

Abstract
Hepatitis B virus (HBV) is the leading cause of liver disease and infects an estimated 240 million people worldwide. It is characterised by a high degree of genetic heterogeneity because of the use of a reverse transcriptase during viral replication. The ten genotypes (A-J) that have been described so far further segregate into a number of subgenotypes which have distinct ethno-geographic distribution. Genotypes A and D are ubiquitous and the most prevalent genotypes in Europe (mainly represented by subgenotypes D1-3 and A2); genotypes B and C are restricted to eastern Asia and Oceania; genotype E to central and western Africa; and genotypes H and F (classified into 4 subgenotypes) to Latin America and Alaska. This review summarises the data obtained by studying the global phylodynamics and phylogeography of HBV genotypes, particularly those concerning the origin and dispersion histories of genotypes A, D, E and F and their subgenotypes. The lack of any consensus concerning the HBV substitution rate and the conflicting data obtained using different calibration approaches make the time of origin and divergence of the various genotypes and subgenotypes largely uncertain. It is hypothesised that HBV evolutionary rates are time dependent, and that the changes depend on the main transmission routes of the genotypes and the dynamics of the infected populations.
Keywords: Hepatitis B virus genotypes and subgenotypes, Phylogeography, Population dynamics and epidemics, Molecular epidemiology, Hepatitis B virus phylodynamics
Core tip: This review describes the main evidence concerning the global phylodynamics and phylogeography of hepatitis B virus (HBV) genotypes and subgenotypes, concentrating particularly on the ubiquitous A and D, and the more restricted E and F genotypes. The lack of consensus on the HBV evolutionary rate make it difficult to reconstruct the timescale of the virus origin. In order to reconcile the possibility of a long evolution and the high evolutionary rate in recent populations, we propose the hypothesis that HBV evolutionary rates are time dependent, and are influenced by the different population dynamics of the viral genotypes.
INTRODUCTION
Hepatitis B virus (HBV) is a major health problem and chronically infects an estimated 240 million people worldwide. In highly endemic areas such as east Asia, sub-Saharan Africa and the Amazon basin, the HBV carrier rate is > 8%. In Europe, the highest prevalence rates are in Turkey, Romania, Bulgaria, Greece, Albania and southern Italy[1,2].
HBV is an enveloped DNA virus that belongs to the Hepadnaviridae family, and infects the hepatocytes of a wide range of animals: the genus avihepadnavirus infects birds (such as ducks, geese, herons, storks, cranes and parrots), and the genus orthohepadnavirus infects mammals (such as squirrels, woodchucks, primates and bats)[3-5].
The viral genome, a circularised, partially double-stranded DNA of about 3.2 kilobases (the minus strand is incomplete within the virion), encompasses four partially overlapping genes (PreS/S, PreC/C, P and X) that encode at least seven proteins: the three surface proteins (the small, large and middle S proteins), two core antigens (HBcAg and HBeAg), the polymerase (encoded by the P gene), and the small regulatory X protein[6].
Despite the constrained nature of its genetic evolution[7], the HBV genome is characterised by considerable variability because of the use of an RNA intermediate and reverse transcriptase during replication. On the basis of the sequence divergence established by analysing the entire viral genome, HBV has been classified into ten genotypes (A-J) and various subgenotypes (indicated by numbers), with a mean nucleotide difference of ≥ 8% between genotypes and 4%-7% between subgenotypes[8-10]. These viral strains partially correspond to the previously described serotypes based on the presence of two pairs of mutually exclusive antigenic determinants (d/y and w/r) in the HBV surface antigen[10].
HBV genotypes have a characteristic ethno-geographic distribution. Some are ubiquitous, such as genotype A, which is present in north-western Europe, North America and central Africa[10], and genotype D, which has been found throughout the world, although its highest prevalence is in the Mediterranean area, the Middle East and southern Asia[10,11]. Genotypes B and C are only present in Asia[12]; genotype E is found in sub-Saharan Africa[10,13], and genotype F in South and Central America[10]. Genotype G has been found in France and the United States[14], whereas genotype H seems to be confined to the northern part of Latin America[8] (Table 1).
Table 1.
Subgenotypes, sub-types and geographical origin of hepatitis B virus
| Subgenotype | Sub-type | Geographical origin | |
| A | A1 (Aa, A’) | adw2, ayw1 | Africa, Asia, South America |
| A2 (Ae, A-A’) | adw2, ayw1 | Northern Europe, North America, South Africa | |
| A3 (Ac) | Cameroon, Gabon, Rwanda | ||
| A4 | Mali, Gambia | ||
| A5 | Nigeria, Rwanda, Cameroon, Haiti (African pop.) | ||
| A6 | Congo, Rwanda | ||
| A7 | ayw1, adw2, ay | Cameroon, Rwanda | |
| B | B1 (Bj) | adw2 | Japan |
| B2 (Ba) | adw2, adw3 | Asia without Japan | |
| B3 | adw2, ayw1 | Indonesia, Philippines | |
| B4 | ayw1, adw2 | Vietnam, Cambodia | |
| B5 | Philippines | ||
| B6 | Alaska, Northern Canada, Greenland | ||
| B7-B9 | Indonesia | ||
| C | C1 (Cs) | adrq+, ayr, adw2, ayw1 | South-east Asia (Vietnam, Myanmar, Thailand, Southern China) |
| C2 (Ce) | adrq+, ayr | Far East (South Korea, Japan, Northern China) | |
| C3 | adrq–, adrq+ | Pacific Islands (Micronesia, Melanesia, Polynesia) | |
| C4 | Australia | ||
| C5 | Philippines, Vietnam | ||
| C6 | Indonesia, Philippines | ||
| C7 | Philippines | ||
| C8-C16 | Indonesia | ||
| D | D1 | ayw2, adw1, ayw1 | Europe, Middle East, Asia, Tunisia, Egypt |
| D2 | ayw3, ayw1 | Europe, Morocco, India | |
| D3 | ayw3, ayw2, ayw4 | South Africa, Asia, Europe, United States, Northern Canada | |
| D4 | ayw2, ayw3 | Australia, Japan, Papua New Guinea | |
| D5 | East India, Japan | ||
| D6 | Indonesia | ||
| D7 | Tunisia | ||
| D8 | Niger | ||
| D9 | Eastern India | ||
| E | ayw4, ayw2 | Sub-Saharan Africa, United Kingdom, France, Saudi Arabia | |
| F | F1a | adw4, ayw4 | South and Central America |
| F1b | adw4 | Argentina, Japan, Venezuela, United States | |
| F2 | adw4 | South America (Brazil, Venezuela, Nicaragua) | |
| F3 | adw4 | Venezuela, Panama, Columbia, Bolivia | |
| F4 | adw4 | Bolivia, France, Argentina | |
| G | adw2 | United States, Germany, Japan, France, Mexico | |
| H | adw4 | United States, Japan, Nicaragua | |
| I | I1 | adw2 | Laos, Vietnam, North-west China |
| I2 | ayw2 | Laos, Vietnam | |
| J | ayw | Japan |
A ninth genotype (I) has recently been proposed after being found in north-western China[15], eastern India[16], Laos[17], and Vietnam[17,18]. Even more recently, a genotype J has been isolated in a Japanese patient with hepatocellular carcinoma[19].
The two genotypes responsible for the majority of infections in Europe are genotype A (mainly subgenotype A2) in the north-west, and genotype D (mainly subgenotypes D1, D2 and D3) in the north-east and the Mediterranean basin[4].
PHYLODYNAMICS AND PHYLOGEOGRAPHY OF VIRUSES
The concept of “phylodynamics” was first introduced by Grenfell in 2004, and describes how microbial genetic variation is modulated by host immunity, transmission bottlenecks and epidemic dynamics that determine the wide variety of pathogen phylogenies observed at scales ranging from an individual host to a population[20].
The use of improved molecular clock models (strict or relaxed) for estimating the evolutionary rates and divergence times of heterochronous sequences (sequences sampled over relatively long periods of time) have recently made it possible to develop coalescence-based methods for studying the dynamics of viral populations at intra- and inter-host level on a calendar time scale. The coalescent describes the relationship between the demographic history of a large population and the genealogical tree of individuals randomly sampled from it[21].
Human demography, such as population subdivisions and movements, play an important role in determining the shape of the genetic structure (phylogeny) of viral populations, which is why phylogeography (the study of the spatial subdivision of viral strains) is a crucial part of the phylodynamic characterisation of an infection[22]. Statistical methods for reconstructing the geographical and spatial spread of a virus on the basis of the sampling locations of viral isolates were originally based on maximum parsimony, which finds the minimum number of migrations justifying the entire phylogeny[23]. More recently, methods that accommodate phylogenetic uncertainties have also been developed. In particular, a Bayesian statistical inference framework incorporating coalescent models allows the simultaneous reconstruction of the temporal and spatial history of an epidemic on the basis of isolates randomly sampled at known times and in different places[24].
These “phylodynamic” and phylogeographical approaches have been used to study the past/recent epidemiology of infections due to highly variable viruses such as human immunodeficiency virus (HIV)[25,26], or hepatitis C virus (HCV)[27,28].
