

Abstract
Molecular virology methods including polymerase chain reaction, cloning and sequencing have revolutionised our understanding of viral genome variation. In the case of hepatitis B virus (HBV), sequencing studies have identified a number of virus variants normally found during the natural course of chronic infection. The appearance of the precore stop codon (with G-for-A substitution at position 1896) and basal core promoter (BCP) (with A-for-T and G-for-A, at positions 1762 and 1764, respectively) variants which reduce or abrogate hepatitis B e antigen (HBeAg) production, heralds the initiation of the seroconversion phase from HBeAg to anti-HBe positivity. The gradual removal of the tolerogenic effect of HBeAg leads to the awakening of the immune response (immune clearance phase). Most patients after HBeAg seroconversion become “inactive HBsAg carriers”. However during the course of infection precore and/or BCP variants may emerge and be selected leading to HBeAg negative chronic hepatitis B (CHB) with high viremia levels (reactivation phase). The prevalence of HBeAg negative CHB has been increasing over the last few decades and has become the commonest type of HBV infection in many countries of the world. This probably reflects the aging of existing HBV carriers and the effective prevention measures restricting new HBV infections. Frequent acute exacerbations accompanied by high viral replication, elevated alanine aminotransferase levels and histological activity are a common feature of HBeAg negative CHB leading to cirrhosis much faster than in HBeAg positive CHB patients.
Keywords: Precore stop codon variants, basal core promoter variants; hepatitis B e antigen negative chronic hepatitis B; Re-activation; Hepatitis B virus-DNA replication
Core tip: Chronic hepatitis B virus infection can in turn go through the immune tolerant, the immune clearance, the inactive or low replicative and the reactivation phases. hepatitis B e antigen (HBeAg) positivity characterises the first two phases. Seroconversion from HBeAg to the corresponding antibody is the result of the appearance of the precore and basal core promoter variants, which either abrogate or lead to reduced HBeAg levels. The absence of HBeAg eliminates its tolerogenic effect on the immune system which thus becomes activated, leading to seroconversion. The reasons why some patients then enter into the reactivation phase during which the variants are the dominant virus present remain unexplained.
INTRODUCTION
It is estimated that worldwide well over 350 million individuals are chronically infected with hepatitis B virus (HBV)[1] and these patients are at increased risk of developing cirrhosis, hepatic decompensation and hepatocellular carcinoma (HCC). Conservative estimates suggest that about 1 million people die per year as a result of HBV-related liver pathologies[2]. HBV infection accounts for 30% of cirrhotic patients and 53% of those with HCC[3].
Following acute HBV infection, the risk of progression to chronicity is age dependent with 5% of adults, and almost 95% of children born to chronically infected mothers, becoming chronic carriers[4-6]. The natural history of chronic hepatitis B (CHB) may progress through four well recognised phases known as the immune tolerant, the immune clearance, the inactive or non-replicative and the re-activation or immune escape phases (Figure 1). These phases do not occur in all individuals and this may depend on the age and route of exposure, and may not be sequential[7].
Figure 1.
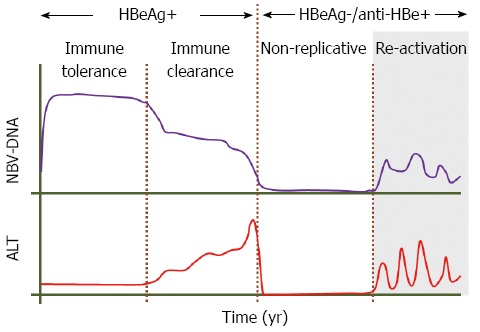
Course of the natural history of hepatitis B virus infection showing the four possible phases of disease. These are referred to as the immune tolerant, the immune clearance, the non-replicative (inactive) and the reactivation phase characterized by increased viral replication and liver damage (shaded area). During the first two phases the patient is hepatitis B e antigen (HBeAg) positive and negative during the last two. Moreover, the wild type hepatitis B virus (HBV) is dominant in the Immune tolerant and immune clearance phases, whilst the precore and basic core promoter variants are the main isolates seen in patients during the reactivation phase, and such patients constitute the HBeAg negative chronic hepatitis B group. Patients in the immune clearance and the reactivation phases are at greater risk of developing cirrhosis and eventually hepatocellular carcinoma. ALT: Alanine aminotransferase.
Patients who are exposed to the virus at a young age first go through the immune tolerant phase, which may last for 20-30 years, and is characterised by hepatitis B e antigen (HBeAg) positivity, high HBV-DNA levels (> 20000 IU/mL), normal or near normal alanine aminotransferase (ALT) levels, normal/minimal histological activity and high hepatitis B surface antigen (HBsAg) levels ranging from 4.5-5 log IU/mL[8-11]. The immune clearance phase is also characterised by HBeAg positivity, but elevated serum ALT levels are associated with histological damage, fluctuating HBV-DNA levels and lower levels of HBsAg (4.3 log IU/mL)[12,13]. These changes are the result of the immune awakening of the host culminating in seroconversion from HBeAg to anti-HBe, selection of the precore and core promoter (BCP) variants mentioned later and entry into the immune control or non-replicative phase. During this phase extremely low or undetectable HBV-DNA, normal ALT levels and normal or near normal liver histology with numerous ground glass hepatocytes are the norm. However for reasons that remain unknown, a subset of patients reactivate viral replication and form the reactivation or immune escape phase characterised by HBV-DNA levels > 2000 IU/mL, fluctuating ALT levels once again and HBsAg levels of around 3.5 log IU/mL. These patients are referred to also as the HBeAg-negative CHB group and nowadays constitute the vast majority of patients seen in outpatient clinics in most countries of the world.
VIROLOGY
Virus and its genome
The mature virion is spherical measuring 45nm in diameter and consists of an envelope comprised of HBsAg and an internal nucleocapsid formed by the core protein (HBcAg). The latter encloses the relaxed circular, partially double stranded DNA genome of 3.2 kb in length. The genome is organised in 4 partially or totally overlapping open reading frames (ORFs, S, C, X and P)[14], which encode for the HBsAg (S), the core protein or HBcAg (C), the X protein (X) which is essential for viral replication and has transactivating potential also, and finally the polymerase (P) which functions as a reverse transcriptase, DNA polymerase and possesses RNase H activity also. HBeAg is a soluble protein derived from its precore/core precursor following proteolytic processing. It is therefore a non-structural protein, is not essential for viral replication, is used as a marker of infectivity and has tolerogenic and immune modulating activity that plays a significant role in viral persistence.
The amino-acid sequence of the S protein has allowed the identification of at least 8 genotypes of the virus plus two additional putative ones, the most common of which are genotypes A-F. Genotype A is seen mostly in Northern Europe, D is prevalent in Southern Europe, the Middle East and Indian subcontinent[15,16], genotypes B and C in the Far East, whilst genotypes E and F are found in Africa and South America respectively[17].
