous (SLE) nephritis in some reports [9,16,23,25]. Meta-analysis of multiple studies suggests that the FcγRIIIa 176F allele does not confer risk for SLE per se but rather confers a 1.2-fold risk for the development of renal disease among lupus patients across ancestry groups [26,27]. In rheumatoid arthritis, this FcγRIIIa V176F is inconsistently associated with disease even when stratified by anti-citrullinated protein antibody (ACPA) seropositivity [5,8,11,13,28–34]. This inconsistency might be due to the differences in sample size, ancestry background, copy number variation (CNV), and technical issues in genotyping given the complexity of the region [34].
FcγRIIIa is of particular interest not only because it is expressed on mononuclear phagocytes and natural killer cells but also because it has a second polymorphic site (rs10127939) in the extracellular domain at amino acid residue 66 which is tri-allelic (L66R/H). This site, originally described by de Haas et al. [35], has not been studied as a contributor to autoimmune phenotypes, and we considered the possibility that it could be influencing disease associations with V176F. Recognizing that there might also be additional as yet unidentified uncommon SNPs which could be influencing receptor biology, we undertook the re-sequencing of the coding region of FCGR3A and analyzed our large cohort of SLE participants and healthy controls to define contributions of FCGR3A variants to lupus risk. Consistent with the meta-analyses of the FcγRIIIa V176F polymorphism, we find that African American persons with the 176F allele tend to develop renal disease. Importantly, this association was strengthened by consideration of the ligand binding properties of the tri-allelic L66RH variants and was prominent in African Americans, but not in Caucasians. Thus, FCGR3A variants contribute to lupus nephritis risk in an ancestry dependent fashion, and the role of alleles with lower affinities for ligand binding suggests that inefficient handling of IgG immune complexes rather than more robust engagement of receptor-mediated inflammatory responses is an important pathophysiologic mechanism.
Materials and Methods
Study participants
A total of 1728 SLE patients (SLE) and 2404 healthy controls (CNTL) included both European Americans (SLE: n=956, CNTL: n=1335) and African Americans (SLE: n=772, CNTL n=1069) provided written informed consent for participation in this study. All patients fulfilled the American College of Rheumatology (ACR) revised criteria for SLE [36,37]. Among the cases, 366 African Americans and 213 European Americans met ACR criteria for SLE with nephritis. These studies were approved by the Institutional Review Board for Human Use.
Reagents
Human IgG (hIgG) and human IgA (hIgA) were purchased from Sigma Chemical Co. (St. Louis, MO). Anti-CD16 mAb 3G8 F(ab')2 was generated by Rockland Immunochemical (Gilbertsville, PA). Goat-anti-human-kappa F(ab')2 was obtained from Southern Biotech Inc. (Birmingham, AL). Phycoerythrin (PE)-conjugated donkey anti-goat IgG, PE-conjugated goat anti-human IgG (H+L), and PE-conjugated goat anti-mouse IgG F(ab')2 antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). Puromycin was obtained from InvivoGen (San Diego, CA) and Geneticin was from Life Technologies, Inc. (Grand Island, NY). QuikChange Site-directed mutagenesis kit was obtained from Stratagene (La Jolla, CA).
Sequencing analysis of FCGR3A exons
Genomic DNA was isolated from peripheral blood of 194 donors using the Puregene (Qiagen) reagent set. Two FCGR3A gene-specific fragments containing five FCGR3A exons (S1, S2, EC1, EC2, and TMC) were generated using gene-specific PCR. The sense primer (5'-CCC CAC CTT TTC TGT GAT CTT TTC AGC C-3') and the antisense primer (5'-CTT TTG TAA GAA CAA AAC AAA ATT TAC AAT-3') were used to amplify a FCGR3A-specific genomic DNA fragment (4480 bps) containing exons coding for S1, S2, and EC1. The sense primer (5'-CTG GTG TTT ACA TTG AGT TCT C-3') and the antisense primer (5'-CTG GCT CTG AGT TCT ATG TTT CCT GCT GCT TGT A-3') were used to amplify a FCGR3A-specific genomic DNA fragment (2606 bps) containing exons of EC2 and TMC. All the PCR products, treated with ExoSAP-IT (Affymetrix, Santa Clara, CA) to remove unincorporated primers and nucleotides according to the manufacturer's instruction, were assessed by direct Sanger sequencing on an ABI 3730xl DNA Analyzer with BigDye v3.1 Sequencing kit (Applied Biosystems, Inc., Foster City, CA). The sequencing primer (5'-TCC TCA CCC CAC ATT ATC TTG -3') in the intron 2 of FCGR3A was used to sequence the FCGR3A EC1 region. The sequencing primer (5'-ACT TTT GGG GAC CTC CTG GTG AT-3') was used to sequence the FCGR3A exon coding for the EC2 region. The primer (5'-TCC CCA CCA TTC CTA CCA CTT GC-3') was used to sequence the transmembrane segment and cytoplasmic domain. The electropherogram data, aligned by the DNASTAR software (DNAStar, Madison, WI) were used for the identification of gene polymorphisms.