This review will concentrate on the phylodynamics and phylogeography of the HBV genotypes for which such studies are available, particularly the two ubiquitous genotypes A and D, and the more restricted genotypes E and F.
PHYLODYNAMICS OF GENOTYPE D
HBV genotype D is the most prevalent genotype in north-eastern Europe, the eastern and central Mediterranean, northern Africa, and the Middle East; furthermore, it is highly prevalent in the Indian sub-continent and a group of islands in the Indian Ocean with high endemic levels of HBV (Nicobare and Andaman)[29], and has also been identified in Oceania[10].
Nine HBV-D subgenotypes (D1-D9) have so far been described (Figure 1): D1 is the most prevalent subgenotype in Greece, Turkey and north Africa[30,31]; D2 in north-eastern Europe (Russia, Belarus, Estonia) and Albania[32,33]; and D3 in Italy and Serbia[34,35]. D4 is the dominant subgenotype in Oceania[10]; D5 in primitive tribes living in India, where a number of different D subgenotypes are also found[36]; D6 in Papua and Indonesia[37], and D7 in Tunisia and Morocco[38,39]. Finally, the recently described D8 and D9 subgenotypes found in Nigeria and India have respectively been recognised as recombinant forms of HBV-D with HBV-E[40] and HBV-C[41].
Figure 1.
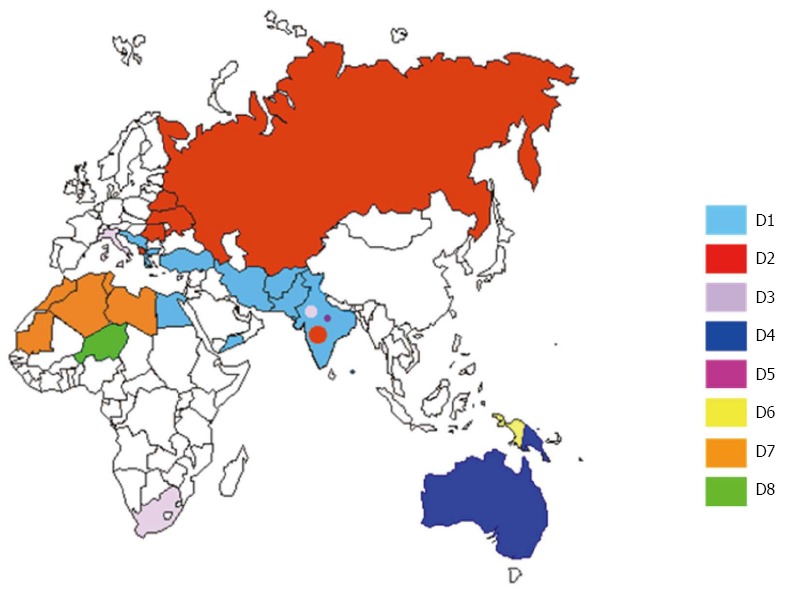
Distribution of hepatitis B virus-D subgenotypes D in Eurasia and the Mediterranean basin. The countries are coloured on the basis of their prevalent subgenotypes.
Two preliminary studies carried out in different places (Italy and Japan), but using similar approaches, suggested a relatively recent origin of genotype D in the early XX century[42,43], and showed the exponential growth of the epidemic due to this genotype in the 1940s and 1950s. Interestingly, both studies recorded a plateau in the number of infections starting in the 1980s, which is in line with the decrease in the incidence of acute HBV infections in developed countries that has been reported since then[43-45]. In Italy, HBV-D3 is associated with parenteral exposure and drug addiction[34,46], and the virus isolated from intravenous drug users (IVDUs) is characterised by a number of S and P gene mutations[34] and has been described also in IVDUs living in other industrialised countries[10,42,47]. Analysis of the dated tree suggested that the ancestor of this strain originated in the 1950s or 1960s, and rapidly spread among IVDUs generating a typical founder effect[43]. Zehender et al[43] also investigated the phylodynamics of the second most important European subgenotype (HBV-A2), which has been shown to be highly prevalent in Italy among people acquiring the infection as a result of sexual transmission, particularly men-having-sex-with-men (MSM). The authors found that the HBV-A epidemic in Italy began later and grew until more recently than that due to HBV-D3. These observations suggested that the spread of HBV in Italy coincided with two distinct epidemic waves: the first occurred at the time of World War II, when the HBV epidemic grew rapidly and was mainly due to subgenotype D3; the second occurred in the 1960s and 1970s, involved subjects at high behavioural risk, and was characterised by the import of new viral strains or the expansion of pre-existing variants (such as the IVDU-associated strain) via different transmission routes.
A further study aimed at investigating the phylodynamics of HBV genotype D in Albania (one of the European countries with the highest level of HBV endemicity) showed that the most prevalent HBV subgenotype was D2 (highly prevalent in north-eastern Europe), and that the epidemic sharply increased between the mid-1980s and the mid-1990s, a period that was characterised by a number of dramatic outflows of Albanians as a result of major socio-economic crises. A recent temporal reconstruction of the epidemiological history of genotypes D and A in Bulgaria[48] estimated a similar period of time for the penetration of subgenotype D1 and showed that, as in Italy, subgenotype A2 entered Bulgaria later than genotype D.
However, a recent and comprehensive reconstruction of the phylogeography of HBV genotype D in Europe and the Mediterranean basin[49] indicates that it originated in the second half of the XIX century in India, and that subgenotype D5 (an indigenous Indian subgenotype) was probably the first to diverge. Then subgenotypes D1-D3 diverged in central Asia and (between the 1930s and 1940s) spread to Europe and the Mediterranean area by means of at least two pathways: a south-western route (mainly due to the diffusion of subgenotype D1) that crossed the Middle East and reached north Africa and the south-eastern Mediterranean; and a second north-western route (closely associated with D2) that crossed the former Soviet Union and reached eastern Europe and the Mediterranean through Albania[50].
This reconstruction makes it possible to hypothesise that the Second World War played a crucial role in the spread of HBV-D from India to the rest of the world. However, the further dispersion of the genotype was probably sustained by the unsafe medical use of injections in medical practice[51-53].
It seems that the epidemiological dichotomy of Europe (which makes genotype D the most prevalent in eastern and southern Europe where HBV is highly endemic, and genotype A the main strain in central and northern Europe where HBV is less widespread) could be due to differences in their main transmission routes[43]: predominantly parenteral transmission in highly endemic areas (unsafe injections, and intra-family transmission), and hetero- and homosexual transmission in less endemic areas[2,54,55].
PHYLODYNAMICS OF GENOTYPE A
Genotype A is the second most ubiquitous HBV genotype, being present in Africa, Asia, Europe, and United States. The isolates have so far been classified into seven distinct evolutionary groups. Subgenotype A1 is widespread in southern and eastern Africa (South Africa, Uganda, Malawi, Tanzania, Congo, Somalia) and southern Asia (India, the Philippines, Bangladesh, Nepal)[10,36,56-58] and South America (Brazil - see above), and subgenotype A2 is the most widespread in Europe and North America[10,56] and has also been isolated in South Africa[59]. Hannoun has suggested that genotype A originated in Africa and hypothesised the import of HBV-A2 from Africa to Europe by Portuguese sailors in the XVI and XVII centuries, and the arrival of A1 in Asia as a consequence of trade and travel between eastern Africa and southern Asia[60]. The more recently described subgenotypes are A3 in Pygmies and Bantus living in Cameroon and Gabon[61,62]; a “tentative A4” from Mali; and subgenotype A5 isolated in patients from Nigeria[63]. Interestingly, HBV-A5 has also been found in Haiti, which suggests that it was the dominant subgenotype in an area near current Nigeria (former Bight of Benin) before the time of the slave trade (between the XVIII and XIX centuries), when it was probably exported to Haiti. It was only recently that HBV-A5 was replaced by genotype E and subgenotype A3, which are now the most prevalent strains in the same area[64]. An HBV-A6 has only been reported in African-Belgian patients[47], and a new “tentative subgenotype A7” has been isolated in Cameroon[65]. It has recently been proposed that A3, “tentative A4”, A5 and “tentative A7” should be classified within a single subgenotype called “quasi subgenotype A3”[47,66].
The first study addressing the origin and population dynamics of HBV genotype A (particularly subgenotype A2)[43] reported the existence of a single phylogenetically homogenous strain that was prevalent among Italian HIV-positive MSM and had a mean time of the most recent common ancestor (tMRCA) going back to 1966, thus suggesting that subgenotype A2 spread largely as a result of sexual transmission between the end of the 1960s and the beginning of the 1980s.