Genomic mutations
Natural stable variants of the virus give rise to well-recognised serological subtypes and genotypes as already mentioned[18]. However, HBV has a higher mutation rate than other DNA viruses (2 × 10-4 base substitutions per site per year)[19], through error prone steps in the replication cycle of the virus. As a result, HBV circulates in blood as a mixture of variants known as quasispecies. A lot of these mutations are lethal to the virus, but those which offer it a replication advantage, facilitate immune escape or cause resistance to antiviral drugs can be preferentially selected. However, two main stable variants have been identified which arise during the natural course of chronic infection and are predominant in patients with HBeAg negative CHB. These are the precore stop codon and BCP variants.
Biology of the precore/core gene
The precore/core ORF has two in frame initiation (AUG) codons, which lead to the synthesis of two co-terminal proteins, namely the precore/core protein which is the precursor of HBeAg (Figure 2A) using the first AUG, and the nucleocapsid or core protein (HBcAg), from the second one. The two proteins are synthesized from different transcripts known as the precore mRNA and pregenomic (pg) RNA, respectively. The first 19 amino acids of the precore/core protein constitute a signal peptide that directs the protein to the endoplasmic reticulum (ER)[20], where cleavage of this peptide frees the remainder of the protein into the ER lumen, where further processing of the carboxy-terminal end results in the formation of HBeAg. This is subsequently released in the blood circulation.
Figure 2.
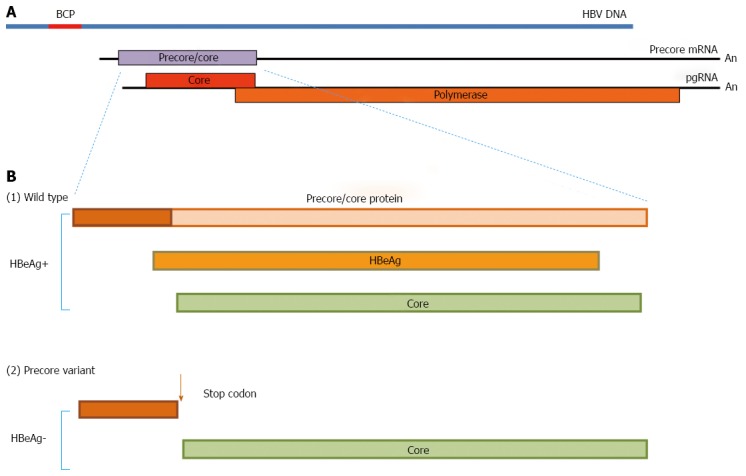
Protein products encoded by the precore/core open reading frames in wild type and precore stop codon variant isolates. A: The 3.2 kb in length HBV DNA genome in linear form (normally circular as cccDNA) and the two longer than genome length precore mRNA and pgRNA, the transcription of which is under the control of the BCP. The transcripts encoding for HBsAg and HBxAg are not shown. The precore mRNA encodes for the precore/core protein (grey open reading frame) and the pgRNA for the core and polymerase (red and orange open reading frames); B: Effect of the precore stop codon mutation on HBeAg production in comparison to that of the wild type virus. The precore/core protein is the precursor of HBeAg and consists of the precore region (in brown) and the core encoding region (in pink). HBeAg is formed through proteolytic cleavage of both its amino- and carboxyl-ends (see text). The presence of the stop codon mutation leads to the abrogation of HBeAg production as a result of pre-mature termination of the synthesis of the precore/core precursor as shown. HBcAg synthesis on the other hand which utilises the pgRNA transcript remains unaffected, thus permitting continued virion production. Please note that the proteins share complete amino acid homology over the overlapped regions. Yet, HBeAg and HBcAg display different antigenic epitopes. ALT: Alanine aminotransferase; HBeAg: Hepatitis B e antigen; HBV: Hepatitis B virus; HBsAg: Hepatitis B surface antigen; BCP: Basic core promoter; HBcAg: Hepatitis B c Antigen; HBxAg: Hepatitis B x Antigen.
Precore variants
The HBeAg-negative phase of hepatitis B viraemia is characterised by the presence of the precore stop codon variant which is selected during seroconversion and becomes dominant, whilst the wild type virus is cleared. This variant has a G to A substitution at position 1986 of the precore region (denoted as G1896A or A1896) (Figure 2B) which converts the codon for tryptophan (TGG, codon 28) to a translational stop codon (TAG)[21]. Other mutations that have the same phenotypic effect are loss of the precore/core protein translation start codon (ATG to ACG or CUG), mutation of the second codon to a stop codon, and frameshifts and deletions resulting in the synthesis of nonsense proteins. A G1899A substitution often accompanies the A1896 change.
The stop codon mutation is not favoured in genotypes A, C and F, whilst genotypes B, C, D and E with a T at this position are more amenable to the emergence of the precore stop codon mutation. This relates to constrains on the secondary structure of the pgRNA which constitutes the replicative intermediate used as template for the synthesis of the negative sense DNA strand by reverse transcription during the replication of the virus. The precore sequence forms the epsilon encapsidation signal, the secondary structure of which is critical for encapsidation but also for binding of the viral polymerase and synthesis of a short DNA primer that initiates DNA synthesis. Genotypes (A, C, F) which possess a C at position 1858 form a canonical Watson-Crick pair with the G at position 1896 as seen in wild-type strains. The remaining genotypes with U at 1856 can form a classical base pair with the precore change A1896 without compromising the epsilon structure.
Biology of the core promoter region: The BCP has a pivotal role in the replication of the virus by directing the synthesis of both the precore and pgRNAs. The BCP overlaps with the 3’ end of the X ORF and the 5’ end of the precore region, and contains cis-acting elements that can independently direct transcription of the precore mRNA and pgRNA, both of which are about 3.5 kb in length[22]. The pgRNA starts 3’ to the precore AUG and is translated into the core and polymerase proteins, but in addition serves as the template for the synthesis of the negative DNA strand of the virus by reverse transcription as stated above (Figure 2)[23]. The precore mRNA, which is slightly longer than the pgRNA and is initiated upstream of the precore start codon, is the template for the translation of the precore/core protein for HBeAg production, as already described. Mutations in the core promoter region may have repercussions on viral gene expression and/or replication, with a concurrent impact on viral pathogenesis.
Basic core promoter variants: A double mutation in the BCP leading to substitutions A1762T and G1764A[24] has been associated either with HBeAg negativity or reduced HBeAg production, as demonstrated by transfection studies[25,26]. There is evidence to suggest that the double BCP mutation results in decreased levels of precore mRNA and therefore diminished production of HBeAg, whilst increased viral replication has been reported as a result of upregulation of pgRNA production, promoting encapsidation and core protein production[27]. The latter finding however remains controversial. The BCP mutations are thought to affect the nucleotide motif of a transcription factor binding site which in turn leads to the differences seen in transcript levels.
The BCP double mutation has been found in patients regardless of HBeAg status. In anti-HBe-positive patients this mutation is often accompanied by a change at position 1753, from T to C or G. Other point mutations upstream and downstream of T1762/A1764 have also been described, occurring either alone or in combination with the double mutation, and in different settings, including chronic hepatitis, fulminant hepatitis B and HCC. Such mutations include T1753C and/or C1766T. Site-directed mutagenesis studies have shown that combined mutations of 1762/1764/1766 and 1753/1762/1764/1766 resulted in higher replication rates and lower HBeAg expression than the 1762/1764 mutations alone[28].