Generation of FcγRIIIa and FcR γ-chain expression constructs
Human FcγRIIIa cDNA from a donor homozygous for thymidine (T) at position 230 and heterozygous at position 559 (SNP 559T>G) was amplified with the sense primer (5'-CGC GGA TCC CAG TGT GGC ATC ATG TGG CAG CTG CTC-3') and anti-sense primer (5'-TTA-TTA GAA TTC TCA TTT GTC TTG AGG GTC CTT TCT-3'). The PCR product was digested with restriction enzyme BamH I and EcoR I and cloned into pcDNA3 plasmid vector (Life Technologies) at BamH I and EcoR I cloning sites. Site-directed mutagenesis was subsequently carried out to create six FcγRIIIa haplotype constructs containing nucleotide substitutions at position 230 using the QuikChange Site-directed Mutagenesis kit. The mutagenesis sense primer FCGR3A 230G allele specific forward primer (5'-TCA CAA TGA GAG CCG230 CAT CTC AAG CCA-3') and antisense primer (5'-TGG CTT GAG ATG C230GG CTC TCA TTG TGA-3') were used for the creation of haplotype constructs containing the FCGR3A 230G allele. The 230A specific forward sense primer (5'-TCA CAA TGA GAG CCA230 CAT CTC AAG CCA-3') and antisense primer (5'- TGG CTT GAG ATG T230GG CTC TCA TTG TGA -3') were used for the generation of constructs containing the FCGR3A 230A allele. Six different pcDNA3 constructs, each containing one of the six FCGR3A haplotype variants, were obtained for expression and functional experiments; the sequences of all constructs were confirmed by direct Sanger sequencing.
The full-length FcR γ-chain (FcRγ) cDNA from a healthy donor was cloned into a pMX-PIE vector that contains a puromycin resistance gene. FcRγ full length cDNA was amplified with the sense primer (5'-CGC GGA TCC CAG CCC AAG ATG ATT CCA GCA GTG GTC-3') and anti-sense primer (5'-CCG GAA TTC CTA AAG CTA CTG TGG TGG TTT CTC ATG-3'). FcRγ cDNA PCR product digested with BamH I and EcoR I was subsequently cloned into the replication defective retroviral vector pMX-PIE as a bicistronic coding unit containing the FcRγ cDNA, followed by an internal ribosomal entry site (IRES) element and the enhanced green fluorescent protein (EGFP). The pMX-PIE construct containing full-length human FcRγ cDNA was confirmed by Sanger sequencing.
Generation of A20 IIA1.6 cell lines stably expressing FcγRIIIa haplotypes
The retroviral pMX-PIE construct containing FcRγ cDNA was transfected into BOSC23 cells and the virus containing supernatant was harvested as described [38]. After passing through a 0.2-μm filter, the supernatant containing virus particles was mixed with polybrene (final concentration of 4 μg /ml) and added to 2 × 106 A20 IIA1.6 cells in 60 mm culture dish. The retrovirally transduced cells were incubated for 4 days in medium containing puromycin (1.5 μg /ml) followed by fluorescence-activated cell sorter to select cells expressing FcRγ (GFP+ cells).
The A20 IIA1.6 cells expressing FcRγ (GFP+ cells), in DMEM medium supplemented with 10% fetal calf serum, were maintained in culture and passaged one day prior to nucleofection. FcRγ expressing cells (2×106) were transfected with each of six FCGR3A haplotype expression constructs using Amaxa® Cell Line Nucleofector® Kit V (Lonza Inc., Basel) and were cultured overnight in DMEM containing 10% FBS. One day after transfection, the culture medium was replaced with fresh medium containing puromycin (1 μg/ml). The cells were subsequently fed with fresh medium containing puromycin (1 μg/ml) every 4 days. The puromycin resistant A20 IIA1.6 cells were expanded and the cells expressing both FcRγ and FcγRIIIa were continuously selected in medium containing puromycin (1.5 μg/ml) and G418 (1 mg/ml). Subsequently, double positive cells (GFP+ for FcRγ and CD16+ for FcγRIIIa) were sorted for comparable levels of FcγRIIIa expression. The transfected cells were maintained in the presence of Geneticin (1 mg/ml) and puromycin (1.5 μg/ml) and were reassessed for quantitative dual expression in each experiment.
IgG binding assays
The binding of monomeric human IgG (hIgG) to cell lines expressing FcγRIIIa alleles was measured by quantitative flow cytometry as previously described [39,40]. Cells expressing equivalent levels of FcγRIIIa were washed with cold PBS containing 1% BSA and subsequently suspended at the density of 106 /ml in round bottomed 96-well plates with 100 μl PBS containing hIgG. A20 IIA1.6 untransfected cells and A20 transfected with empty vector were used as controls for the binding assay. The cells, in triplicates, were incubated on ice for 30 minutes. Human IgA (15 μg/ml) was also used as a negative control for the binding assay. After being washed three times, cells were incubated with 100 μl of PE-conjugated goat anti-human IgG (H+L) antibody (1 μg/ml) on ice for 30 min. The cells were washed and fixed with 1% formaldehyde before being analyzed with flow cytometry.
The binding of model immune complexes (ICs) to cell lines expressing FcγRIIIa alleles, untransfected A20 cells, and A20 transfected with empty vector were also measured by flow cytometry. An equimolar mixture of hIgG and goat-anti-human-kappa F(ab')2 was incubated at 37 °C for 2 hours to generate model IgG immune complexes [39]. An equimolar mixture of hIgA and goat-anti-human-kappa F(ab')2 were incubated to generate IgA specific ICs. The complexes were centrifuged at 10,000×g for 10 minutes to eliminate larger aggregates and the supernatants were harvested for experiments. A20 IIA1.6 cells (1×105/well) were added to a 96-well round bottom plate. Serial dilutions (0, 1, 3, 5, 7 μg/ml) of ICs were made with ice cold PBS containing 1% BSA. The ICs were added to each well in triplicates and incubated for 30 min on ice. The cells were then washed three times with cold PBS containing 1% BSA before being stained with 100 μl of phycoerythrin conjugated-donkey anti-goat IgG antibody (1:200 dilutions, final concentration 5 μg/ml) on ice for 30 min. Following further washes, the cells were fixed with 1% formaldehyde and analyzed by quantitative flow cytometry.