Interestingly, a single HBV genotype A2 variant has also been isolated from MSM living in The Netherlands[67], a country with a low level of HBV endemicity (0.3%-0.5%) in which sexual transmission is the main route of infection[55]. Using a Bayesian skyline approach, the authors observed that, after a considerable decrease in the incidence of HBV in the early 1980s, there was no subsequent change up to 2003. This trend, which is similar to that of HIV during the same period, probably reflects changes in sexual behaviours due to the HIV/AIDS epidemic[55]. A further study performed in The Netherlands is particularly important because it represents the first attempt to apply the phylodynamic approach to monitoring the efficacy of a targeted vaccination programme for MSM living in Amsterdam[68]. The authors claimed that the use of coalescent analysis provided important information for assessing the impact of vaccinations but, while recognising that the study opened up new ways of assessing the effectiveness of intervention programmes, Halloran et al[69] pointed out that the study was limited by its small sample size and a possible sampling bias. A more recent study designed to assess the efficacy of the selective vaccination programme in The Netherlands extended the analysis to nationwide population-based surveillance between 2004 and 2010, and observed a significant decrease in the effective number of infections due to subgenotype A2 among the MSM, which was corroborated by a parallel decrease in the incidence of acute HBV infections[70].
It has also been found that HBV-A2 is highly prevalent and shows relatively low genetic diversity among MSM in Japan[71], thus suggesting a distinct and recent worldwide HBV-A2 epidemic among MSM.
There is still a lack of phylogeographical studies of the spatial distribution of genotype A. The only exception is a recent study by Kramvis and Paraskevis[72], who used a maximum parsimony method to reconstruct the worldwide dispersion of subgenotype A1. They concluded that HBV-A1 most probably originated in Africa, and that the slave trade and colonisation played a major role in its global dispersion: in particular, the Arabian East African slave trade from Africa to India (until the late XIX century), the Belgian colonisation of the Congo (in the first half of the XX century), and the European slave trade (until the XIX century).
PHYLODYNAMICS OF GENOTYPE E
Genotype A is widespread in southern and eastern Africa, but genotype E is the most prevalent strain in central and western Africa (Figure 2), and is essentially an African genotype insofar as it has only been isolated from people born in Africa[73]. However, although it is found over a large area, it is interesting to note that it has a very low degree of genetic diversity: the isolates studied by means of phylogenetic analysis do not segregate into distinct subgenotypes, but are included into a single monophyletic group[74]. This observation suggests that it has a relatively recent evolutionary history among humans and, despite the forced immigration of western African slaves[74], the absence of any significant spread among Afro-Americans indicates that it was probably rare in west Africa at the time of the slave trade and before the XIX century. The only one documented finding of its presence in America is that of Alvarado Mora, who in 2010 identified nine HBV-infected subjects carrying genotype E in the relatively isolated Afro-American community of Quibdò, Colombia[75]; all of these strains were identified by means of their two-nucleotide synapomorphy in the S region, thus forming a highly significant monophyletic group. The time-scaled phylogeny reconstructed by directly estimating the evolutionary rate (a mean of 3.2 × 10-4 s/s per year) using 51 dated sequences retrieved from public databases indicated a tMRCA of about 29 years, thus suggesting the very recent introduction of HBV-E within this community[75]. The use of a slower evolutionary rate (1.5 × 10-5 s/s per year) previously calculated by others in HBeAg-positive carriers[76] gave a tMRCA for the Colombian clade within a period consistent with the transatlantic slave trade (between 257 and 738 years ago), but this evolutionary rate was the lowest limit estimate based on a retrospective comparison of mother to child isolates in cases of presumed vertical transmission[77,78].
Figure 2.
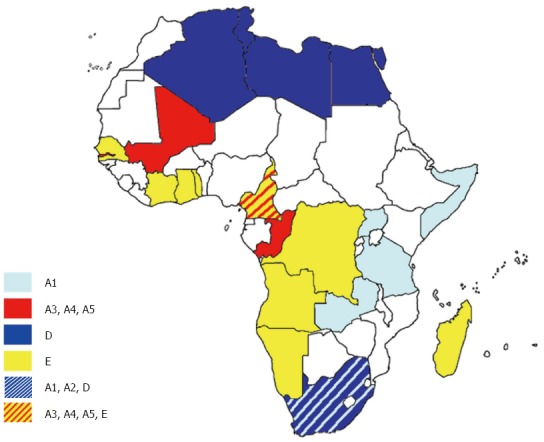
Distribution of hepatitis B virus genotypes in Africa. The countries are coloured on the basis of their prevalent genotypes/subgenotypes.
On the contrary, HBV genotype A is widely represented among Afro-American communities[79,80] and is more genetically diverse than genotype E, which suggests it has been present in western Africa for longer[79]. Forbi et al[81] found a high prevalence of HBV infection (mainly due to HBV-E) in two remote Nigerian communities. They found a high degree of intra-host heterogeneity and a high frequency of inter-host variant sharing. This suggests the relatively recent introduction of HBV-E and support the view that the explosive spread of HBV-E in Africa must have been due to a new and highly efficient route of transmission, probably the unsafe use of needles during numerous mass-vaccination campaigns (against yaws, sleeping sickness, smallpox and measles), which were particularly frequent in west and central Africa between the 1920s and 1960s[79,81].
PHYLODYNAMICS OF GENOTYPE F
Genotype F is indigenous to America, and the most prevalent HBV genotype in Central and South America, and among the Amerindians of the Amazon basin[82,83]. Genotype F is classified into four subgenotypes (F1-F4) that are further sub-divided into different clades. As shown in Figure 3, F1 is highly prevalent in Central America (clade F1a), Alaska and south-east America (clade F1b)[84,85]; F2 is highly prevalent in Venezuela (clades F2a and b), and is also present in Brazil (only clade F2a)[84]; F3 is present in central (Panama) and northern Latin America (Colombia and Venezuela); and F4 is present in Bolivia and Argentina (where it co-circulates with F1b)[86].
Figure 3.
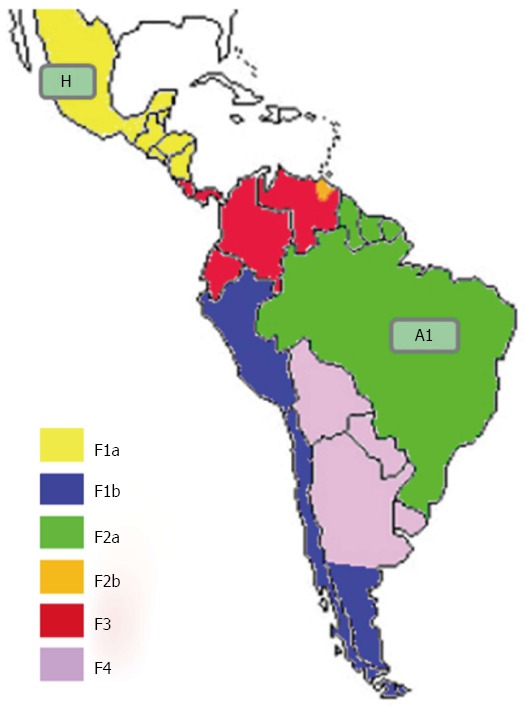
Distribution of hepatitis B virus-F subgenotypes in Latin America. The countries are coloured on the basis of their prevalent genotypes/subgenotypes.
The main problem in reconstructing the phylodynamics of the HBV-F genotype is the lack of agreement concerning its substitution rate. Torres et al[86] estimated it using a Bayesian molecular clock and obtained a mean value of 1.67 × 10-4 (Table 2), with a root tMRCA of 284 years, and tMRCAs for the different subgenotypes and clades ranging from 23 (F1b) to 70 years (F2). Nevertheless, coalescent analysis based on this timescale showed a very recent increase in the number of infections starting about 15 years ago. Given the presence of HBV-F among the Amerindian population, which suggests the long evolution of this strain, the authors considered the estimated evolutionary time scale epidemiologically highly unrealistic and decided to use different mean substitution rates covering the range of those previously estimated (from 0.6 × 10-6 to 7.7 × 10-4 s/s per year). In particular, the authors chose an arbitrary “intermediate” value of 1 × 10-5 s/s per year, and obtained a tMRCA of 4400 years for the entire tree and 700-800 years for the different F subgenotypes/clades. Under these conditions, the exponential increase in the number of infections would have started about 300 years ago which suggests the pre-Columbian origin of HBV-F and its subgenotypes, some of which would have disappeared during the conquests of the XVI and XVII century as a result of the extreme decline in population numbers. The epidemic would then have expanded because of the rapid increase in the Latin American population since the XVIII century[86].
Table 2.
