

Abstract
At glutamatergic synapses, local endocytic recycling of AMPA receptors (AMPARs) is important for the supply of a mobile pool of AMPARs required for synaptic potentiation. This local recycling of AMPARs critically relies on the presence of an endocytic zone (EZ) near the postsynaptic density (PSD). The precise mechanisms that couple the EZ to the PSD still remain largely elusive, with the large GTPase Dynamin-3 and the multimeric PSD adaptor protein Homer1 as the two main players identified. Here, we demonstrate that a physical interaction between the X-linked mental retardation protein oligophrenin-1 (OPHN1) and Homer1b/c is crucial for the positioning of the EZ adjacent to the PSD, and present evidence that this interaction is important for OPHN1's role in controlling activity-dependent strengthening of excitatory synapses in the rat hippocampus. Disruption of the OPHN1-Homer1b/c interaction causes a displacement of EZs from the PSD, along with impaired AMPAR recycling and reduced AMPAR accumulation at synapses, in both basal conditions and conditions that can induce synaptic potentiation. Together, our findings unveil a novel role for OPHN1 as an interaction partner of Homer1b/c in spine EZ positioning, and provide new mechanistic insight into how genetic deficits in OPHN1 can lead to impaired synapse maturation and plasticity.
Keywords: endocytic zone, mental retardation, Rho-GAP, synaptic potentiation
Introduction
Activity-dependent changes in the strength of excitatory synapses are crucial for normal brain function, particularly during the development of synaptic connections and memory formation. One major mechanism that regulates synaptic strength involves the regulated trafficking of AMPA receptors (AMPARs) into and out of synapses, with increases in synaptic AMPARs typically resulting in long-term potentiation (LTP) of synaptic strength and their removal in long-term depression (LTD; Kessels and Malinow, 2009; Huganir and Nicoll, 2013). Increasing evidence indicates that synaptic delivery and retrieval of AMPARs, both during basal activity and synaptic plasticity, involve an intricate interplay between endo/exocytic AMPAR recycling events and lateral diffusion of surface AMPARs; the latter likely constitutes an obligatory step between intracellular pools of AMPARs and the synapse (Makino and Malinow, 2009; Henley et al., 2011; Anggono and Huganir, 2012; Choquet and Triller, 2013). How all these processes are precisely interconnected remains under intense investigation.
Interestingly, recent studies reported that endocytic zones (EZs), stably positioned sites of clathrin adjacent to the postsynaptic density (PSD), play a key role in capturing AMPARs diffusing out of the synapse, allowing for localized endocytosis and recycling of AMPARs (Blanpied et al., 2002; Lu et al., 2007). This localized endocytosis/recycling of AMPARs, which is sustained by the presence of EZs near the PSD, was subsequently shown to be pivotal for maintaining a mobile pool of surface AMPARs required for synaptic potentiation (Petrini et al., 2009). While the precise mechanisms that position the EZ near the PSD remain to be resolved, Ehlers et al. (Lu et al., 2007) demonstrated it to be dependent on a physical interaction between large GTPase Dynamin-3, a component of the clathrin endocytic machinery, and postsynaptic scaffold protein Homer1, which acts as an adaptor for multiple protein interactions at the PSD.
In a previous study, we had found that the X-linked mental retardation protein OPHN1 (Billuart et al., 1998), a Rho GTPase-activating protein (Rho-GAP), interacts with Homer1b/c and that the two proteins colocalize at excitatory synapses (Govek et al., 2004). OPHN1, at the hippocampal CA3-CA1 synapse, plays an important role in activity-dependent maturation and plasticity of excitatory synapses (Nadif Kasri et al., 2009), and both OPHN1's Rho-GAP activity and interaction with Homer1b/c are required for its role in regulating basal synaptic function and plasticity (Nadif Kasri et al., 2009, 2011; see Fig. 1, below). While OPHN1 through its Rho-GAP activity was reported to control AMPAR stabilization by locally modulating RhoA activity and actin dynamics (Nadif Kasri et al., 2009), it remained unclear how OPHN1 via its interaction with Homer1b/c contributes to synapse strengthening. Here, we demonstrate that a key mechanism involves the positioning of EZs near the PSD. Indeed, disruption of the OPHN1–Homer1b/c interaction causes a displacement of EZs from the PSD, along with impaired AMPAR recycling and reduced AMPAR accumulation at synapses. Thus, our data unveil a novel function for OPHN1 in spine EZ positioning and provide further insight into how loss-of-OPHN1 function leads to impaired synapse maturation and plasticity.
Figure 1.