Statistical analysis
The binding data were analyzed by paired Student's t test. Pairwise linkage disequilibrium was determined for the two FCGR3A SNPs using HaploView [42]. The frequency distribution of alleles between SLE and control subjects and between SLE and SLE nephritis subjects were compared using the generalized Cochran-Mantel-Haenszel test statistics, specifically the means score test. Haplotype analyses comparing the distribution of haplotypes in healthy controls versus SLE cases and in SLE cases without nephritis versus cases with nephritis were conducted using Haploview and with SAS 9.3, utilizing a Type I error rate of 0.05. Logistic regression was used to determine whether there was an association of increasing FCGR3A copy number and SLE risk, SLE severity, or alleles of the rs396991 and rs10127939 SNPs. European American and African American populations were analyzed separately for all tests.
For haplotype analysis, donors, homozygous at both the 66 and 176 sites and therefore homozygous for a single haplotype, were assigned unambiguously to one of the three categories based on their predicted FcγRIIIa ligand binding capabilities: “High”, “Mid” and “Low” (Table 1). Based on IgG binding data, donors with high-binding alleles (R or H) at 66 and homozygous 176V were classified as “High.” Donors homozygous for 66L and 176V were classified as “Mid.” Donors homozygous for 176F were classified as “Low.” The chi-square p-value was calculated with Cochran Mantel-Haenszel statistics using SAS.
Table 1.
Binding affinity-based FcyRIHa categorization.
| FcγRIIIa-66 Alleles | FcγRIIIa-176 Alleles |
||
|---|---|---|---|
| W | VF | FF | |
| (R/H)(R/H) | High | High | Low |
| L(R/H) | High | Mid | Low |
| LL | Mid | Low | Low |
Donors were assigned to one of the three categories based on their FcγRIIIa predicted ligand binding capabilities. Donors assigned to the “High” group had at least one copy of FcγRIIIa of 176V with 66R/H. Donors assigned to the “Low” group had either all copies of FcγRIIIa-176F or at least one copy of FcγRIIIa-176F and no chance to have a copy of FcγRIIIa-176V and FcγRIIIa-66R or 66H. All other donors were assigned to the “Mid” category.
Second, to include all donors with two copies of FCGR3A, haplotype analysis was done with Haploview [42]. For the haplotype association analysis, the chi-square test statistic was obtained by summing the fractional likelihoods of each individual for each haplotype estimated from the Expectation-Maximization (EM) algorithm. Thus, if a particular individual has been determined by the EM have a 40% likelihood of haplotype A and 60% likelihood of haplotype B, 0.4 and 0.6 would be added to the counts for A and B respectively.
Third, to include all donors, donors were assigned to one of the three “High”, “Mid” and “Low” categories used for the double-homozygote donors based on their genotype and the probability of having certain haplotypes. Donors assigned to the “High” group were 66R or 66H for all copies of FcγRIIIa, and had at least one copy of FcγRIIIa of 176V. Donors assigned to the “Low” group were 176F for all copies of FcγRIIIa or were 176F for one copy of FcγRIIIa and neither 66H nor 66R for all copies of FcγRIIIa. Our results showed that FcγRIIIa with 176F had lower binding capacity regardless of the allele at 66. All of these donors therefore have at least one copy of FcγRIIIa that has low binding capacity and no copies of FcγRIIIa with high binding capacity. All other donors with different combinations of 66 and 176 alleles were assigned to the “Mid” category. The donor assignment scheme is shown in Table 1.
Results
Identification of novel SNPs in the coding region of FCGR3A gene
We re-sequenced the entire coding region of FCGR3A from 194 individuals, including 97 participants with SLE. In addition to the known non-synonymous polymorphisms at nucleotide positions 230, 559 and a synonymous polymorphism at nt 249 (rs114535887; C249T) [15,35], we found a rare non-synonymous SNP that changes amino acid position 232 from asparagine to tyrosine in the FcγRIIIa cytoplasmic domain (Figure 1). Two healthy donors out of 194 (~1%) were heterozygous carriers for the SNP 727A>T that results in a transversion from 727A to 727T (rs115866423).
Figure 1. Identification of a rare FCGR3A SNP.

A rare SNP was identified at nucleotide position 727 that changes amino acid codon 232 from asparagine (N) to tyrosine (Y). The sequence traces of the 232N homozygous donor (upper panel) and the 232N/Y heterozygous donor (lower panel) are shown.