Hepatitis B virus genotype substitution rates estimated using different methods
| Genotype | Evolutionary rate | 95%HPD | TMRCA | 95%HPD | Ref. |
| A | 0.90 × 10-4 | 0.12 × 10-4-1.7 × 10-4 | 237 | 29-455 | [43] |
| 8.6 × 10-4 | 4.3 × 10-4-14.3 × 10-4 | [106] | |||
| A2 | 0.91 × 10-4 | 0.11 × 10-4-1.7 × 10-4 | 62 | 12-112 | [43] |
| A2 | 3.23 × 10-5 | 5.6 × 10-8-9.0 × 10-5 | [71] | ||
| 2.72 × 10-4 | 1.73 × 10-4-5.8 × 10-4 | [48] | |||
| C | 2 × 10-4 | 5.4 × 10-4-3.61 × 10-4 | [106] | ||
| 3.73 × 10-4 | 96 | 23-271 | [107] | ||
| D | 6.3 × 10-4 | 4.8 × 10-4-7.8 × 10-4 | 84 | 55-110 | [49] |
| 4.4 × 10-4 | 2.6 × 10-4-6.2 × 10-4 | 128 | 66-210 | [49] | |
| 1.21 × 10-4 | 1.83 × 10-4-2.27 × 10-4 | [106] | |||
| 4.30 × 10-4 | 1.16 × 10-4-7.26 × 10-4 | 66 | 26-140 | [107] | |
| 3.67 × 10-4 | 2.8 × 10-4-4.5 × 10-4 | 85 | 67-103 | [43] | |
| D3 | 3.29 × 10-4 | 2.9 × 10-4-3.7 × 10-4 | 70 | 61-79 | [43] |
| 5.4 × 10-5 | 4.0 × 10-5-7.21 × 10-5 | 104 | 139-79 | [42] | |
| D1 | 4.2 × 10-4 | 3 × 10-4-6.6 × 10-4 | 46 | 20-67 | [48] |
| E | 9.29 × 10-4 | 0.18 × 10-4-20.2 × 10-4 | [106] | ||
| 2.4 × 10-4 | 0.55 × 10-4-5.54 × 10-4 | 58 | 40-83 | [81] | |
| 3.2 × 10-4 | 2.2 × 10-4-4.5 × 10-4 | 29.6 | 16-52 | [75] | |
| F | 1.67 × 10-4 | 0.94 × 10-4-2.37 × 10-4 | 284 | 120-501 | [86] |
| 1.67 × 10-4 | 0.94 × 10-4-2.37 × 10-4 | 255 | 224-282 | [89] | |
| 2.60 × 10-4-1.5 × 10-5 | 150.9-2418.4 | [87] | |||
| Root | 7.7 × 10-4 | 0.84 × 10-4-8.61 × 10-4 | 229 | 64-580 | [107] |
| 2.2 × 10-6 | 1.5 × 10-6-3 × 10-6 | 33600 | 22000-47000 | [94] |
Evolutionary rates are expressed as mean substitutions/site per year (s/s per year), with confidence intervals as 95%HPD. The times of the most recent common ancestor (tMRCA) are also indicated with confidence limits.
In their study of the molecular epidemiology and evolutionary dynamics of HBV-F in Colombia, Alvarado Mora et al[87] found that HBV-F3 was the most prevalent subgenotype in Colombia, and suggested it originated in Venezuela and was probably the oldest F subgenotype as it is closely related to genotype H[84,87].
Mello et al[88] studied the phylodynamics of HBV-F in Brazil, and found that subgenotype F2a was highly prevalent. They concluded that, like F3[87], F2a most probably originated in Venezuela, and reached Brazil following a north-to-south viral flow.
Godoy et al[89] have recently studied the selection pressure acting on the viral proteins of the complete genome of 126 HBV-F and HBV-H isolates retrieved from public databases, and observed an excess of synonymous mutations in the younger branches of the tree that may explain the high evolutionary rates measured in recent samples.
HBV ORIGIN AND EVOLUTION
A number of conflicting hypotheses have been put forward concerning the origin of HBV. It has been proposed that it originated in the New World and then spread globally as a result of European colonisation over the last 400 years[90], but this conflicts with the observation of its widespread distribution among wild Old World apes (chimpanzees, orang-utans and gibbons). A second hypothesis proposes a co-divergence of HBV and its primate hosts over a period of 10-35 million years[91], but this implies a very slow evolutionary rate (about 3-4 × 10-9 s/s per year) that is incompatible with current molecular clock estimates indicating a faster rate of evolution[43,77,92,93]. Interestingly, the viruses isolated from non-human primates have the same divergence as that observed among human genotypes, and their phylogenetic patterns show that some human genotypes are more closely related to non-human viruses than to other human genotypes, thus suggesting different viral leaps from primates to humans and vice versa and excluding the idea of virus/primate co-divergence[22]. A third hypothesis is that HBV was present in anatomically modern humans and spread as a result of their migrations over the last 100000 years or so[9,13].
These conflicting theories raise a fundamental doubt: how fast is the evolution of HBV and its genotypes? Is HBV a lazy but dangerous ancient travelling companion of primate evolution (co-divergence), or a recent and unexpected encounter between a rapidly evolving parasite and an evolutionarily stable primate host? As said above, the main obstacle to inferring the time course of the evolution of HBV is the lack of agreement concerning its substitution rate. Studies based on external calibration approaches[76,94] suggest that HBV is a slowly evolving virus, whereas those based on internal calibration indicate that it is a highly variable virus that evolves at a rate comparable with that of other retroviruses. The use of inappropriate calibration procedures may cause errors in time-scale reconstructions[89]. Table 2 shows the mean values and confidence intervals of the substitution rates obtained by various authors using different approaches, and the root tMRCA estimates for the different HBV genotypes. Most of these methods (using heterochronous sequences or pedigree estimates) agree on a relatively high evolutionary rate and mean genotype tMRCAs not exceeding 300 years.
One recent study[94] using an external calibration approach, correlated the pattern of HBV dispersion with human migrations, and estimated its time-scale on the basis of this hypothesis. The authors calibrated the root of genotypes F and H (the Amerindian-specific HBV strains) to the coalescence times of the Amerindian population (estimated to be 13-20 kiloyears ago, KYA)[94], and suggested that the virus originated about 33600 years ago (95%HPD 22-47.1 KYA) and estimated a mean evolutionary rate of 2.2 × 10-6 s/s per year (95%HPD: 1.5 × 10-6-3.0 × 10-6), one of the slowest ever proposed for HBV (Table 2).
The phylogeographical reconstruction of the history of genotype D suggested an ancient origin (7-16 KYA) of this genotype and dated the spread of the currently circulating subgenotypes (D1-D7) to about 5-6 KYA , which conflicts with our previously described spatio-temporal reconstruction of the past epidemiological history of the same genotype[49]. The considerable difference between these two reconstructions is probably due to the fact that the rate of evolution calculated on the basis of heterochronous sequences (4.4 × 10-4-95%HPD: 2.6-6.2 × 10-4 s/s per year) is at least two logs faster than that calculated by means of external calibration. For this reason, we decided to investigate the possibility of using a different evolutionary time-scale based on external calibration and running a new analysis of our original data set with the addition of a group of HBV-D3 isolates from Italian IVDUs that form a highly significant sub-clade[43]. To this end, we used the previously estimated tMRCA of subgenotype D4 in remote Oceania (median estimate 6.2 KYA; 95%HPD 2.4-11.0 KYA) and the tMRCA of the root of the entire HBV-D tree (median 10.7 KYA; 95%HPD: 6.6-16.4 KYA)[94]. As shown in Figure 4, we obtained tMRCA estimates for the origin of genotype D and its subgenotypes ranging from about 9000 to about 5000 years ago (95%HPD: 14269-3804 for the tree root), whereas the mean tMRCA of the IVDU-associated sub-clade dated back to 2841 years ago (95%HPD: 4918-885). As mentioned above, our original analysis indicated that the ancestor of the IVDU-associated strain probably emerged in the 1950s and 1960s, and subsequently spread rapidly within the IVDU population[43], thus reflecting the known history of the IVDU epidemic. On the contrary, it is very difficult to explain the nature of this sub-clade (which mainly involves IVDUs) assuming its origin more than 2000 years ago.
Figure 4.
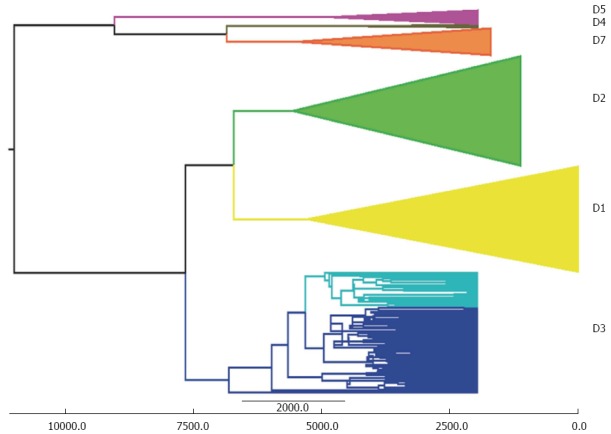
Maximum clade credibility tree of hepatitis B virus-D P gene sequences. The numbers on the internal nodes represent posterior probabilities, and the scale at the bottom of the bottom of the tree the number of years before the present. The clades corresponding to the main hepatitis B virus-D subgenotypes (D1-D5 and D7) are highlighted. The intravenous drug users-associated D3 sub-clade is coloured pale blue.
In an attempt to account for the apparent discrepancies in the evolutionary rate estimates, it is possible to hypothesise that the rapid evolutionary dynamics characterising modern HBV do not reflect its evolution on a deeper time scale. All of the methods used to date the tree presume that the evolutionary rate does not change over time; however, this is not true and what we call the evolutionary rate is an average of the values existing at different times.