CLINICAL SIGNIFICANCE OF HBeAg NEGATIVE VARIANTS
Precore stop codon variant
Epidemiology: In the early 1980s, Hadziyannis et al[29] reported that HBV could replicate in the absence of HBeAg. This finding was subsequently attributed to the presence of the T1896A mutation in the precore region of the viral genome[21]. The precore variant frequency varies worldwide and is determined by the prevailing virus genotype[30]. In Mediterranean countries where HBV genotype D is predominant, practically all HBeAg negative CHB patients harbour the precore variant[31-33]. In China, where genotypes B and C predominate, 38% of the HBeAg patients harbour the precore variant, 42% the BCP variant and 12% both[34]. The prevalence of HBeAg negative CHB has been increasing over the last few decades probably reflecting the aging of existing HBV carriers and the effective prevention measures restricting new HBV infections[35,36]. In a previous study from Italy, HBeAg negative CHB prevalence rose from 41% during the 1975-1985 period to 90% during the 1990s[33,37]. The predominance of HBeAg negative CHB nowadays has also been supported by a French study[38].
The precore variant emerges during the course of HBV infection particularly in patients infected at birth or in the early years of life from infected family members[30,39]. Thus, the development of HBeAg negative CHB caused by the precore variant is a multivariate process depending on the age and mode of transmission, duration of the infection and sex[30,40]. In Greece, the age of acquisition of HBV infection cannot easily be defined, but it seems that intrafamilial spread during the early years of life is the most common mode of transmission[30]. In this respect, 40% of siblings of HBeAg negative CHB patients are also HBsAg/anti-HBe positive, while drug addicts, homosexuals and multiply transfused patients with a different mode of transmission are rarely HBsAg/anti-HBe positive[31]. In HBeAg negative CHB patients, male sex predominates with male to the female ratio varying between 4.6-17[30].
Epidemiological studies have shown that HBeAg negative CHB with the precore variant can take years or decades after HBeAg seroconversion to become predominant[30,41]. Hence the age of HBeAg negative CHB patients ranges from 40 to 55 years[42,43], much older than HBeAg positive CHB patients from the same region. The epidemiology of precore and basal core promoter variants and their correlations with distribution of HBV genotypes are shown in Table 1.
Table 1.
Epidemiology of precore and basal core promoter variants and their correlation with hepatitis B virus genotype distribution[15-18,30]
| Genotype | Region | Precore variant | Basal core promoter variant |
| B, C | East and South East Asia | Yes | Yes |
| A | Northern Europe, North America, Central Africa | No | Yes |
| D | Southern Europe, Middle East, Indian subcontinent | Yes | No |
| E | West and Sub-Saharan Africa | Yes | No |
| F | Indian Population of Central and South America | No | No |
Emergence and selection of precore mutants: The precore variant is either co-transmitted in acute infection or it emerges and then coexists in very small amounts with the predominant wild type virus during the latter period of the HBeAg positive phase (immune tolerant phase) of the disease[30,40,44]. It is selected at, or after, seroconversion to anti-HBe (immune clearance or seroconversion phase), whilst the HBeAg-producing strain gradually diminishes and is eventually cleared. This process can take years, during which time a mixture of both strains is usually seen[45,46]. Most patients after HBeAg seroconversion become “inactive HBsAg carriers” (immune control or “inactive HBsAg carrier” phase)[47]. However, some of these patients after HBeAg seroconversion develop high viraemia accompanied by necro-inflammatory activity on liver biopsy (reactivation phase)[48].
It is noteworthy that in patients with high viral replication and normal aminotransferases during the HBeAg positive phase, the precore/core encoding sequence on analysis shows only very few, if any mutations[49-51]. Core protein amino-acid substitutions within T helper cell epitopes are involved in the control of the infection, while those in B cell epitopes may influence the re-emergence of the precore variant and reactivation of the infection[52]. During the reactivation phase of HBV infection, amino acid substitutions in the core protein are frequent[53-55]. Some investigators have suggested that an important selection pressure in HBeAg negative CHB is the presence of anti-HBe in the absence of an adequate CTL response, that renders the precore variant less vulnerable to immune clearance compared to the wild type virus[56]. However, others have demonstrated increased overall CD4+ T cell responses in patients with HBeAg negative CHB, comparable to those of acute hepatitis B[57].
Clinical presentation and prognosis of HBeAg negative CHB patients: HBeAg negative CHB patients are usually asymptomatic. CHB is diagnosed incidentally either after screening for HBsAg for blood donation or after diagnosis of CHB in a family member or because of detection of elevated AST/ALT values. Some patients are not aware of their CHB status until they have developed cirrhosis with complications or hepatocellular carcinoma. Clinically overt exacerbations with jaundice may also occur[30,48].
The definition of “inactive HBsAg carrier” requires the presence of serum HBV DNA levels below 2000 IU/mL and persistently normal serum aminotransferases (below 40 IU/mL) at least every 3-4 mo for 1 year[58]. The “inactive carrier” state may persist indefinitely and has excellent prognosis for survival. However, long-term longitudinal studies of adult inactive carriers have reported that loss of the inactive state and progression to CHB, usually HBeAg-negative, may occur with a cumulative rate varying from 10% to more than 30% with an annual frequency ranging from 0.9% to 3%[59-65]. The frequencies of reactivation during follow-up vary according to the endemicity of HBV in a particular area, age of spontaneous HBeAg seroconversion, mode and age of transmission, HBV genotype and viraemia levels at entry in the study[39,47]. The duration of the inactive HBsAg carrier state usually lasts for years to decades but some patients progress directly from HBeAg positive to HBeAg negative CHB[62]. The HBeAg negative CHB stage is also characterized by older age as it represents the later phase in the course of chronic liver disease, high HBV DNA levels but lower than HBeAg positive CHB, liver necroinflammation and positive HBcAg staining in the liver[66]. Many of them have cirrhosis at the time of first presentation[7,62,37,64].
During the natural course of chronic HBV infection, HBsAg serum levels decline progressively from the immune tolerant to the low replicative phase[10,11]. A positive correlation was observed between HBsAg titer and serum HBV DNA or intrahepatic cccDNA levels in HBeAg positive patients. In HBeAg negative patients all three parameters decrease compared to HBeAg positive patients. However the correlation between HBsAg titer and serum HBV DNA or intrahepatic cccDNA levels weakens in HBeAg negative patients because in such patients HBsAg is either virion-associated, forms subviral HBsAg particles or is produced from integrated sequences[67]. Thus additional markers such as HBV DNA are used in combination with HBsAg quantification to differentiate inactive carriers from patients with chronic hepatitis. Genotypes may also play a role in HBsAg levels and it is evident that B/C genotypes have lower HBsAg levels than A/D ones[68].
Differentiating HBeAg negative patients into two groups i.e., those with chronic hepatitis and inactive carriers, is very important as the former could benefit from treatment. Recently, many investigators by using HBsAg quantification alone or in combination with HBV DNA levels, have tried to diagnose accurately inactive carriers from active disease and identify those who are going to clear HBsAg[12].