FcγRIIIa haplotypes influence the binding of human IgG
Although its affinity for IgG is less than that for the high affinity FcγRIa (CD64), FcγRIIIa has a higher affinity for IgG than either FcγRIIa or FcγRIIIb. This affinity is sufficient to allow binding of monomer IgG at physiological concentrations [15]. To determine whether both the FcγRIIIa-66L/H/R and 176F/V polymorphisms can influence the binding of monomeric human IgG, we compared the IgG binding capacities of the six haplotypes containing the six combinations of FcγRIIIa-66L/H/R and 176F/V genotypes transfected into A20 IIA1.6 cells, which lack endogenous FcγR expression, and sorted for equal expression of FcγRIIIa. A20 untransfected and A20 transfected with empty vector were used as controls. The levels of binding were analyzed by flow cytometry using human IgG and PE-conjugated goat anti-human IgG F(ab)'2; mAb 3G8 F(ab)'2 was used to confirm identical levels of FcγRIIIa expressed on the A20 IIA1.6 cell surface (Figure 2A). We observed that hIgG or hIgA did not bind to A20 untransfected cells or A20 transfected with empty vector (data not shown). As shown in Figure 2B, FcγRIIIa -66R-176V had the highest binding for human monomeric IgG, and both 66H-176V and 66R-176V haplotypes had significantly higher binding of monomeric IgG than 66L-176V (p < 0.05), suggesting a contribution of the allele 66L/H/R allele to the FcγRIIIa avidity for hIgG. The FcγRIIIa 66L-176V demonstrated an intermediate level of hIgG binding at 15 μg/ml of IgG. Independent of the FcγRIIIa-66L/H/R, the FcγRIIIa-176F bound significantly less hIgG, which is consistent with previous reports [15,24]. There was no significant difference between 66L and 66R and 66H in the context of 176F. Thus, our data demonstrate that both the tri-allelic FcγRIIIa-66L/H/R and biallelic FcγRIIIa-176F/V contribute to the binding capacity of receptor for monomeric human IgG. Interestingly, the 66H and 66R alleles are in strong linkage equilibrium with the 176V allele, which has been reported as the only contributor for IgG binding difference between FcγRIIIa alleles [15,24]. Based on the frequency of individuals who are homozygous 66L and homozygous 176V (AA: 10.1%, EA: 4.1%; Supplemental Table 1) the population frequency of the 66L-176V haplotype is approximately 31% in African Americans and 20% in European Americans. Estimated by Haploview using 1822 African American controls and 2242 European American controls, the haplotype frequencies of 66L-176V are 32.1% and 21.9% respectively.
Figure 2. FCGR3A haplotypes affect the hIgG binding.
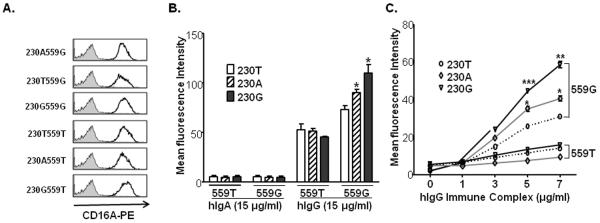
A). Expression of CD16A on A20 IIA1.6 cells. IIA1.6 cell lines expressed equivalent levels of CD16A on the cell surface as detected by anti-CD16-PE using flow cytometry. The shaded histogram represents the mIgG1-PE isotype control. Cell number is plotted on the Y axis. B). FCGR3A haplotypes significantly affect monomeric hIgG binding to the receptor. The bars represent the mean fluorescence intensities (± SEM). Asterisks indicate statistically significant differences in binding of hlgG (paired Student's t test, p<0.05). The haplotype 66R-176V had the highest binding capacity for monomeric hIgG. No significant differences in binding to monomeric hIgG were found among haplotype 66L-176F, 66H-176F, and 66R-176F. A20 IIA1.6 cells, untransfected or transfected with empty vector, showed no binding of hIgG (<3 MFI units). C). FCGR3A haplotypes significantly affect hIgG immune complex binding to the receptor. The haplotype 66R-176V and 66H-176V had significantly more binding capacity for hIgG ICs than the haplotype 66L-176V (p < 0.05). There were no significant differences in binding to IgG immune complexes (ICs) at various concentrations (0, 1, 3, 5, 7 μg/ml) among three 176F-containing haplotypes (66L-176F, 66H-176F, and 66R-176F). Three independent experiments showed the same results. A20 IIA1.6 cells, untransfected or transfected with empty vector, showed no binding of ICs (<3 MFI units).
Binding of immune complex
FcγRIIIa plays an important role in immune complex handling. The binding of human IgG immune complexes to different FcγRIIIa allelic forms was further investigated to determine the consequences of FcγRIIIa polymorphism on the relative binding avidity. Human IgG was complexed with goat anti-human-kappa F(ab')2 and then incubated with A20 IIA1.6 cell lines expressing different FcγRIIIa allelic forms. A phycoerythrin-conjugated donkey anti-goat IgG was subsequently added into the cells, as described in the “Materials and Methods.” As shown in Figure 2C, all FcγRIIIa haplotypes containing 176F bound significantly less ICs than did 176V and there were no significant differences between the 66L/H/R alleles in the context of 176F. However, in the context of the higher binding FcγRIIIa-176V allele, receptors with 66R and 66H bound more ICs compared to the common allele 66L (p = 0.005, p = 0.03, respectively). HIgG or hIgA ICs did not bind to untransfected or empty vector transfected A20 cells (data not shown).
Association of FcγRIIIa SNPs with lupus phenotypes
In our cohort, we determined the copy number of FCGR3A and of the rs396991 and rs10127939 alleles (Supplementary Table 2). The distribution of FCGR3A CNV was not significantly different between controls and patients with and without nephritis (p > 0.10).
An association between the low affinity FcγRIIIa-176F allele and risk for SLE nephritis has been reported in different ethnic groups without considering the possible contribution of 66L/R/H polymorphism. In our cohort we identified a trend supporting this association in African Americans SLE nephritis (p = 0.0609, OR = 1.25, 95% CI, 0.99, 1.57) (Table 2) but not in European Americans.
Table 2.