It is well known that substitution rate estimates change frequently depending on the calibration approach: they are faster when recent calibration points are used over a period of a few years (as in the case of heterochronous sampling), and slower when based on more remote events (such as fossil or paleoanthropological data)[89,95]. This time dependency of evolutionary rates[96] may be due to calibration errors, model mis-specifications or mutational saturation[97], and particularly to the fact that not all of the currently existing mutants will remain fixed in the population. The current substitution rate is higher than that measured over a longer time span, because it also includes mutants that are destined to become extinct as a result of purifying selection or genetic drift. For the same reason, the substitution rates measured over very long times are underestimated when applied to the analysis of more recent evolutionary events. These disparities can obviously affect our estimates of tMRCAs, leading to a possible underestimate of deeper nodes in the case of heterochronous sampling calibration[89], and a consequent overestimate of short-term nodes when fossil calibration is used for organisms with a high rate of evolution. It is therefore inappropriate to use a short-term evolutionary rate to study events that are distant in time (such as the origin of viral genotypes in humans or other animals), and similarly inappropriate to use long-term substitution rates to calculate the time-scale of recent events such as intra-genotype evolution. As pointed out by Ho et al[98], molecular ecological and epidemiological studies deal with intra-specific (intra-genotype) data that have evolved on the genealogical rather than the longer phylogenetic time-scale, and using deep fossil calibration and inter-specific data could therefore lead to erroneous conclusions.
Another reason for the differences in the rate of virus evolution at different times depends on the demographic history of the viral population, which frequently reflects the dynamics of the host populations and the ecological/epidemiological characteristics of the infection.
The main support for the hypothesis of the long co-evolution of human HBV genotypes comes from HBV-F and its subgenotypes. These are distributed in different areas and aboriginal tribes[99] in which the prevalence of infection is very high (up to 30%)[100,101], thus suggesting that they have been generated by viral evolution in small and isolated populations. On the basis of genetic studies, all aboriginal Americans came from a population that originated in the region between the Altai mountains and the Amur river, and reached Beringia 30-22 KYA and North America 16.6 KYA[102]. This founding population probably consisted of fewer than 5000 people[103], divided into bands of probably less than 100, who reached North America in successive waves over a period of 1500 years or more[104]. In some of these small, numerically stable and isolated groups, HBV might have easily become hyper-endemic and prevalently transmitted vertically. The resulting immunotolerance at population level due to prevalent immature children infections and to the high prevalence of HBeAg positivity, which is a prerequisite for efficient vertical transmission[105] and is associated with slower viral evolutionary rates[106], may support the ethno-anthropological hypothesis of the pre-Columbian introduction of HBV into the Americas.
On the contrary, the penetration of HBV into large, fast-changing, highly mobile and susceptible populations depends on other (mainly horizontal) routes of transmission, which may also be responsible for higher evolutionary rates. Its main circulation in large populations of immunocompetent adults would explain the more rapid evolution of the virus due to stronger selective pressure and the reduced effects of genetic drift.
CONCLUSION
In order to reconcile a long evolution of HBV in humans with the evidence indicating a high evolutionary rate in recent populations, it is possible to advance a two-(or multiple-) speed hypothesis according to which the evolutionary rate of HBV changed over time depending on particular contingencies. In particular, it would be slowed in small, numerically stable, isolated and hyper-endemic populations in which vertical transmission prevails, and accelerated in large and rapidly growing populations in which the virus is mainly transmitted horizontally.
On the basis of all of these observations, it can be hypothesised that some HBV genotypes are widespread in and between continents and have an “epidemic” distribution: this may be due to rapid and mainly parenteral transmission leading to a high prevalence of infection in the general population and among risk groups (genotypes D and E), or the result of sexual transmission in hypo-endemic countries in which a sufficient number of adults are still susceptible to infection (subgenotype A2).
Other strains, such as genotype F, have an “endemic” pattern. They are found in geographically more restricted areas, have circulated for longer in their respective populations, and mainly spread as a result of vertical or intra-familiar transmission.
Footnotes
P- Reviewers: Sagnelli E, Satapathy SK S- Editor: Zhai HH L- Editor: O’Neill M E- Editor: Wang CH
References
- 1.Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 2.Rantala M, van de Laar MJ. Surveillance and epidemiology of hepatitis B and C in Europe - a review. Euro Surveill. 2008;13:pii 18880. doi: 10.2807/ese.13.21.18880-en. [DOI] [PubMed] [Google Scholar]
- 3.Piasecki T, Harkins GW, Chrząstek K, Julian L, Martin DP, Varsani A. Avihepadnavirus diversity in parrots is comparable to that found amongst all other avian species. Virology. 2013;438:98–105. doi: 10.1016/j.virol.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Schaefer S. Hepatitis B virus genotypes in Europe. Hepatol Res. 2007;37:S20–S26. doi: 10.1111/j.1872-034X.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- 5.He B, Fan Q, Yang F, Hu T, Qiu W, Feng Y, Li Z, Li Y, Zhang F, Guo H, et al. Hepatitis virus in long-fingered bats, Myanmar. Emerg Infect Dis. 2013;19:638–640. doi: 10.3201/eid1904.121655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zoulim F, Saputelli J, Seeger C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol. 1994;68:2026–2030. doi: 10.1128/jvi.68.3.2026-2030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizokami M, Orito E, Ohba K, Ikeo K, Lau JY, Gojobori T. Constrained evolution with respect to gene overlap of hepatitis B virus. J Mol Evol. 1997;44 Suppl 1:S83–S90. doi: 10.1007/pl00000061. [DOI] [PubMed] [Google Scholar]
- 8.Arauz-Ruiz P, Norder H, Robertson BH, Magnius LO. Genotype H: a new Amerindian genotype of hepatitis B virus revealed in Central America. J Gen Virol. 2002;83:2059–2073. doi: 10.1099/0022-1317-83-8-2059. [DOI] [PubMed] [Google Scholar]
- 9.Magnius LO, Norder H. Subtypes, genotypes and molecular epidemiology of the hepatitis B virus as reflected by sequence variability of the S-gene. Intervirology. 1995;38:24–34. doi: 10.1159/000150411. [DOI] [PubMed] [Google Scholar]
- 10.Norder H, Couroucé AM, Coursaget P, Echevarria JM, Lee SD, Mushahwar IK, Robertson BH, Locarnini S, Magnius LO. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology. 2004;47:289–309. doi: 10.1159/000080872. [DOI] [PubMed] [Google Scholar]
- 11.Amini-Bavil-Olyaee S, Sarrami-Forooshani R, Mahboudi F, Sabahi F, Adeli A, Noorinayer B, Azizi M, Reza Zali M. Genotype characterization and phylogenetic analysis of hepatitis B virus isolates from Iranian patients. J Med Virol. 2005;75:227–234. doi: 10.1002/jmv.20261. [DOI] [PubMed] [Google Scholar]
- 12.Okamoto H, Tsuda F, Sakugawa H, Sastrosoewignjo RI, Imai M, Miyakawa Y, Mayumi M. Typing hepatitis B virus by homology in nucleotide sequence: comparison of surface antigen subtypes. J Gen Virol. 1988;69(Pt 10):2575–2583. doi: 10.1099/0022-1317-69-10-2575. [DOI] [PubMed] [Google Scholar]