Thus, HBsAg levels < 1000 IU/mL and HBV DNA < 2000 IU/mL at a single time point assessed in Italian patients with genotype D identified inactive carriers with a positive predictive value of 88%[69]. In addition, HBsAg levels < 100 IU/mL predicted clearance of HBsAg with high specificity and sensitivity during follow-up[68]. In a recent report where 129 HBeAg negative CHB patients with normal ALT at baseline and genotypes A-E were included, applying combined HBsAg levels > 1000 IU/mL and HBV DNA > 200 IU/mL cutoffs at a single time point, accurately differentiated patients who reactivated[70]. The utility of HBsAg quantification with or without HBV DNA level determinations in asymptomatic HBeAg negative CHB in predicting either those who are going to seroclear HBsAg or those who are going to reactivate are listed in Table 2.
Table 2.
Clinical importance of hepatitis B surface antigen quantification in hepatitis B surface antigen negative patients in either predicting hepatitis B surface antigen seroclearance or differentiating inactive carriers from chronic hepatitis patients
| Ref. |
Studies predicting HBsAg seroclearance |
||
| Study design | HBsAg levels | Reliability of prediction | |
| Chan et al[68] | Genotype B/C, longitudinal study for 11 yr | HBsAg < 100 IU/mL | 75% sensitivity and 91% specificity |
| Chan et al[85] | Longitudinal study for 99 ± 16 mo | HBsAg levels < 1000 IU/mL and HBV DNA < 2000 IU/mL | Cumulative probability of 9% and 21% at 5 and 8 yr respectively |
| Tseng et al[86] | Follow-up at 1 yr after spontaneous HBeAg seroclearance | HBsAg < 100 IU/mL vs 100-999 IU/mL | Hazard ratio 24.3 vs 4.4 for HBsAg seroclearance |
| Tseng et al[87] | Genotype B/C follow-up of 11.6 yr | HBV DNA < 2000 IU/mL and HBsAg < 10 IU/mL | Adjusted hazard ratio of HBsAg loss was 13.2 |
| Martinot-Peignoux et al[70] | Follow-up of 1 yr | HBsAg < 1000 IU/mL, annual decrease > 0.3 log IU/mL | 95%NPV and 89% PPV |
| Differentiation of inactive disease from chronic hepatitis | |||
| Brunetto et al[69] | Genotype D, Follow-up for 34.5 mo | HBsAg levels < 1000 IU/mL and HBV DNA < 2000 IU/mL | 88% NPV and 97% PPV to identify inactive carriers |
| Martinot-Peignoux et al[70] | Follow-up of 1 yr | HBsAg levels > 1000 IU/mL and HBV DNA > 200 IU/mL | 96% NPV and 92% sensitivity to identify reactivation |
| Larsson et al[88] | Single time point evaluation of ALT, histological score | HBsAg levels < 1000 IU/mL and HBV DNA < 10000 IU/mL | 96% PV to identify inactive carriers |
| Park et al[89] | Genotype C follow-up > 48 mo | HBsAg levels > 850 IU/mL and HBV DNA > 850 IU/mL | 85% diagnostic accuracy to identify reactivation |
HBV: Hepatitis B virus; NPV: Negative predictive value; HBsAg: Hepatitis B surface antigen; PPV: Positive predictive value.
During follow-up, there are two patterns of HBeAg negative CHB activity: (1) persistently high viral replication and elevated ALT/AST without periods of remission; and (2) episodes of exacerbation and remission. During periods of exacerbation, AST/ALT elevation is preceded by a rise in HBV DNA to high levels. AST/ALT elevations usually resemble acute hepatitis episodes and are accompanied by jaundice and high anti-HBc IgM levels[30,48]. In the absence of a previous CHB diagnosis these episodes may therefore be misconceived as de novo acute hepatitis B. The periods of remission may be short or long lasting. In the latter case, the patient may be misclassified as an “inactive HBsAg carrier”. The 20%-30% of patients with histologically documented CHB have normal ALT/AST at the time of presentation[7]. However spontaneous sustained remissions of the disease activity are rare[64].
The incidence rate of cirrhosis in HBeAg negative CHB patients is 2.8 and 9.7 per 100 person-years in East Asian and European countries respectively and the corresponding 5-year cumulative risk for cirrhosis is 13% and 38%[60,62-64]. The corresponding rates for HBeAg positive CHB are 1.6 and 3.8 per 100 person-years and the corresponding 5-year cumulative risk for cirrhosis 8% and 17%[62,64,69,70]. The higher rates of cirrhosis development in HBeAg negative CHB reflect the advanced stage of liver disease at presentation and diagnosis.
Basal core promoter variants
The biological significance of the double A1762T/G1764A mutation is still under investigation, particularly in relation to the precore stop-codon mutation. The presence of the double mutation is clearly associated with down-regulation of HBeAg production[71-73], as demonstrated by transfection studies. The double BCP mutation results in decreased levels of the precore mRNA and therefore diminished production of HBeAg[27,74]. Despite the low levels of precore mRNA, increased viral replication as a result of upregulation of pgRNA production, promoting encapsidation and core protein production have been described[26]. HBeAg negativity with the BCP mutations has been associated with severe liver disease[75-78]. In addition BCP mutations are more frequently associated with genotype C than genotype B[77,78] and this finding partly explains the association between genotype C and more severe liver histological findings[79]. The double mutation is associated with increased risk of developing hepatocellular carcinoma[70] independently of HBV genotype[78,80,81] and viral load[81,82]. The overlapping of the BCP A1762T/G1764A mutations with the HBx protein encoding region may explain this association as the protein has oncogenic properties[83,84]. A combination of the A1896 mutation, BCP A1762T/G1764A mutations and pre-S deletions has been found with high prevalence in patients with chronic liver disease[80].
CONCLUSION
HBeAg negative chronic hepatitis B constitutes the predominant patient group with chronic hepatitis B nowadays. The ‘‘inactive carrier” and reactivation state are two sides of the same coin as the former may persist indefinitely or progress to chronic hepatitis B during long-term carriage. The differentiation of inactive carriers from HBeAg negative chronic hepatitis B patients is important as the former have a low incidence of cirrhosis and hepatocellular carcinoma while the latter are associated with a high risk of developing cirrhosis and hepatocellular carcinoma and therefore may benefit more from timely antiviral treatment.