Distributions of FcγRIIIa haplotypes in controls, SLE participants, and SLE participants stratified by nephritis phenotype.
| FcγRIIIa Haplotype | FcγRIIIa-176F/V only | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| High (66R/H-176V) | Mid (66L-176V) | Low (66L/R/H-176F) | X2p | VV | VF | FF | X2p | ||
| EA | CNTL | 15 | 46 | 517 | 104 | 564 | 517 | ||
| SLE all | 9 | 36 | 392 | 0.7542 | 72 | 370 | 392 | 0.2219 | |
| SLE neph (−) | 8 | 26 | 308 | 56 | 282 | 308 | |||
| SLE neph (+) | 1 | 10 | 84 | 0.9377 | 16 | 88 | 84 | 0.5924 | |
|
| |||||||||
| AA | CNTL | 2 | 97 | 413 | 109 | 431 | 413 | ||
| SLE all | 2 | 64 | 289 | 0.8392 | 76 | 283 | 289 | 0.7762 | |
| SLE neph (−) | 2 | 38 | 145 | 49 | 147 | 145 | |||
| SLE neph (+) | 0 | 26 | 144 | 0.0318 | 27 | 136 | 144 | 0.0609 | |
EA: European Americans; AA: African Americans; SLE neph (−): SLE patients negative for nephritis; SLE neph (+): SLE positive for nephritis.
Because our binding assays showed that the amino acid residue at position 66 significantly affects the binding capacity of FcγRIIIa in the context of the 176V allele, we determined whether there was the association between FcγRIIIa binding capability, defined by these alleles, and both systemic lupus per se and SLE nephritis. For the analysis of both polymorphic sites, donors were stratified by ethnicity and assigned to one of three categories based on their predicted FcγRIIIa ligand binding capabilities as shown in Table 1. As a first step, we considered only doubly homozygous donors, as their FCGR3A haplotypes could be determined unambiguously. The less common 66R and 66H alleles were combined because they both enhance the binding of FcγRIIIa-176V. There was no significant association between FcγRIIIa haplotypes and overall SLE in African Americans and European Americans (Table 2). In case-case analysis, the trend of enrichment for the low binding FcγRIIIa-176F in African American SLE participants with nephritis was enhanced by considering the combination of both FcγRIIIa-66 and FcγRIIIa-176 alleles (Table 2). In donors with unambiguous haplotypes we observed an association of the high-binding haplotype (66R or 66H with 176V) with SLE without nephritis in African Americans (p = 0.0318, OR = 1.819, 95% CI 1.047–3.157).
We also used Haploview to compare haplotype likelihoods in all donors with no FCGR3A CNV. For this analysis the two high-binding 66 alleles (R and H) were combined into a single H allele, due to their comparable biological effects. Again, we saw no association of any haplotypes with SLE compared to controls in either ethnicity (p > 0.10). Similar to our results with unambiguous haplotypes, in case-case analysis we saw an association of the low binding 66L-176F haplotype with SLE with nephritis (p = 0.030). As in the analyses with unambiguous donors, the association was only seen in African Americans. Interestingly, the high-binding 66H-176V haplotype was enriched in SLE without nephritis (p = 0.0133).
To include all of the individuals in our cohort, we extended our FcγRIIIa binding subtyping based on the likelihood of having high and low binding FcγRIIIa haplotypes (Table 1). Donors with one copy of FCGR3A were categorized as homozygous for the 66 and 176 polymorphisms and heterozygous donors with three or four copies of FCGR3A were categorized as VF for the 176 polymorphism and/or L (R/H) for the position 66 polymorphism, because, like diploid heterozygotes, they have at least one copy of each allele. In African Americans, the high binding group was significantly associated with SLE without nephritis, and the low binding group with SLE with nephritis (p = 0.002, OR = 1.83, 95% CI 1.23 – 2.72). This association was not seen in European Americans in case-control analyses (Table 3).
Table 3.
Distributions of FcγRIIIa categories in controls, SLE participants, and SLE participants stratified by nephritis phenotype.
| FcγRIIIa-66(R/H/L)+176(F/V) |
|||||
|---|---|---|---|---|---|
| High | Mid | Low | X2p | ||
| European Americans | Control | 58 | 237 | 890 | |
| SLE all | 36 | 172 | 626 | 0.8301 | |
| SLE neph (−) | 30 | 130 | 486 | ||
| SLE neph (+) | 6 | 43 | 139 | 0.9722 | |
|
| |||||
| African Americans | Control | 12 | 128 | 813 | |
| SLE all | 12 | 84 | 552 | 0.7297 | |
| SLE neph (−) | 11 | 51 | 279 | ||
| SLE neph (+) | 1 | 33 | 273 | 0.0024 | |
SLE neph (−): SLE patients negative for nephritis; SLE neph (+): SLE positive for nephritis.
Discussion
Because of the role of autoantibodies and immune complexes in the pathogenesis of SLE, there has been substantial interest in the role of naturally occurring, functionally significant polymorphisms in the family of receptors binding immunoglobulin in disease pathogenesis. Through both large array-based studies and more focused candidate gene studies, variants in several of these receptors have been implicated in disease. For example, the FcγRIIIa (CD16A) variant in the second Ig-like domain (encoding 176F) alters the affinity of ligand binding and has been associated with lupus nephritis in several different ethnic groups [9,15,23–25]. However, structural variation in FCGR3A includes a second non-synonymous SNP in the extracellular domain as well as copy number variation, each of which might influence net effective receptor function. Therefore, we have taken our SLE cohort (PROFILE) to examine the contribution of these variants to SLE risk and among SLE patients, risk of SLE nephritis.