- 13.Norder H, Couroucé AM, Magnius LO. Complete genomes, phylogenetic relatedness, and structural proteins of six strains of the hepatitis B virus, four of which represent two new genotypes. Virology. 1994;198:489–503. doi: 10.1006/viro.1994.1060. [DOI] [PubMed] [Google Scholar]
- 14.Stuyver L, De Gendt S, Van Geyt C, Zoulim F, Fried M, Schinazi RF, Rossau R. A new genotype of hepatitis B virus: complete genome and phylogenetic relatedness. J Gen Virol. 2000;81:67–74. doi: 10.1099/0022-1317-81-1-67. [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Yuan Q, Ge SX, Wang HY, Zhang YL, Chen QR, Zhang J, Chen PJ, Xia NS. Molecular and phylogenetic analyses suggest an additional hepatitis B virus genotype “I”. PLoS One. 2010;5:e9297. doi: 10.1371/journal.pone.0009297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arankalle VA, Gandhe SS, Borkakoty BJ, Walimbe AM, Biswas D, Mahanta J. A novel HBV recombinant (genotype I) similar to Vietnam/Laos in a primitive tribe in eastern India. J Viral Hepat. 2010;17:501–510. doi: 10.1111/j.1365-2893.2009.01206.x. [DOI] [PubMed] [Google Scholar]
- 17.Olinger CM, Jutavijittum P, Hübschen JM, Yousukh A, Samountry B, Thammavong T, Toriyama K, Muller CP. Possible new hepatitis B virus genotype, southeast Asia. Emerg Infect Dis. 2008;14:1777–1780. doi: 10.3201/eid1411.080437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tran TT, Trinh TN, Abe K. New complex recombinant genotype of hepatitis B virus identified in Vietnam. J Virol. 2008;82:5657–5663. doi: 10.1128/JVI.02556-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatematsu K, Tanaka Y, Kurbanov F, Sugauchi F, Mano S, Maeshiro T, Nakayoshi T, Wakuta M, Miyakawa Y, Mizokami M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J Virol. 2009;83:10538–10547. doi: 10.1128/JVI.00462-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, Holmes EC. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–332. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- 21.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 22.Holmes EC. Evolutionary history and phylogeography of human viruses. Annu Rev Microbiol. 2008;62:307–328. doi: 10.1146/annurev.micro.62.081307.162912. [DOI] [PubMed] [Google Scholar]
- 23.Pybus OG, Rambaut A. Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet. 2009;10:540–550. doi: 10.1038/nrg2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Comput Biol. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai A, Ciccozzi M, Franzetti M, Simonetti FR, Bozzi G, Binda F, Rosi A, Bonora S, De Luca A, Balotta C, et al. Local and global spatio-temporal dynamics of HIV-1 subtype F1. J Med Virol. 2014;86:186–192. doi: 10.1002/jmv.23783. [DOI] [PubMed] [Google Scholar]
- 26.Lewis F, Hughes GJ, Rambaut A, Pozniak A, Leigh Brown AJ. Episodic sexual transmission of HIV revealed by molecular phylodynamics. PLoS Med. 2008;5:e50. doi: 10.1371/journal.pmed.0050050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciccozzi M, Zehender G, Polat C, Lai A, Kayman T, Sorrentino C, Ebranati E, Cella E, Lo Presti A, Abacioglu H. Phylogenetic analysis of HCV-4d in Turkey: the curious case of Kayseri province. J Med Virol. 2014;86:454–460. doi: 10.1002/jmv.23787. [DOI] [PubMed] [Google Scholar]
- 28.Zehender G, Sorrentino C, Lai A, Ebranati E, Gabanelli E, Lo Presti A, Vujoševic D, Lauševic D, Terzić D, Shkjezi R, et al. Reconstruction of the evolutionary dynamics of hepatitis C virus subtypes in Montenegro and the Balkan region. Infect Genet Evol. 2013;17:223–230. doi: 10.1016/j.meegid.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 29.Murhekar MV, Murhekar KM, Sehgal SC. Epidemiology of hepatitis B virus infection among the tribes of Andaman and Nicobar Islands, India. Trans R Soc Trop Med Hyg. 2008;102:729–734. doi: 10.1016/j.trstmh.2008.04.044. [DOI] [PubMed] [Google Scholar]
- 30.Bozdayi G, Türkyilmaz AR, Idilman R, Karatayli E, Rota S, Yurdaydin C, Bozdayi AM. Complete genome sequence and phylogenetic analysis of hepatitis B virus isolated from Turkish patients with chronic HBV infection. J Med Virol. 2005;76:476–481. doi: 10.1002/jmv.20386. [DOI] [PubMed] [Google Scholar]
- 31.Garmiri P, Rezvan H, Abolghasemi H, Allain JP. Full genome characterization of hepatitis B virus strains from blood donors in Iran. J Med Virol. 2011;83:948–952. doi: 10.1002/jmv.21772. [DOI] [PubMed] [Google Scholar]
- 32.Tallo T, Tefanova V, Priimägi L, Schmidt J, Katargina O, Michailov M, Mukomolov S, Magnius L, Norder H. D2: major subgenotype of hepatitis B virus in Russia and the Baltic region. J Gen Virol. 2008;89:1829–1839. doi: 10.1099/vir.0.83660-0. [DOI] [PubMed] [Google Scholar]
- 33.Zehender G, Ebranati E, Bernini F, Lo Presti A, Rezza G, Delogu M, Galli M, Ciccozzi M. Phylogeography and epidemiological history of West Nile virus genotype 1a in Europe and the Mediterranean basin. Infect Genet Evol. 2011;11:646–653. doi: 10.1016/j.meegid.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 34.De Maddalena C, Giambelli C, Tanzi E, Colzani D, Schiavini M, Milazzo L, Bernini F, Ebranati E, Cargnel A, Bruno R, et al. High level of genetic heterogeneity in S and P genes of genotype D hepatitis B virus. Virology. 2007;365:113–124. doi: 10.1016/j.virol.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 35.Lazarevic I, Cupic M, Delic D, Svirtlih NS, Simonovic J, Jovanovic T. Distribution of HBV genotypes, subgenotypes and HBsAg subtypes among chronically infected patients in Serbia. Arch Virol. 2007;152:2017–2025. doi: 10.1007/s00705-007-1031-0. [DOI] [PubMed] [Google Scholar]
- 36.Banerjee A, Kurbanov F, Datta S, Chandra PK, Tanaka Y, Mizokami M, Chakravarty R. Phylogenetic relatedness and genetic diversity of hepatitis B virus isolates in Eastern India. J Med Virol. 2006;78:1164–1174. doi: 10.1002/jmv.20677. [DOI] [PubMed] [Google Scholar]
- 37.Lusida MI, Nugrahaputra VE, Soetjipto R, Nagano-Fujii M, Sasayama M, Utsumi T, Hotta H. Novel subgenotypes of hepatitis B virus genotypes C and D in Papua, Indonesia. J Clin Microbiol. 2008;46:2160–2166. doi: 10.1128/JCM.01681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitab B, El Feydi AE, Afifi R, Derdabi O, Cherradi Y, Benazzouz M, Rebbani K, Brahim I, Salih Alj H, Zoulim F, et al. Hepatitis B genotypes/subgenotypes and MHR variants among Moroccan chronic carriers. J Infect. 2011;63:66–75. doi: 10.1016/j.jinf.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 39.Meldal BH, Moula NM, Barnes IH, Boukef K, Allain JP. A novel hepatitis B virus subgenotype, D7, in Tunisian blood donors. J Gen Virol. 2009;90:1622–1628. doi: 10.1099/vir.0.009738-0. [DOI] [PubMed] [Google Scholar]
- 40.Abdou Chekaraou M, Brichler S, Mansour W, Le Gal F, Garba A, Dény P, Gordien E. A novel hepatitis B virus (HBV) subgenotype D (D8) strain, resulting from recombination between genotypes D and E, is circulating in Niger along with HBV/E strains. J Gen Virol. 2010;91:1609–1620. doi: 10.1099/vir.0.018127-0. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh S, Banerjee P, Deny P, Mondal RK, Nandi M, Roychoudhury A, Das K, Banerjee S, Santra A, Zoulim F, et al. New HBV subgenotype D9, a novel D/C recombinant, identified in patients with chronic HBeAg-negative infection in Eastern India. J Viral Hepat. 2013;20:209–218. doi: 10.1111/j.1365-2893.2012.01655.x. [DOI] [PubMed] [Google Scholar]
- 42.Michitaka K, Tanaka Y, Horiike N, Duong TN, Chen Y, Matsuura K, Hiasa Y, Mizokami M, Onji M. Tracing the history of hepatitis B virus genotype D in western Japan. J Med Virol. 2006;78:44–52. doi: 10.1002/jmv.20502. [DOI] [PubMed] [Google Scholar]
- 43.Zehender G, De Maddalena C, Giambelli C, Milazzo L, Schiavini M, Bruno R, Tanzi E, Galli M. Different evolutionary rates and epidemic growth of hepatitis B virus genotypes A and D. Virology. 2008;380:84–90. doi: 10.1016/j.virol.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 44.Coppola N, Masiello A, Tonziello G, Pisapia R, Pisaturo M, Sagnelli C, Messina V, Iodice V, Sagnelli E. Factors affecting the changes in molecular epidemiology of acute hepatitis B in a Southern Italian area. J Viral Hepat. 2010;17:493–500. doi: 10.1111/j.1365-2893.2009.01201.x. [DOI] [PubMed] [Google Scholar]