Footnotes
P- Reviewers: Liu CJ, Rajeshwari K S- Editor: Qi Y L- Editor: A E- Editor: Wang CH
References
- 1.Lee WM. Hepatitis B virus infection. N Engl J Med. 1997;337:1733–1745. doi: 10.1056/NEJM199712113372406. [DOI] [PubMed] [Google Scholar]
- 2.Jiang D, Guo H, Xu C, Chang J, Gu B, Wang L, Block TM, Guo JT. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J Virol. 2008;82:1665–1678. doi: 10.1128/JVI.02113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45:529–538. doi: 10.1016/j.jhep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Coursaget P, Yvonnet B, Chotard J, Vincelot P, Sarr M, Diouf C, Chiron JP, Diop-Mar I. Age- and sex-related study of hepatitis B virus chronic carrier state in infants from an endemic area (Senegal) J Med Virol. 1987;22:1–5. doi: 10.1002/jmv.1890220102. [DOI] [PubMed] [Google Scholar]
- 5.McMahon BJ, Alward WL, Hall DB, Heyward WL, Bender TR, Francis DP, Maynard JE. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis. 1985;151:599–603. doi: 10.1093/infdis/151.4.599. [DOI] [PubMed] [Google Scholar]
- 6.Tassopoulos NC, Papaevangelou GJ, Sjogren MH, Roumeliotou-Karayannis A, Gerin JL, Purcell RH. Natural history of acute hepatitis B surface antigen-positive hepatitis in Greek adults. Gastroenterology. 1987;92:1844–1850. doi: 10.1016/0016-5085(87)90614-7. [DOI] [PubMed] [Google Scholar]
- 7.Hadziyannis SJ, Papatheodoridis GV. Hepatitis B e antigen-negative chronic hepatitis B: natural history and treatment. Semin Liver Dis. 2006;26:130–141. doi: 10.1055/s-2006-939751. [DOI] [PubMed] [Google Scholar]
- 8.Lai CL, Lok AS, Lin HJ, Wu PC, Yeoh EK, Yeung CY. Placebo-controlled trial of recombinant alpha 2-interferon in Chinese HBsAg-carrier children. Lancet. 1987;2:877–880. doi: 10.1016/s0140-6736(87)91371-7. [DOI] [PubMed] [Google Scholar]
- 9.Lok AS, Lai CL, Wu PC, Lau JY, Leung EK, Wong LS. Treatment of chronic hepatitis B with interferon: experience in Asian patients. Semin Liver Dis. 1989;9:249–253. doi: 10.1055/s-2008-1040518. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen T, Desmond P, Locarnini S. The role of quantitative hepatitis B serology in the natural history and management of chronic hepatitis B. Hepatol Int. 2009;3:5–15. doi: 10.1007/s12072-009-9149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaroszewicz J, Calle Serrano B, Wursthorn K, Deterding K, Schlue J, Raupach R, Flisiak R, Bock CT, Manns MP, Wedemeyer H, et al. Hepatitis B surface antigen (HBsAg) levels in the natural history of hepatitis B virus (HBV)-infection: a European perspective. J Hepatol. 2010;52:514–522. doi: 10.1016/j.jhep.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 12.Perrillo RP. Treatment of chronic hepatitis B with interferon: experience in western countries. Semin Liver Dis. 1989;9:240–248. doi: 10.1055/s-2008-1040517. [DOI] [PubMed] [Google Scholar]
- 13.Karayiannis P. Serum hepatitis B surface antigen levels and their utility as a predictor of sustained virological response after antiviral treatment. Hepat Mon. 2012;12:420–422. doi: 10.5812/hepatmon.6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Norder H, Couroucé AM, Magnius LO. Complete genomes, phylogenetic relatedness, and structural proteins of six strains of the hepatitis B virus, four of which represent two new genotypes. Virology. 1994;198:489–503. doi: 10.1006/viro.1994.1060. [DOI] [PubMed] [Google Scholar]
- 16.Lindh M, Andersson AS, Gusdal A. Genotypes, nt 1858 variants, and geographic origin of hepatitis B virus--large-scale analysis using a new genotyping method. J Infect Dis. 1997;175:1285–1293. doi: 10.1086/516458. [DOI] [PubMed] [Google Scholar]
- 17.Naumann H, Schaefer S, Yoshida CF, Gaspar AM, Repp R, Gerlich WH. Identification of a new hepatitis B virus (HBV) genotype from Brazil that expresses HBV surface antigen subtype adw4. J Gen Virol. 1993;74(Pt 8):1627–1632. doi: 10.1099/0022-1317-74-8-1627. [DOI] [PubMed] [Google Scholar]
- 18.Magnius LO, Norder H. Subtypes, genotypes and molecular epidemiology of the hepatitis B virus as reflected by sequence variability of the S-gene. Intervirology. 1995;38:24–34. doi: 10.1159/000150411. [DOI] [PubMed] [Google Scholar]
- 19.Mimms L. Hepatitis B virus escape mutants: “pushing the envelope” of chronic hepatitis B virus infection. Hepatology. 1995;21:884–887. doi: 10.1002/hep.1840210341. [DOI] [PubMed] [Google Scholar]
- 20.Ou JH, Laub O, Rutter WJ. Hepatitis B virus gene function: the precore region targets the core antigen to cellular membranes and causes the secretion of the e antigen. Proc Natl Acad Sci USA. 1986;83:1578–1582. doi: 10.1073/pnas.83.6.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carman WF, Jacyna MR, Hadziyannis S, Karayiannis P, McGarvey MJ, Makris A, Thomas HC. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet. 1989;2:588–591. doi: 10.1016/s0140-6736(89)90713-7. [DOI] [PubMed] [Google Scholar]
- 22.Yu X, Mertz JE. Promoters for synthesis of the pre-C and pregenomic mRNAs of human hepatitis B virus are genetically distinct and differentially regulated. J Virol. 1996;70:8719–8726. doi: 10.1128/jvi.70.12.8719-8726.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nassal M, Schaller H. Hepatitis B virus replication--an update. J Viral Hepat. 1996;3:217–226. doi: 10.1111/j.1365-2893.1996.tb00047.x. [DOI] [PubMed] [Google Scholar]
- 24.Protzer U, Nassal M, Chiang PW, Kirschfink M, Schaller H. Interferon gene transfer by a hepatitis B virus vector efficiently suppresses wild-type virus infection. Proc Natl Acad Sci USA. 1999;96:10818–10823. doi: 10.1073/pnas.96.19.10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckwold VE, Xu Z, Chen M, Yen TS, Ou JH. Effects of a naturally occurring mutation in the hepatitis B virus basal core promoter on precore gene expression and viral replication. J Virol. 1996;70:5845–5851. doi: 10.1128/jvi.70.9.5845-5851.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moriyama K, Okamoto H, Tsuda F, Mayumi M. Reduced precore transcription and enhanced core-pregenome transcription of hepatitis B virus DNA after replacement of the precore-core promoter with sequences associated with e antigen-seronegative persistent infections. Virology. 1996;226:269–280. doi: 10.1006/viro.1996.0655. [DOI] [PubMed] [Google Scholar]