Consistent with other studies [16], we found FcγRIIIa-176F to be associated with nephritis in an independent population of African Americans (Table 2). The strength of this association did not reach the same level of statistical significance as suggested by Karassa, et al. [26], in a multi-ethnic meta-analysis, and this may reflect, in part, the larger total patient numbers included in that study. Surprisingly, we found no trend for association in European Americans, despite our population size. One study did find such an association [23], and the basis for that finding may lie in the allele frequencies in the control group which were different from those found both in our study and other studies [26,27].
To determine if there might be other variants which could influence both receptor function and disease associations, we re-sequenced 194 individuals, including 97 SLE patients. We did see a variation at nt727 in two healthy donors (~1%), suggesting that this is a rare variant. Because the rare SNP occurred in healthy controls, it unlikely has significant contribution to the SLE disease process.
Because our in vitro studies implicated a second non-synonymous SNP at nucleotide position 230, encoding residues at amino acid position 66, in influencing ligand binding, we considered the possibility that both the nt 559 and the nt 230 polymorphisms might lead to a strong association with disease phenotype. Indeed, the two taken together showed a strong association with lupus nephritis in African Americans (p = 0.002) but not in European Americans. The mechanism of the 66L/R/H polymorphism contribution is not immediately evident. Alterations in glycosylation can influence ligand binding by FcγRIIIa [40,43,44]. The SNP 230 is located at +1 position of the second canonical N-glycosylation site in the first extracellular domain and might alter FcγRIIIa glycosylation as the +1 position of N-glycosylation motif could significantly affect the oligosaccharide transfer efficiency in the process of N-glycosylation [45]. Therefore, the substitution of leucine with arginine or histidine may inhibit the N-glycosylation. Alternatively, crystal structure analysis indicates that the binding site includes F176V, but the same analysis would suggest that 66L/R/H is not directly involved. Given the location of 66L/R/H on the C-C' strand of the N-terminal D1 domain [46] and the predicted flexibility in molecular modeling, this residue may alter the C'-strand structure of the D1 domain that is involved in inter-molecular interactions such as those suggested between CR3 and FcγRIII [47,48]. Such a mechanism, while speculative, would be particularly interesting given the strong contributions of CR3 polymorphisms to disease risk [1,49].
The association of low binding variants of FcγRIIIa with nephritis suggests that less efficient immune complex handling is an important contributor to lupus pathogenesis. This impact most likely affects all ancestry groups although the evidence in European Americans is somewhat variable. The combined effect of both the rs10127939 and rs396991 suggests that the effects of FcγRIIIa polymorphisms may be underestimated in the literature because typically only the rs396991 variants, encoding the V176F polymorphism, have been studied. The disease associations suggest that targeting FcγRIIIa in persons with the 176F for enhanced receptor expression and function may be an important avenue for therapeutic discovery.
Supplementary Material
ACKNOWLEDGMENTS
We thank Deborrah McDuffie and Qiao Shang for technical assistance and Dr. Andrew Gibson for helpful discussion.
This work was supported by grants from NIH (R01 AR043727, K24 AR002138, P60 2AR30692, UL1 RR025741, P60 AR048095, P01 AR049084, P30 AR048311 and UL1 TR00165).
Footnotes
The authors have no relevant disclosures.
References
- 1.International Consortium for Systemic Lupus Erythematosus G. Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khor CC, Davila S, Breunis WB, Lee YC, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. 2011;43:1241–6. doi: 10.1038/ng.981. [DOI] [PubMed] [Google Scholar]
- 3.Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet. 2012;44:517–21. doi: 10.1038/ng.2220. [DOI] [PubMed] [Google Scholar]
- 4.Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet. 2009;41:1313–8. doi: 10.1038/ng.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radstake TR, Petit E, Pierlot C, van de Putte LB, Cornelis F, Barrera P. Role of Fcgamma receptors IIA, IIIA, and IIIB in susceptibility to rheumatoid arthritis. J Rheumatol. 2003;30:926–33. [PubMed] [Google Scholar]
- 6.Su K, Wu J, Edberg JC, Li X, Ferguson P, Cooper GS, et al. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcgammaRIIb alters receptor expression and associates with autoimmunity. I. Regulatory FCGR2B polymorphisms and their association with systemic lupus erythematosus. J Immunol. 2004;172:7186–91. doi: 10.4049/jimmunol.172.11.7186. [DOI] [PubMed] [Google Scholar]
- 7.Chen JY, Wang CM, Ma CC, Luo SF, Edberg JC, Kimberly RP, et al. Association of a transmembrane polymorphism of Fcgamma receptor IIb (FCGR2B) with systemic lupus erythematosus in Taiwanese patients. Arthritis Rheum. 2006;54:3908–17. doi: 10.1002/art.22220. [DOI] [PubMed] [Google Scholar]