OPHN1–Homer interaction is critical for LTP. A, B, LTP was induced in CA1 neurons infected with indicated lentivirus (A) or incubated with indicated peptides (B) using a pairing protocol. Top, Representative traces of AMPAR-mediated EPSCs before (1) and after (2) LTP induction. Calibration: 20 pA, 20 ms. Bottom, Normalized AMPAR-mediated EPSCs before and after LTP induction for induced pathway (closed symbols) and control pathway (open symbols). Error bars represent SEM; n = 7 (for A and B), *p < 0.05 measured between 40 and 45 min, Mann–Whitney test.
Materials and Methods
DNA/RNAi constructs, peptides, antibodies.
Plasmids expressing clathrin-DsRed and PSD-95-GFP were obtained from M. Ehlers and pCI-SEP-GluA1 from R. Malinow. cDNAs encoding Homer1b/c were cloned into pEGFP-C1 and pcDNA3.1 with addition of an N-terminal mCherry epitope-tag. Lentiviral vectors (pFUGW) expressing OPHN1#2 shRNA alone, or together with RNAi-resistant OPHN1Hom, were described previously (Nadif Kasri et al., 2011). Sequences of pep-OPHN1Hom, pep-contHom, and pep-OPHN1Endo were described previously (Nadif Kasri et al., 2011). HIV-1 Tat sequence was fused to peptides to aid delivery into cells. Primary antibodies used: anti-GluA1 (Millipore; catalog #MAB2263, RRID:AB_1977459), anti-synapsin-1 (Millipore; catalog #AB1543, RRID:AB_2200400), anti-Clathrin (Thermo Fisher; catalog #MA1-065, RRID:AB_2083179), and anti-PSD-95 (Millipore; catalog #MAB1596, RRID:AB_2092365).
Hippocampal slice cultures and dissociated neuronal cultures.
Dissociated hippocampal neurons were prepared from E18 rats of either sex, and transfected by calcium phosphate method after 14 d in vitro (14 DIV). Three to five days post transfection, neurons were incubated with peptides for 24 h (see Figs. 3A–D, 4), and imaged as indicated. Hippocampal slice cultures were prepared from P7 to P9 rats. Slices were infected after 1 DIV or treated with peptides after 7–9 DIV and used for electrophysiology at 8–10 DIV (Fig. 1), or biolistically transfected at 7–9 DIV, and used for imaging experiments 3–4 d after RNAi delivery or 24 h after peptide delivery (see Fig. 2A–E). Peptides were delivered by bath application. All animal care protocols were approved by Cold Spring Harbor Laboratory.
Figure 3.

Disruption of OPHN1–Homer interaction reduces the number of newly exocytosed AMPARs at synaptic sites in basal synaptic conditions. A, Representative pseudocolor images of SEP-GluA1 fluorescence in dendrites of cultured hippocampal neurons before (1), just after (2), and 20 min after (3) exposure to large bleach in boxed region. Arrow depicts example of SEP fluorescence return in spine after bleach. Scale bars, Left, 10 μm; right, 2 μm. B, Schematic of the experiment. C, Quantification of SEP-GluA1 fluorescence at synapses of neurons expressing SEP-GluA1 and mCherry-Homer and pretreated with no pep, pep-contHom, or pep-OPHN1Hom, before and after bleach at indicated time points. D, Quantitative analysis of SEP-GluA1 exocytosis at synapses 20 min after bleach. Error bars represent SEM; n = 237–645 synapses (for C and D), ***p < 0.001 against no pep (one-way ANOVA). E, Representative images of surface GluA1 in neurons pretreated with indicated peptides before antibody labeling assay. Synapsin-1 was used to mark synapses. Bottom, Enlarged view of dendritic segment boxed at the top. Scale bars, Top, 10 μm; bottom, 2 μm. F, Quantification of number of surface GluA1 puncta at synapses; n = 32–35 neurons from 3 cultures/condition. Error bar indicates SEM, ***p < 0.001 against no pep (one-way ANOVA).
Figure 4.