- 27.Laras A, Koskinas J, Hadziyannis SJ. In vivo suppression of precore mRNA synthesis is associated with mutations in the hepatitis B virus core promoter. Virology. 2002;295:86–96. doi: 10.1006/viro.2001.1352. [DOI] [PubMed] [Google Scholar]
- 28.Parekh S, Zoulim F, Ahn SH, Tsai A, Li J, Kawai S, Khan N, Trépo C, Wands J, Tong S. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J Virol. 2003;77:6601–6612. doi: 10.1128/JVI.77.12.6601-6612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hadziyannis SJ, Lieberman HM, Karvountzis GG, Shafritz DA. Analysis of liver disease, nuclear HBcAg, viral replication, and hepatitis B virus DNA in liver and serum of HBeAg Vs. anti-HBe positive carriers of hepatitis B virus. Hepatology. 1983;3:656–662. doi: 10.1002/hep.1840030505. [DOI] [PubMed] [Google Scholar]
- 30.Hadziyannis SJ, Vassilopoulos D. Hepatitis B e antigen-negative chronic hepatitis B. Hepatology. 2001;34:617–624. doi: 10.1053/jhep.2001.27834. [DOI] [PubMed] [Google Scholar]
- 31.Laras A, Koskinas J, Avgidis K, Hadziyannis SJ. Incidence and clinical significance of hepatitis B virus precore gene translation initiation mutations in e antigen-negative patients. J Viral Hepat. 1998;5:241–248. doi: 10.1046/j.1365-2893.1998.00109.x. [DOI] [PubMed] [Google Scholar]
- 32.Grandjacques C, Pradat P, Stuyver L, Chevallier M, Chevallier P, Pichoud C, Maisonnas M, Trépo C, Zoulim F. Rapid detection of genotypes and mutations in the pre-core promoter and the pre-core region of hepatitis B virus genome: correlation with viral persistence and disease severity. J Hepatol. 2000;33:430–439. doi: 10.1016/s0168-8278(00)80279-2. [DOI] [PubMed] [Google Scholar]
- 33.Rizzetto M, Volpes R, Smedile A. Response of pre-core mutant chronic hepatitis B infection to lamivudine. J Med Virol. 2000;61:398–402. [PubMed] [Google Scholar]
- 34.Chan HL, Hussain M, Lok AS. Different hepatitis B virus genotypes are associated with different mutations in the core promoter and precore regions during hepatitis B e antigen seroconversion. Hepatology. 1999;29:976–984. doi: 10.1002/hep.510290352. [DOI] [PubMed] [Google Scholar]
- 35.Funk ML, Rosenberg DM, Lok AS. World-wide epidemiology of HBeAg-negative chronic hepatitis B and associated precore and core promoter variants. J Viral Hepat. 2002;9:52–61. doi: 10.1046/j.1365-2893.2002.00304.x. [DOI] [PubMed] [Google Scholar]
- 36.Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology. 2006;43:S173–S181. doi: 10.1002/hep.20956. [DOI] [PubMed] [Google Scholar]
- 37.Gaeta GB, Stornaiuolo G, Precone DF, Lobello S, Chiaramonte M, Stroffolini T, Colucci G, Rizzetto M. Epidemiological and clinical burden of chronic hepatitis B virus/hepatitis C virus infection. A multicenter Italian study. J Hepatol. 2003;39:1036–1041. doi: 10.1016/s0168-8278(03)00470-7. [DOI] [PubMed] [Google Scholar]
- 38.Zarski JP, Marcellin P, Leroy V, Trepo C, Samuel D, Ganne-Carrie N, Barange K, Canva V, Doffoel M, Cales P. Characteristics of patients with chronic hepatitis B in France: predominant frequency of HBe antigen negative cases. J Hepatol. 2006;45:355–360. doi: 10.1016/j.jhep.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Hadziyannis SJ. Natural history of chronic hepatitis B in Euro-Mediterranean and African countries. J Hepatol. 2011;55:183–191. doi: 10.1016/j.jhep.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 40.Chan HL, Hui Y, Leung NW, Ching JY, Chan FK, Sung JJ. Risk factors for active liver disease in HBeAg-negative chronic hepatitis B virus-infected patients. Am J Gastroenterol. 2000;95:3547–3551. doi: 10.1111/j.1572-0241.2000.03373.x. [DOI] [PubMed] [Google Scholar]
- 41.Brunetto MR, Giarin MM, Oliveri F, Chiaberge E, Baldi M, Alfarano A, Serra A, Saracco G, Verme G, Will H. Wild-type and e antigen-minus hepatitis B viruses and course of chronic hepatitis. Proc Natl Acad Sci USA. 1991;88:4186–4190. doi: 10.1073/pnas.88.10.4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lok AS, Hussain M, Cursano C, Margotti M, Gramenzi A, Grazi GL, Jovine E, Benardi M, Andreone P. Evolution of hepatitis B virus polymerase gene mutations in hepatitis B e antigen-negative patients receiving lamivudine therapy. Hepatology. 2000;32:1145–1153. doi: 10.1053/jhep.2000.19622. [DOI] [PubMed] [Google Scholar]
- 43.Tassopoulos NC, Volpes R, Pastore G, Heathcote J, Buti M, Goldin RD, Hawley S, Barber J, Condreay L, Gray DF. Efficacy of lamivudine in patients with hepatitis B e antigen-negative/hepatitis B virus DNA-positive (precore mutant) chronic hepatitis B. Lamivudine Precore Mutant Study Group. Hepatology. 1999;29:889–896. doi: 10.1002/hep.510290321. [DOI] [PubMed] [Google Scholar]
- 44.Chang MH, Hsu HY, Ni YH, Tsai KS, Lee PI, Chen PJ, Hsu YL, Chen DS. Precore stop codon mutant in chronic hepatitis B virus infection in children: its relation to hepatitis B e seroconversion and maternal hepatitis B surface antigen. J Hepatol. 1998;28:915–922. doi: 10.1016/s0168-8278(98)80337-1. [DOI] [PubMed] [Google Scholar]
- 45.Loriot MA, Marcellin P, Talbodec N, Guigonis V, Gigou M, Boyer N, Bezeaud A, Erlinger S, Benhamou JP. Low frequency of precore hepatitis B virus mutants in anti-hepatitis B e-positive reactivation after loss of hepatitis B e antigen in patients with chronic hepatitis B. Hepatology. 1995;21:627–631. [PubMed] [Google Scholar]
- 46.Lok AS, Akarca US, Greene S. Predictive value of precore hepatitis B virus mutations in spontaneous and interferon-induced hepatitis B e antigen clearance. Hepatology. 1995;21:19–24. [PubMed] [Google Scholar]
- 47.Hadziyannis SJ. Unrevealing the natural course of the so-called “inactive HBsAg or HBV carrier state”. Hepatol Int. 2007;1:281–284. doi: 10.1007/s12072-007-9004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hadziyannis SJ, Vassilopoulos D. Immunopathogenesis of hepatitis B e antigen negative chronic hepatitis B infection. Antiviral Res. 2001;52:91–98. doi: 10.1016/s0166-3542(01)00173-5. [DOI] [PubMed] [Google Scholar]