- 8.Chen JY, Wang CM, Wu JM, Ho HH, Luo SF. Association of rheumatoid factor production with FcgammaRIIIa polymorphism in Taiwanese rheumatoid arthritis. Clin Exp Immunol. 2006;144:10–6. doi: 10.1111/j.1365-2249.2006.03021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koene HR, Kleijer M, Swaak AJ, Sullivan KE, Bijl M, Petri MA, et al. The Fc gammaRIIIA-158F allele is a risk factor for systemic lupus erythematosus. Arthritis Rheum. 1998;41:1813–8. doi: 10.1002/1529-0131(199810)41:10<1813::AID-ART13>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 10.Lee YH, Ji JD, Song GG. Fcgamma receptor IIB and IIIB polymorphisms and susceptibility to systemic lupus erythematosus and lupus nephritis: a meta-analysis. Lupus. 2009;18:727–34. doi: 10.1177/0961203309104020. [DOI] [PubMed] [Google Scholar]
- 11.Morgan AW, Griffiths B, Ponchel F, Montague BM, Ali M, Gardner PP, et al. Fcgamma receptor type IIIA is associated with rheumatoid arthritis in two distinct ethnic groups. Arthritis Rheum. 2000;43:2328–34. doi: 10.1002/1529-0131(200010)43:10<2328::AID-ANR21>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 12.Niederer HA, Clatworthy MR, Willcocks LC, Smith KG. FcgammaRIIB, FcgammaRIIIB, and systemic lupus erythematosus. Ann N Y Acad Sci. 2010;1183:69–88. doi: 10.1111/j.1749-6632.2009.05132.x. [DOI] [PubMed] [Google Scholar]
- 13.Nieto A, Caliz R, Pascual M, Mataran L, Garcia S, Martin J. Involvement of Fcgamma receptor IIIA genotypes in susceptibility to rheumatoid arthritis. Arthritis Rheum. 2000;43:735–9. doi: 10.1002/1529-0131(200004)43:4<735::AID-ANR3>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 14.Shrestha S, Wiener HW, Olson AK, Edberg JC, Bowles NE, Patel H, et al. Functional FCGR2B gene variants influence intravenous immunoglobulin response in patients with Kawasaki disease. J Allergy Clin Immunol. 2011;128:677–80. doi: 10.1016/j.jaci.2011.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. 1997;100:1059–70. doi: 10.1172/JCI119616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edberg JC, Langefeld CD, Wu J, Moser KL, Kaufman KM, Kelly J, et al. Genetic linkage and association of Fcgamma receptor IIIA (CD16A) on chromosome 1q23 with human systemic lupus erythematosus. Arthritis Rheum. 2002;46:2132–40. doi: 10.1002/art.10438. [DOI] [PubMed] [Google Scholar]
- 17.Hong CH, Lee JS, Lee HS, Bae SC, Yoo DH. The association between FcgammaRIIIB polymorphisms and systemic lupus erythematosus in Korea. Lupus. 2005;14:346–50. doi: 10.1191/0961203305lu2086oa. [DOI] [PubMed] [Google Scholar]
- 18.Karassa FB, Trikalinos TA, Ioannidis JP. The role of FcgammaRIIA and IIIA polymorphisms in autoimmune diseases. Biomed Pharmacother. 2004;58:286–91. doi: 10.1016/j.biopha.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Lee YH, Ji JD, Song GG. Associations between FCGR3A polymorphisms and susceptibility to rheumatoid arthritis: a metaanalysis. J Rheumatol. 2008;35:2129–35. doi: 10.3899/jrheum.080186. [DOI] [PubMed] [Google Scholar]
- 20.Marques RB, Thabet MM, White SJ, Houwing-Duistermaat JJ, Bakker AM, Hendriks GJ, et al. Genetic variation of the Fc gamma receptor 3B gene and association with rheumatoid arthritis. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan AW, Barrett JH, Griffiths B, Subramanian D, Robinson JI, Keyte VH, et al. Analysis of Fcgamma receptor haplotypes in rheumatoid arthritis: FCGR3A remains a major susceptibility gene at this locus, with an additional contribution from FCGR3B. Arthritis Res Ther. 2006;8:R5. doi: 10.1186/ar1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morris DL, Roberts AL, Witherden AS, Tarzi R, Barros P, Whittaker JC, et al. Evidence for both copy number and allelic (NA1/NA2) risk at the FCGR3B locus in systemic lupus erythematosus. Eur J Hum Genet. 2010;18:1027–31. doi: 10.1038/ejhg.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seligman VA, Suarez C, Lum R, Inda SE, Lin D, Li H, et al. The Fcgamma receptor IIIA-158F allele is a major risk factor for the development of lupus nephritis among Caucasians but not non-Caucasians. Arthritis Rheum. 2001;44:618–25. doi: 10.1002/1529-0131(200103)44:3<618::AID-ANR110>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 24.Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood. 1997;90:1109–14. [PubMed] [Google Scholar]
- 25.Salmon JE, Ng S, Yoo DH, Kim TH, Kim SY, Song GG. Altered distribution of Fcgamma receptor IIIA alleles in a cohort of Korean patients with lupus nephritis. Arthritis Rheum. 1999;42:818–9. doi: 10.1002/1529-0131(199904)42:4<818::aid-anr28>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 26.Karassa FB, Trikalinos TA, Ioannidis JP. The Fc gamma RIIIA-F158 allele is a risk factor for the development of lupus nephritis: a meta-analysis. Kidney Int. 2003;63:1475–82. doi: 10.1046/j.1523-1755.2003.00873.x. [DOI] [PubMed] [Google Scholar]
- 27.Li LH, Yuan H, Pan HF, Li WX, Li XP, Ye DQ. Role of the Fcgamma receptor IIIA-V/F158 polymorphism in susceptibility to systemic lupus erythematosus and lupus nephritis: a meta-analysis. Scand J Rheumatol. 2010;39:148–54. doi: 10.3109/03009740903292304. [DOI] [PubMed] [Google Scholar]