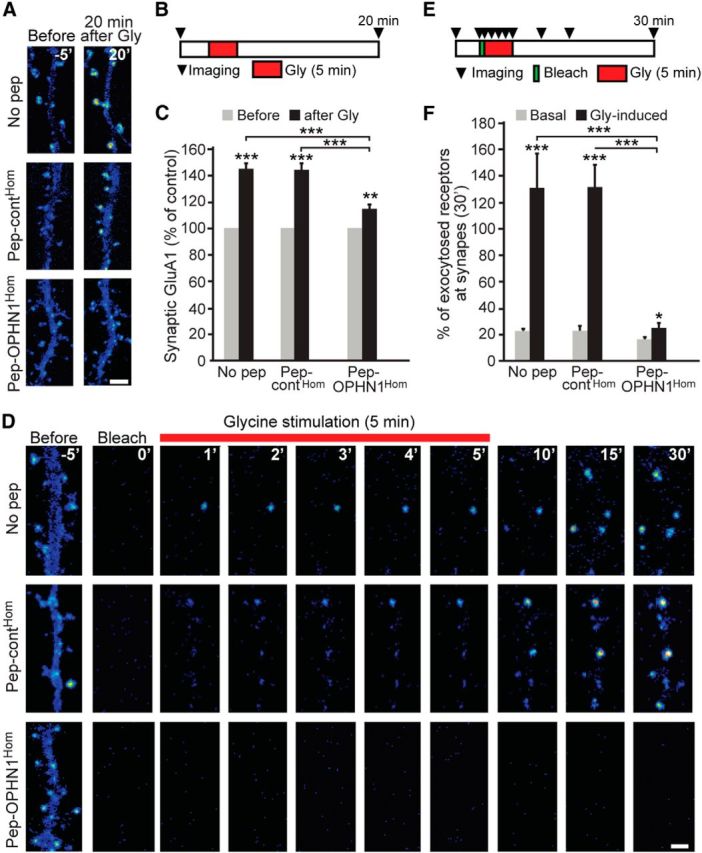
OPHN1–Homer interaction is essential for glycine-induced increase in receptor recycling/exocytosis and synaptic AMPAR number. A, Representative pseudocolor images of SEP-GluA1-expressing neurons pretreated with indicated peptides, taken 5 min before and 20 min after glycine stimulation. Scale bar, 2 μm. B, Schematic diagram for experiment in A. C, Quantification of surface synaptic SEP-GluA1 before and 20 min after glycine stimulation. Error bars represent SEM; n = 142–267 spines/synapses, **p < 0.01, ***p < 0.001 (t test, unpaired). D, Representative pseudocolor images of dendrites of SEP-GluA1-expressing neurons pretreated with indicated peptides, taken before bleach, just after bleach, and following glycine stimulation at indicated time points. Scale bar, 2 μm. In A and D, mCherry-Homer or tDimer was coexpressed to label synapses/spines; not depicted for clarity. E, Schematic diagram for experiment in D. F, Quantitative analysis of glycine-induced SEP-GluA1 exocytosis at synapses 30 min after bleach. Error bars represent SEM; n = 101–220 spines/synapses, *p < 0.05, ***p < 0.001 (t test, unpaired).
Figure 2.

OPHN1–Homer interaction is required for positioning EZ near PSD. A, Representative images of CA1 neurons expressing PSD-95-GFP and clathrin-DsRed together with indicated shRNA, or treated with indicated peptides. Scale bars, 2 μm. Arrowhead depicts EZ-negative synapse. B, Top, Domain structure of OPHN1. Homer1b/c and Endo2/3 binding sites are indicated. Bottom, Quantitative analysis of EZ-negative synapses. Error bars represent SEM; n = 268–414 (shRNA-experiment) and 504–1045 (peptide-experiment) spines/synapses, ***p < 0.001 against scr#1 for shRNA experiment (Student's t test) and against no pep for peptide experiment (one-way ANOVA). C, Representative images of CA1 neurons expressing Homer-GFP and clathrin-DsRed and treated with indicated peptides. Scale bar, 2 μm. D, Quantitative analysis of EZ-negative synapses. Error bars represent SEM; n = 778–839 spines/synapses, ***p < 0.001 against no pep (one-way ANOVA). E, Quantitative analysis of clathrin puncta. Error bars represent SEM; n = 6–8 neurons/condition, p > 0.05 against scr#1 or pep-contHom (Student's t test). F, Representative images of dissociated hippocampal neurons (18–20 DIV) treated with indicated peptides and immunostained for endogenous PSD-95 and clathrin heavy chain. G, Quantitative analysis of EZ-negative synapses. Error bars represent SEM; n = 828–912 spines/synapses, ***p < 0.001 against no pep (one-way ANOVA).
AMPAR surface labeling.
Hippocampal neuronal cultures (18–21 DIV) were pre-incubated with peptides for 48 h (see Fig. 3E,F) and live labeled with anti-GluA1 antibody for 10 min. Surface GluA1 was visualized with Alexa 488-conjugated goat anti-mouse secondary antibody. For costaining with synapsin-1, cells were permeabilized with 0.2% Triton X-100, followed by incubation with anti-synapsin-1 and Alexa 594-conjugated goat anti-rabbit secondary antibody. GluA1-immunoreactive puncta were defined as discrete points along a dendrite within 50 μm from soma. GluA1 puncta were analyzed in two to three dendrites for each neuron. The number of GluA1 puncta for each dendrite was averaged, which represents the value of that cell, and this value equals an n of 1; 32–35 cells were analyzed per condition. Data were collected from three independent experiments.