- 49.Bozkaya H, Akarca US, Ayola B, Lok AS. High degree of conservation in the hepatitis B virus core gene during the immune tolerant phase in perinatally acquired chronic hepatitis B virus infection. J Hepatol. 1997;26:508–516. doi: 10.1016/s0168-8278(97)80415-1. [DOI] [PubMed] [Google Scholar]
- 50.Akarca US, Lok AS. Naturally occurring hepatitis B virus core gene mutations. Hepatology. 1995;22:50–60. [PubMed] [Google Scholar]
- 51.Hannoun C, Horal P, Lindh M. Long-term mutation rates in the hepatitis B virus genome. J Gen Virol. 2000;81:75–83. doi: 10.1099/0022-1317-81-1-75. [DOI] [PubMed] [Google Scholar]
- 52.Carman WF, Boner W, Fattovich G, Colman K, Dornan ES, Thursz M, Hadziyannis S. Hepatitis B virus core protein mutations are concentrated in B cell epitopes in progressive disease and in T helper cell epitopes during clinical remission. J Infect Dis. 1997;175:1093–1100. doi: 10.1086/516447. [DOI] [PubMed] [Google Scholar]
- 53.Alexopoulou A, Karayiannis P, Hadziyannis SJ, Aiba N, Thomas HC. Emergence and selection of HBV variants in an anti-HBe positive patient persistently infected with quasi-species. J Hepatol. 1997;26:748–753. doi: 10.1016/s0168-8278(97)80238-3. [DOI] [PubMed] [Google Scholar]
- 54.Alexopoulou A, Owsianka AM, Kafiri G, Dourakis SP, Carman WF, Hadziyannis SJ. Core variability does not affect response to interferon alpha in HBeAg negative chronic hepatitis B. J Hepatol. 1998;29:345–351. doi: 10.1016/s0168-8278(98)80050-0. [DOI] [PubMed] [Google Scholar]
- 55.Alexopoulou A, Baltayiannis G, Eroglu C, Nastos T, Dourakis SP, Archimandritis AJ, Karayiannis P. Core mutations in patients with acute episodes of chronic HBV infection are associated with the emergence of new immune recognition sites and the development of high IgM anti-HBc index values. J Med Virol. 2009;81:34–41. doi: 10.1002/jmv.21337. [DOI] [PubMed] [Google Scholar]
- 56.Maruyama T, Mitsui H, Maekawa H, Yamada H, Hirayama M, Iino S, Yasuda K, Koike K, Kimura S, Milich DR. Emergence of the precore mutant late in chronic hepatitis B infection correlates with the severity of liver injury and mutations in the core region. Am J Gastroenterol. 2000;95:2894–2904. doi: 10.1111/j.1572-0241.2000.03201.x. [DOI] [PubMed] [Google Scholar]
- 57.Vassilopoulos D, Rapti I, Nikolaou M, Hadziyannis E, Hadziyannis SJ. Cellular immune responses in hepatitis B virus e antigen negative chronic hepatitis B. J Viral Hepat. 2008;15:817–826. doi: 10.1111/j.1365-2893.2008.00996.x. [DOI] [PubMed] [Google Scholar]
- 58.European Association For The Study Of The Liver. EASL clinical practice guidelines: Management of chronic hepatitis B virus infection. J Hepatol. 2012;57:167–185. doi: 10.1016/j.jhep.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 59.Papatheodoridis GV, Chrysanthos N, Hadziyannis E, Cholongitas E, Manesis EK. Longitudinal changes in serum HBV DNA levels and predictors of progression during the natural course of HBeAg-negative chronic hepatitis B virus infection. J Viral Hepat. 2008;15:434–441. doi: 10.1111/j.1365-2893.2007.00957.x. [DOI] [PubMed] [Google Scholar]
- 60.Manno M, Cammà C, Schepis F, Bassi F, Gelmini R, Giannini F, Miselli F, Grottola A, Ferretti I, Vecchi C, et al. Natural history of chronic HBV carriers in northern Italy: morbidity and mortality after 30 years. Gastroenterology. 2004;127:756–763. doi: 10.1053/j.gastro.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 61.Manesis EK, Papatheodoridis GV, Sevastianos V, Cholongitas E, Papaioannou C, Hadziyannis SJ. Significance of hepatitis B viremia levels determined by a quantitative polymerase chain reaction assay in patients with hepatitis B e antigen-negative chronic hepatitis B virus infection. Am J Gastroenterol. 2003;98:2261–2267. doi: 10.1111/j.1572-0241.2003.07715.x. [DOI] [PubMed] [Google Scholar]
- 62.Hsu YS, Chien RN, Yeh CT, Sheen IS, Chiou HY, Chu CM, Liaw YF. Long-term outcome after spontaneous HBeAg seroconversion in patients with chronic hepatitis B. Hepatology. 2002;35:1522–1527. doi: 10.1053/jhep.2002.33638. [DOI] [PubMed] [Google Scholar]
- 63.Chu CM, Hung SJ, Lin J, Tai DI, Liaw YF. Natural history of hepatitis B e antigen to antibody seroconversion in patients with normal serum aminotransferase levels. Am J Med. 2004;116:829–834. doi: 10.1016/j.amjmed.2003.12.040. [DOI] [PubMed] [Google Scholar]
- 64.Fattovich G, Bortolotti F, Donato F. Natural history of chronic hepatitis B: special emphasis on disease progression and prognostic factors. J Hepatol. 2008;48:335–352. doi: 10.1016/j.jhep.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 65.Fattovich G, Olivari N, Pasino M, D’Onofrio M, Martone E, Donato F. Long-term outcome of chronic hepatitis B in Caucasian patients: mortality after 25 years. Gut. 2008;57:84–90. doi: 10.1136/gut.2007.128496. [DOI] [PubMed] [Google Scholar]
- 66.Hadziyannis SJ, Bramou T, Alexopoulou A, Makris A. Immunopathogenesis and natural course of anti-HBe positive chronic hepatitis and natural course of anti-HBe positive chronic hepatitis with replicating B virus. In: Hollinger FB, Lemon SM, Margolis H, editors. Viral Hepatitis and Liver Disease. Baltimore: Williams and Wilkins; 1991. pp. 673–676. [Google Scholar]
- 67.Manesis EK, Papatheodoridis GV, Tiniakos DG, Hadziyannis ES, Agelopoulou OP, Syminelaki T, Papaioannou C, Nastos T, Karayiannis P. Hepatitis B surface antigen: relation to hepatitis B replication parameters in HBeAg-negative chronic hepatitis B. J Hepatol. 2011;55:61–68. doi: 10.1016/j.jhep.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 68.Chan HL, Wong GL, Tse CH, Chan HY, Wong VW. Viral determinants of hepatitis B surface antigen seroclearance in hepatitis B e antigen-negative chronic hepatitis B patients. J Infect Dis. 2011;204:408–414. doi: 10.1093/infdis/jir283. [DOI] [PubMed] [Google Scholar]