- 28.Brun JG, Madland TM, Vedeler CA. Immunoglobulin G Fc-receptor (FcgammaR) IIA, IIIA, and IIIB polymorphisms related to disease severity in rheumatoid arthritis. J Rheumatol. 2002;29:1135–40. [PubMed] [Google Scholar]
- 29.Kastbom A, Ahmadi A, Soderkvist P, Skogh T. The 158V polymorphism of Fc gamma receptor type IIIA in early rheumatoid arthritis: increased susceptibility and severity in male patients (the Swedish TIRA project) Rheumatology (Oxford) 2005;44:1294–8. doi: 10.1093/rheumatology/kei010. [DOI] [PubMed] [Google Scholar]
- 30.Kyogoku C, Tsuchiya N, Matsuta K, Tokunaga K. Studies on the association of Fc gamma receptor IIA, IIB, IIIA and IIIB polymorphisms with rheumatoid arthritis in the Japanese: evidence for a genetic interaction between HLA-DRB1 and FCGR3A. Genes Immun. 2002;3:488–93. doi: 10.1038/sj.gene.6363921. [DOI] [PubMed] [Google Scholar]
- 31.Milicic A, Misra R, Agrawal S, Aggarwal A, Brown MA, Wordsworth BP. The F158V polymorphism in FcgammaRIIIA shows disparate associations with rheumatoid arthritis in two genetically distinct populations. Ann Rheum Dis. 2002;61:1021–3. doi: 10.1136/ard.61.11.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan AW, Keyte VH, Babbage SJ, Robinson JI, Ponchel F, Barrett JH, et al. FcgammaRIIIA-158V and rheumatoid arthritis: a confirmation study. Rheumatology (Oxford) 2003;42:528–33. doi: 10.1093/rheumatology/keg169. [DOI] [PubMed] [Google Scholar]
- 33.Stewart-Akers AM, Cunningham A, Wasko MC, Morel PA. Fc gamma R expression on NK cells influences disease severity in rheumatoid arthritis. Genes Immun. 2004;5:521–9. doi: 10.1038/sj.gene.6364121. [DOI] [PubMed] [Google Scholar]
- 34.Thabet MM, Huizinga TW, Marques RB, Stoeken-Rijsbergen G, Bakker AM, Kurreeman FA, et al. Contribution of Fcgamma receptor IIIA gene 158V/F polymorphism and copy number variation to the risk of ACPA-positive rheumatoid arthritis. Ann Rheum Dis. 2009;68:1775–80. doi: 10.1136/ard.2008.099309. [DOI] [PubMed] [Google Scholar]
- 35.de Haas M, Koene HR, Kleijer M, de Vries E, Simsek S, van Tol MJ, et al. A triallelic Fc gamma receptor type IIIA polymorphism influences the binding of human IgG by NK cell Fc gamma RIIIa. J Immunol. 1996;156:2948–55. [PubMed] [Google Scholar]
- 36.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 37.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 38.Ehrhardt GR, Davis RS, Hsu JT, Leu CM, Ehrhardt A, Cooper MD. The inhibitory potential of Fc receptor homolog 4 on memory B cells. Proc Natl Acad Sci U S A. 2003;100:13489–94. doi: 10.1073/pnas.1935944100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armour KL, Smith CS, Clark MR. Expression of human FcgammaRIIIa as a GPI-linked molecule on CHO cells to enable measurement of human IgG binding. J Immunol Methods. 2010;354:20–33. doi: 10.1016/j.jim.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Drescher B, Witte T, Schmidt RE. Glycosylation of FcgammaRIII in N163 as mechanism of regulating receptor affinity. Immunology. 2003;110:335–40. doi: 10.1046/j.1365-2567.2003.01743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mueller M, Barros P, Witherden AS, Roberts AL, Zhang Z, Schaschl H, et al. Genomic pathology of SLE-associated copy-number variation at the FCGR2C/FCGR3B/FCGR2B locus. Am J Hum Genet. 2013;92:28–40. doi: 10.1016/j.ajhg.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 43.Edberg JC, Kimberly RP. Cell type-specific glycoforms of Fc gamma RIIIa (CD16): differential ligand binding. J Immunol. 1997;159:3849–57. [PubMed] [Google Scholar]
- 44.Yamamoto K, Sugita N, Kobayashi T, Okuda K, Van De Winkel JG, Yoshie H. Evidence for a novel polymorphism affecting both N-linked glycosylation and ligand binding of the IgG receptor IIIB (CD16) Tissue Antigens. 2001;57:363–6. doi: 10.1034/j.1399-0039.2001.057004363.x. [DOI] [PubMed] [Google Scholar]
- 45.Igura M, Kohda D. Selective control of oligosaccharide transfer efficiency for the N-glycosylation sequon by a point mutation in oligosaccharyltransferase. J Biol Chem. 2011;286:13255–60. doi: 10.1074/jbc.M110.213900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Boesen CC, Radaev S, Brooks AG, Fridman WH, Sautes-Fridman C, et al. Crystal structure of the extracellular domain of a human Fc gamma RIII. Immunity. 2000;13:387–95. doi: 10.1016/s1074-7613(00)00038-8. [DOI] [PubMed] [Google Scholar]
- 47.Zhou M, Todd RF, 3rd, van de Winkel JG, Petty HR. Cocapping of the leukoadhesin molecules complement receptor type 3 and lymphocyte function-associated antigen-1 with Fc gamma receptor III on human neutrophils. Possible role of lectin-like interactions. J Immunol. 1993;150:3030–41. [PubMed] [Google Scholar]
- 48.Zhou Y, Wu J, Kucik DF, White NB, Redden DT, Szalai AJ, et al. Multiple lupus associated ITGAM variants alter mac-1 function on neutrophils. Arthritis Rheum. 2013;65:2907–16. doi: 10.1002/art.38117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nath SK, Han S, Kim-Howard X, Kelly JA, Viswanathan P, Gilkeson GS, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. 2008;40:152–4. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.