- 45.Wasley A, Samandari T, Bell BP. Incidence of hepatitis A in the United States in the era of vaccination. JAMA. 2005;294:194–201. doi: 10.1001/jama.294.2.194. [DOI] [PubMed] [Google Scholar]
- 46.Zehender G, De Maddalena C, Milazzo L, Piazza M, Galli M, Tanzi E, Bruno R. Hepatitis B virus genotype distribution in HIV-1 coinfected patients. Gastroenterology. 2003;125:1559–160; author reply 1660. doi: 10.1016/j.gastro.2003.03.006. [DOI] [PubMed] [Google Scholar]
- 47.Pourkarim MR, Amini-Bavil-Olyaee S, Lemey P, Maes P, Van Ranst M. Are hepatitis B virus “subgenotypes” defined accurately? J Clin Virol. 2010;47:356–360. doi: 10.1016/j.jcv.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 48.Ciccozzi M, Babakir-Mina M, Lo Presti A, Salpini R, Cella E, Gabanelli E, Teoharov P, Kevorkyan A, Perno CF, Zehender G, et al. Molecular analysis of hepatitis B virus in Bulgaria. J Med Virol. 2013;85:49–54. doi: 10.1002/jmv.23432. [DOI] [PubMed] [Google Scholar]
- 49.Zehender G, Ebranati E, Gabanelli E, Shkjezi R, Lai A, Sorrentino C, Lo Presti A, Basho M, Bruno R, Tanzi E, et al. Spatial and temporal dynamics of hepatitis B virus D genotype in Europe and the Mediterranean Basin. PLoS One. 2012;7:e37198. doi: 10.1371/journal.pone.0037198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zehender G, Shkjezi R, Ebranati E, Gabanelli E, Abazaj Z, Tanzi E, Kraja D, Bino S, Ciccozzi M, Galli M. Reconstruction of the epidemic history of hepatitis B virus genotype D in Albania. Infect Genet Evol. 2012;12:291–298. doi: 10.1016/j.meegid.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 51.Dentinger C, Pasat L, Popa M, Hutin YJ, Mast EE. Injection practices in Romania: progress and challenges. Infect Control Hosp Epidemiol. 2004;25:30–35. doi: 10.1086/502288. [DOI] [PubMed] [Google Scholar]
- 52.Hutin YJ, Harpaz R, Drobeniuc J, Melnic A, Ray C, Favorov M, Iarovoi P, Shapiro CN, Woodruff BA. Injections given in healthcare settings as a major source of acute hepatitis B in Moldova. Int J Epidemiol. 1999;28:782–786. doi: 10.1093/ije/28.4.782. [DOI] [PubMed] [Google Scholar]
- 53.Hutin YJ, Hauri AM, Armstrong GL. Use of injections in healthcare settings worldwide, 2000: literature review and regional estimates. BMJ. 2003;327:1075. doi: 10.1136/bmj.327.7423.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Custer B, Sullivan SD, Hazlet TK, Iloeje U, Veenstra DL, Kowdley KV. Global epidemiology of hepatitis B virus. J Clin Gastroenterol. 2004;38:S158–S168. doi: 10.1097/00004836-200411003-00008. [DOI] [PubMed] [Google Scholar]
- 55.van Houdt R, Bruisten SM, Geskus RB, Bakker M, Wolthers KC, Prins M, Coutinho RA. Ongoing transmission of a single hepatitis B virus strain among men having sex with men in Amsterdam. J Viral Hepat. 2010;17:108–114. doi: 10.1111/j.1365-2893.2009.01158.x. [DOI] [PubMed] [Google Scholar]
- 56.Bowyer SM, van Staden L, Kew MC, Sim JG. A unique segment of the hepatitis B virus group A genotype identified in isolates from South Africa. J Gen Virol. 1997;78(Pt 7):1719–1729. doi: 10.1099/0022-1317-78-7-1719. [DOI] [PubMed] [Google Scholar]
- 57.Kramvis A, Weitzmann L, Owiredu WK, Kew MC. Analysis of the complete genome of subgroup A’ hepatitis B virus isolates from South Africa. J Gen Virol. 2002;83:835–839. doi: 10.1099/0022-1317-83-4-835. [DOI] [PubMed] [Google Scholar]
- 58.Sugauchi F, Kumada H, Acharya SA, Shrestha SM, Gamutan MT, Khan M, Gish RG, Tanaka Y, Kato T, Orito E, et al. Epidemiological and sequence differences between two subtypes (Ae and Aa) of hepatitis B virus genotype A. J Gen Virol. 2004;85:811–820. doi: 10.1099/vir.0.79811-0. [DOI] [PubMed] [Google Scholar]
- 59.Kimbi GC, Kramvis A, Kew MC. Distinctive sequence characteristics of subgenotype A1 isolates of hepatitis B virus from South Africa. J Gen Virol. 2004;85:1211–1220. doi: 10.1099/vir.0.19749-0. [DOI] [PubMed] [Google Scholar]
- 60.Hannoun C, Söderström A, Norkrans G, Lindh M. Phylogeny of African complete genomes reveals a West African genotype A subtype of hepatitis B virus and relatedness between Somali and Asian A1 sequences. J Gen Virol. 2005;86:2163–2167. doi: 10.1099/vir.0.80972-0. [DOI] [PubMed] [Google Scholar]
- 61.Kurbanov F, Tanaka Y, Fujiwara K, Sugauchi F, Mbanya D, Zekeng L, Ndembi N, Ngansop C, Kaptue L, Miura T, et al. A new subtype (subgenotype) Ac (A3) of hepatitis B virus and recombination between genotypes A and E in Cameroon. J Gen Virol. 2005;86:2047–2056. doi: 10.1099/vir.0.80922-0. [DOI] [PubMed] [Google Scholar]
- 62.Makuwa M, Souquière S, Telfer P, Apetrei C, Vray M, Bedjabaga I, Mouinga-Ondeme A, Onanga R, Marx PA, Kazanji M, et al. Identification of hepatitis B virus subgenotype A3 in rural Gabon. J Med Virol. 2006;78:1175–1184. doi: 10.1002/jmv.20678. [DOI] [PubMed] [Google Scholar]
- 63.Olinger CM, Venard V, Njayou M, Oyefolu AO, Maïga I, Kemp AJ, Omilabu SA, le Faou A, Muller CP. Phylogenetic analysis of the precore/core gene of hepatitis B virus genotypes E and A in West Africa: new subtypes, mixed infections and recombinations. J Gen Virol. 2006;87:1163–1173. doi: 10.1099/vir.0.81614-0. [DOI] [PubMed] [Google Scholar]
- 64.Andernach IE, Nolte C, Pape JW, Muller CP. Slave trade and hepatitis B virus genotypes and subgenotypes in Haiti and Africa. Emerg Infect Dis. 2009;15:1222–1228. doi: 10.3201/eid1508.081642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hübschen JM, Mbah PO, Forbi JC, Otegbayo JA, Olinger CM, Charpentier E, Muller CP. Detection of a new subgenotype of hepatitis B virus genotype A in Cameroon but not in neighbouring Nigeria. Clin Microbiol Infect. 2011;17:88–94. doi: 10.1111/j.1469-0691.2010.03205.x. [DOI] [PubMed] [Google Scholar]
- 66.Pourkarim MR, Amini-Bavil-Olyaee S, Lemey P, Maes P, Van Ranst M. HBV subgenotype misclassification expands quasi-subgenotype A3. Clin Microbiol Infect. 2011;17:947–949. doi: 10.1111/j.1469-0691.2010.03374.x. [DOI] [PubMed] [Google Scholar]
- 67.van Steenbergen JE, Niesters HG, Op de Coul EL, van Doornum GJ, Osterhaus AD, Leentvaar-Kuijpers A, Coutinho RA, van den Hoek JA. Molecular epidemiology of hepatitis B virus in Amsterdam 1992-1997. J Med Virol. 2002;66:159–165. doi: 10.1002/jmv.2125. [DOI] [PubMed] [Google Scholar]
- 68.van Ballegooijen WM, van Houdt R, Bruisten SM, Boot HJ, Coutinho RA, Wallinga J. Molecular sequence data of hepatitis B virus and genetic diversity after vaccination. Am J Epidemiol. 2009;170:1455–1463. doi: 10.1093/aje/kwp375. [DOI] [PubMed] [Google Scholar]
- 69.Halloran ME, Holmes EC. Invited commentary: Evaluating vaccination programs using genetic sequence data. Am J Epidemiol. 2009;170:1464–146; discussion 1464-146;. doi: 10.1093/aje/kwp366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hahné S, van Houdt R, Koedijk F, van Ballegooijen M, Cremer J, Bruisten S, Coutinho R, Boot H. Selective hepatitis B virus vaccination has reduced hepatitis B virus transmission in the Netherlands. PLoS One. 2013;8:e67866. doi: 10.1371/journal.pone.0067866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujisaki S, Yokomaku Y, Shiino T, Koibuchi T, Hattori J, Ibe S, Iwatani Y, Iwamoto A, Shirasaka T, Hamaguchi M, et al. Outbreak of infections by hepatitis B virus genotype A and transmission of genetic drug resistance in patients coinfected with HIV-1 in Japan. J Clin Microbiol. 2011;49:1017–1024. doi: 10.1128/JCM.02149-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kramvis A, Paraskevis D. Subgenotype A1 of HBV--tracing human migrations in and out of Africa. Antivir Ther. 2013;18:513–521. doi: 10.3851/IMP2657. [DOI] [PubMed] [Google Scholar]
- 73.Kramvis A, Kew MC. Epidemiology of hepatitis B virus in Africa, its genotypes and clinical associations of genotypes. Hepatol Res. 2007;37:S9–S19. doi: 10.1111/j.1872-034X.2007.00098.x. [DOI] [PubMed] [Google Scholar]
- 74.Mulders MN, Venard V, Njayou M, Edorh AP, Bola Oyefolu AO, Kehinde MO, Muyembe Tamfum JJ, Nebie YK, Maiga I, Ammerlaan W, et al. Low genetic diversity despite hyperendemicity of hepatitis B virus genotype E throughout West Africa. J Infect Dis. 2004;190:400–408. doi: 10.1086/421502. [DOI] [PubMed] [Google Scholar]