- 69.Brunetto MR, Oliveri F, Colombatto P, Moriconi F, Ciccorossi P, Coco B, Romagnoli V, Cherubini B, Moscato G, Maina AM, et al. Hepatitis B surface antigen serum levels help to distinguish active from inactive hepatitis B virus genotype D carriers. Gastroenterology. 2010;139:483–490. doi: 10.1053/j.gastro.2010.04.052. [DOI] [PubMed] [Google Scholar]
- 70.Martinot-Peignoux M, Lapalus M, Laouénan C, Lada O, Netto-Cardoso AC, Boyer N, Ripault MP, Carvalho-Filho R, Asselah T, Marcellin P. Prediction of disease reactivation in asymptomatic hepatitis B e antigen-negative chronic hepatitis B patients using baseline serum measurements of HBsAg and HBV-DNA. J Clin Virol. 2013;58:401–407. doi: 10.1016/j.jcv.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 71.Kurosaki M, Enomoto N, Asahina Y, Sakuma I, Ikeda T, Tozuka S, Izumi N, Marumo F, Sato C. Mutations in the core promoter region of hepatitis B virus in patients with chronic hepatitis B. J Med Virol. 1996;49:115–123. doi: 10.1002/(SICI)1096-9071(199606)49:2<115::AID-JMV8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 72.Sato S, Suzuki K, Akahane Y, Akamatsu K, Akiyama K, Yunomura K, Tsuda F, Tanaka T, Okamoto H, Miyakawa Y, et al. Hepatitis B virus strains with mutations in the core promoter in patients with fulminant hepatitis. Ann Intern Med. 1995;122:241–248. doi: 10.7326/0003-4819-122-4-199502150-00001. [DOI] [PubMed] [Google Scholar]
- 73.Takahashi K, Aoyama K, Ohno N, Iwata K, Akahane Y, Baba K, Yoshizawa H, Mishiro S. The precore/core promoter mutant (T1762A1764) of hepatitis B virus: clinical significance and an easy method for detection. J Gen Virol. 1995;76(Pt 12):3159–3164. doi: 10.1099/0022-1317-76-12-3159. [DOI] [PubMed] [Google Scholar]
- 74.Buckwold VE, Xu Z, Yen TS, Ou JH. Effects of a frequent double-nucleotide basal core promoter mutation and its putative single-nucleotide precursor mutations on hepatitis B virus gene expression and replication. J Gen Virol. 1997;78(Pt 8):2055–2065. doi: 10.1099/0022-1317-78-8-2055. [DOI] [PubMed] [Google Scholar]
- 75.Kidd-Ljunggren K, Oberg M, Kidd AH. Hepatitis B virus X gene 1751 to 1764 mutations: implications for HBeAg status and disease. J Gen Virol. 1997;78(Pt 6):1469–1478. doi: 10.1099/0022-1317-78-6-1469. [DOI] [PubMed] [Google Scholar]
- 76.Lindh M, Gustavson C, Mårdberg K, Norkrans G, Dhillon AP, Horal P. Mutation of nucleotide 1,762 in the core promoter region during hepatitis B e seroconversion and its relation to liver damage in hepatitis B e antigen carriers. J Med Virol. 1998;55:185–190. doi: 10.1002/(sici)1096-9071(199807)55:3<185::aid-jmv1>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 77.Orito E, Mizokami M, Sakugawa H, Michitaka K, Ishikawa K, Ichida T, Okanoue T, Yotsuyanagi H, Iino S. A case-control study for clinical and molecular biological differences between hepatitis B viruses of genotypes B and C. Japan HBV Genotype Research Group. Hepatology. 2001;33:218–223. doi: 10.1053/jhep.2001.20532. [DOI] [PubMed] [Google Scholar]
- 78.Kao JH, Chen PJ, Lai MY, Chen DS. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology. 2003;124:327–334. doi: 10.1053/gast.2003.50053. [DOI] [PubMed] [Google Scholar]
- 79.Yuen MF, Tanaka Y, Ng IO, Mizokami M, Yuen JC, Wong DK, Yuan HJ, Sum SM, Chan AO, Lai CL. Hepatic necroinflammation and fibrosis in patients with genotypes Ba and C, core-promoter and precore mutations. J Viral Hepat. 2005;12:513–518. doi: 10.1111/j.1365-2893.2005.00629.x. [DOI] [PubMed] [Google Scholar]
- 80.Chen BF, Liu CJ, Jow GM, Chen PJ, Kao JH, Chen DS. High prevalence and mapping of pre-S deletion in hepatitis B virus carriers with progressive liver diseases. Gastroenterology. 2006;130:1153–1168. doi: 10.1053/j.gastro.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 81.Liu CJ, Chen BF, Chen PJ, Lai MY, Huang WL, Kao JH, Chen DS. Role of hepatitis B virus precore/core promoter mutations and serum viral load on noncirrhotic hepatocellular carcinoma: a case-control study. J Infect Dis. 2006;194:594–599. doi: 10.1086/505883. [DOI] [PubMed] [Google Scholar]
- 82.Liu CJ, Chen BF, Chen PJ, Lai MY, Huang WL, Kao JH, Chen DS. Role of hepatitis B viral load and basal core promoter mutation in hepatocellular carcinoma in hepatitis B carriers. J Infect Dis. 2006;193:1258–1265. doi: 10.1086/502978. [DOI] [PubMed] [Google Scholar]
- 83.Hsia CC, Yuwen H, Tabor E. Hot-spot mutations in hepatitis B virus X gene in hepatocellular carcinoma. Lancet. 1996;348:625–626. doi: 10.1016/S0140-6736(05)64851-9. [DOI] [PubMed] [Google Scholar]
- 84.Zheng Y, Li J, Ou JH. Regulation of hepatitis B virus core promoter by transcription factors HNF1 and HNF4 and the viral X protein. J Virol. 2004;78:6908–6914. doi: 10.1128/JVI.78.13.6908-6914.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chan HL, Wong VW, Wong GL, Tse CH, Chan HY, Sung JJ. A longitudinal study on the natural history of serum hepatitis B surface antigen changes in chronic hepatitis B. Hepatology. 2010;52:1232–1241. doi: 10.1002/hep.23803. [DOI] [PubMed] [Google Scholar]
- 86.Tseng TC, Liu CJ, Su TH, Wang CC, Chen CL, Chen PJ, Chen DS, Kao JH. Serum hepatitis B surface antigen levels predict surface antigen loss in hepatitis B e antigen seroconverters. Gastroenterology. 2011;141:517–525, 525.e1-2. doi: 10.1053/j.gastro.2011.04.046. [DOI] [PubMed] [Google Scholar]
- 87.Tseng TC, Liu CJ, Yang HC, Su TH, Wang CC, Chen CL, Kuo SF, Liu CH, Chen PJ, Chen DS, et al. Determinants of spontaneous surface antigen loss in hepatitis B e antigen-negative patients with a low viral load. Hepatology. 2012;55:68–76. doi: 10.1002/hep.24615. [DOI] [PubMed] [Google Scholar]
- 88.Larsson SB, Eilard A, Malmström S, Hannoun C, Dhillon AP, Norkrans G, Lindh M. HBsAg quantification for identification of liver disease in chronic hepatitis B virus carriers. Liver Int. 2013:Epub ahead of print. doi: 10.1111/liv.12345. [DOI] [PubMed] [Google Scholar]
- 89.Park H, Lee JM, Seo JH, Kim HS, Ahn SH, Kim do Y, Han KH, Chon CY, Park JY. Predictive value of HBsAg quantification for determining the clinical course of genotype C HBeAg-negative carriers. Liver Int. 2012;32:796–802. doi: 10.1111/j.1478-3231.2011.02693.x. [DOI] [PubMed] [Google Scholar]