- 75.Alvarado Mora MV, Romano CM, Gomes-Gouvêa MS, Gutierrez MF, Carrilho FJ, Pinho JR. Molecular epidemiology and genetic diversity of hepatitis B virus genotype E in an isolated Afro-Colombian community. J Gen Virol. 2010;91:501–508. doi: 10.1099/vir.0.015958-0. [DOI] [PubMed] [Google Scholar]
- 76.Simmonds P, Midgley S. Recombination in the genesis and evolution of hepatitis B virus genotypes. J Virol. 2005;79:15467–15476. doi: 10.1128/JVI.79.24.15467-15476.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hannoun C, Horal P, Lindh M. Long-term mutation rates in the hepatitis B virus genome. J Gen Virol. 2000;81:75–83. doi: 10.1099/0022-1317-81-1-75. [DOI] [PubMed] [Google Scholar]
- 78.Okamoto H, Imai M, Kametani M, Nakamura T, Mayumi M. Genomic heterogeneity of hepatitis B virus in a 54-year-old woman who contracted the infection through materno-fetal transmission. Jpn J Exp Med. 1987;57:231–236. [PubMed] [Google Scholar]
- 79.Andernach IE, Hübschen JM, Muller CP. Hepatitis B virus: the genotype E puzzle. Rev Med Virol. 2009;19:231–240. doi: 10.1002/rmv.618. [DOI] [PubMed] [Google Scholar]
- 80.Araujo NM, Mello FC, Yoshida CF, Niel C, Gomes SA. High proportion of subgroup A’ (genotype A) among Brazilian isolates of Hepatitis B virus. Arch Virol. 2004;149:1383–1395. doi: 10.1007/s00705-003-0269-4. [DOI] [PubMed] [Google Scholar]
- 81.Forbi JC, Vaughan G, Purdy MA, Campo DS, Xia GL, Ganova-Raeva LM, Ramachandran S, Thai H, Khudyakov YE. Epidemic history and evolutionary dynamics of hepatitis B virus infection in two remote communities in rural Nigeria. PLoS One. 2010;5:e11615. doi: 10.1371/journal.pone.0011615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bertolini D, Moreira R, Soares M, Bensabath G, Lemos M, MELLO IMVGC PJ. Genotyping of hepatitis B virus in indigenous populations from Amazon region, Brazil. Virus Rev Res. 2000;5(2-suppl 1):101. [Google Scholar]
- 83.Castilho Mda C, Oliveira CM, Gimaque JB, Leão JD, Braga WS. Epidemiology and molecular characterization of hepatitis B virus infection in isolated villages in the Western Brazilian Amazon. Am J Trop Med Hyg. 2012;87:768–774. doi: 10.4269/ajtmh.2012.12-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alvarado-Mora MV, Pinho JR. Distribution of HBV genotypes in Latin America. Antivir Ther. 2013;18:459–465. doi: 10.3851/IMP2599. [DOI] [PubMed] [Google Scholar]
- 85.Venegas M, Alvarado-Mora MV, Villanueva RA, Rebello Pinho JR, Carrilho FJ, Locarnini S, Yuen L, Brahm J. Phylogenetic analysis of hepatitis B virus genotype F complete genome sequences from Chilean patients with chronic infection. J Med Virol. 2011;83:1530–1536. doi: 10.1002/jmv.22129. [DOI] [PubMed] [Google Scholar]
- 86.Torres C, Piñeiro y Leone FG, Pezzano SC, Mbayed VA, Campos RH. New perspectives on the evolutionary history of hepatitis B virus genotype F. Mol Phylogenet Evol. 2011;59:114–122. doi: 10.1016/j.ympev.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 87.Alvarado Mora MV, Romano CM, Gomes-Gouvêa MS, Gutierrez MF, Botelho L, Carrilho FJ, Pinho JR. Molecular characterization of the Hepatitis B virus genotypes in Colombia: a Bayesian inference on the genotype F. Infect Genet Evol. 2011;11:103–108. doi: 10.1016/j.meegid.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 88.Mello FC, Araujo OC, Lago BV, Motta-Castro AR, Moraes MT, Gomes SA, Bello G, Araujo NM. Phylogeography and evolutionary history of hepatitis B virus genotype F in Brazil. Virol J. 2013;10:236. doi: 10.1186/1743-422X-10-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Godoy BA, Alvarado-Mora MV, Gomes-Gouvêa MS, Pinho JR, Fagundes N. Origin of HBV and its arrival in the Americas--the importance of natural selection on time estimates. Antivir Ther. 2013;18:505–512. doi: 10.3851/IMP2600. [DOI] [PubMed] [Google Scholar]
- 90.Bollyky PL, Rambaut A, Grassly N, Carman WF, Holmes EC. Hepatitis B virus has a recent new world evolutionary origin. Hepatology. 1997;28:765. [Google Scholar]
- 91.MacDonald DM, Holmes EC, Lewis JC, Simmonds P. Detection of hepatitis B virus infection in wild-born chimpanzees (Pan troglodytes verus): phylogenetic relationships with human and other primate genotypes. J Virol. 2000;74:4253–4257. doi: 10.1128/jvi.74.9.4253-4257.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fares MA, Holmes EC. A revised evolutionary history of hepatitis B virus (HBV) J Mol Evol. 2002;54:807–814. doi: 10.1007/s00239-001-0084-z. [DOI] [PubMed] [Google Scholar]
- 93.Orito E, Mizokami M, Ina Y, Moriyama EN, Kameshima N, Yamamoto M, Gojobori T. Host-independent evolution and a genetic classification of the hepadnavirus family based on nucleotide sequences. Proc Natl Acad Sci USA. 1989;86:7059–7062. doi: 10.1073/pnas.86.18.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Paraskevis D, Magiorkinis G, Magiorkinis E, Ho SY, Belshaw R, Allain JP, Hatzakis A. Dating the origin and dispersal of hepatitis B virus infection in humans and primates. Hepatology. 2013;57:908–916. doi: 10.1002/hep.26079. [DOI] [PubMed] [Google Scholar]
- 95.Ho SY, Lanfear R, Bromham L, Phillips MJ, Soubrier J, Rodrigo AG, Cooper A. Time-dependent rates of molecular evolution. Mol Ecol. 2011;20:3087–3101. doi: 10.1111/j.1365-294X.2011.05178.x. [DOI] [PubMed] [Google Scholar]
- 96.Ho SY, Phillips MJ, Cooper A, Drummond AJ. Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol Biol Evol. 2005;22:1561–1568. doi: 10.1093/molbev/msi145. [DOI] [PubMed] [Google Scholar]
- 97.Ho SY, Shapiro B, Phillips MJ, Cooper A, Drummond AJ. Evidence for time dependency of molecular rate estimates. Syst Biol. 2007;56:515–522. doi: 10.1080/10635150701435401. [DOI] [PubMed] [Google Scholar]
- 98.Ho SY, Saarma U, Barnett R, Haile J, Shapiro B. The effect of inappropriate calibration: three case studies in molecular ecology. PLoS One. 2008;3:e1615. doi: 10.1371/journal.pone.0001615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Devesa M, Pujol FH. Hepatitis B virus genetic diversity in Latin America. Virus Res. 2007;127:177–184. doi: 10.1016/j.virusres.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 100.Braga WS. [Hepatitis B and D virus infection within Amerindians ethnic groups in the Brazilian Amazon: epidemiological aspects] Rev Soc Bras Med Trop. 2004;37 Suppl 2:9–13. doi: 10.1590/s0037-86822004000700002. [DOI] [PubMed] [Google Scholar]
- 101.Echevarría JM, León P. Epidemiology of viruses causing chronic hepatitis among populations from the Amazon Basin and related ecosystems. Cad Saude Publica. 2003;19:1583–1591. doi: 10.1590/s0102-311x2003000600003. [DOI] [PubMed] [Google Scholar]
- 102.Tamm E, Kivisild T, Reidla M, Metspalu M, Smith DG, Mulligan CJ, Bravi CM, Rickards O, Martinez-Labarga C, Khusnutdinova EK, et al. Beringian standstill and spread of Native American founders. PLoS One. 2007;2:e829. doi: 10.1371/journal.pone.0000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kitchen A, Miyamoto MM, Mulligan CJ. A three-stage colonization model for the peopling of the Americas. PLoS One. 2008;3:e1596. doi: 10.1371/journal.pone.0001596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Goebel T, Waters MR, O’Rourke DH. The late Pleistocene dispersal of modern humans in the Americas. Science. 2008;319:1497–1502. doi: 10.1126/science.1153569. [DOI] [PubMed] [Google Scholar]
- 105.Hadziyannis SJ. Natural history of chronic hepatitis B in Euro-Mediterranean and African countries. J Hepatol. 2011;55:183–191. doi: 10.1016/j.jhep.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 106.Harrison A, Lemey P, Hurles M, Moyes C, Horn S, Pryor J, Malani J, Supuri M, Masta A, Teriboriki B, et al. Genomic analysis of hepatitis B virus reveals antigen state and genotype as sources of evolutionary rate variation. Viruses. 2011;3:83–101. doi: 10.3390/v3020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhou Y, Holmes EC. Bayesian estimates of the evolutionary rate and age of hepatitis B virus. J Mol Evol. 2007;65:197–205. doi: 10.1007/s00239-007-0054-1. [DOI] [PubMed] [Google Scholar]