Imaging of newly exocytosed and synaptic receptors.
Cultured neurons coexpressing SEP-GluA1 and tDimer or mCherry-Homer on coverslips were mounted in a custom-built imaging chamber and imaged using a spinning-disk confocal microscope (PerkinElmer). Photobleach was performed with a laser 488 nm at 70% power. For glycine-induced receptor exocytosis, right after bleach, neurons were treated with modified aCSF containing the following (in mm): 0.2 glycine, 0.1 picrotoxin, 3 CaCl2, 0 MgCl2, and 0.001 strychnine for 5 min, and returned to regular aCSF for the rest of the imaging session. Exocytosed receptors at synapses/spines were measured as SEP fluorescence in bleached regions at indicated time points using Volocity 6.1 software. “Optical barriers” were applied to prevent lateral diffusion of nonbleached surface receptors from neighboring regions into bleached ones (Petrini et al., 2009). For quantification of spontaneous and activity-driven receptor exocytosis, SEP fluorescence detected after bleach was normalized to that measured at the beginning of the imaging session (baseline), setting to zero the residual fluorescence signal right after bleach. Of note, the amount of receptors undergoing exocytosis may be underestimated, as during photobleaching some bleached receptors may endocytose and later exocytose again–their contribution to exocytosis cannot be detected because the signal has been bleached. For glycine-induced synaptic GluA1 incorporation, the intensity of SEP-GluA1 was measured before and after chemical LTP induction, and values were normalized to baseline.
Electrophysiology.
Whole-cell recordings were obtained with Multiclamp 700B amplifiers, as described previously (Nadif Kasri et al., 2009). LTP was induced by pairing presynaptic stimulation at 3 Hz with depolarization of the postsynaptic neuron at 0 mV for 3 min; recordings were maintained for at least 40 min after pairing. One stimulating pathway was used to induce LTP whereas the other pathway was a control pathway. EPSC amplitude was normalized to the average baseline amplitude before pairing. To avoid contribution of NMDAR deficiency in OPHN1Hom mutant conditions, brain slices were cultured in high MgCl2 (Nadif Kasri et al., 2009).
Results
OPHN1–Homer interaction is required for LTP
We previously demonstrated that OPHN1's interaction with Homer1b/c (henceforth referred to as Homer) is important for its role in regulating basal synaptic function (Nadif Kasri et al., 2011). Whether this interaction is also required for OPHN1's role in controlling functional changes during LTP remained to be determined. To assess this, we took advantage of a previously described OPHN1 mutant (OPHN1Hom), defective in Homer binding (Nadif Kasri et al., 2011), and examined the effects of replacing endogenous OPHN1 with OPHN1Hom on LTP induction and expression at the hippocampal CA3-CA1 pathway. Specifically, CA1 neurons in cultured hippocampal slices were infected with a lentivirus that coexpresses OPHN1 short-hairpin (sh) RNA (OPHN1#2) and RNAi-resistant OPHN1Hom fused to EGFP (Nadif Kasri et al., 2011), and the magnitude of LTP in control uninfected and infected cells was examined 7 d post infection. Whereas LTP-inducing stimuli produced a robust and long-lasting potentiation in control neurons, only an initial, transient increase in synaptic transmission was observed in simultaneously recorded neurons coexpressing OPHN1#2 shRNA and OPHN1Hom (Fig. 1A). Notably, a similar deficit in LTP was observed in cells expressing OPHN1#2 shRNA alone, which was completely rescued upon coexpression of RNAi-resistant OPHN1WT (Nadif Kasri et al., 2009). These data imply that disruption of the OPHN1–Homer interaction impairs LTP. To corroborate these findings, we investigated the impact of a peptide consisting of an OPHN1 sequence that contains the Homer ligand domain (pep-OPHN1Hom) on LTP in hippocampal brain slices. This peptide specifically disrupts the OPHN1–Homer interaction; complex formation between Homer and other binding partners, including Dynamin-3 and mGluR5, is not affected (Nadif Kasri et al., 2011). Brain slices were incubated for 24 h with pep-OPHN1Hom or control peptide (pep-contHom) containing three amino acid substitutions in the binding motif, and the magnitude of LTP was examined. We found that pep-OPHN1Hom, but not pep-contHom, significantly reduced LTP (Fig. 1B). Thus, these data indicate that OPHN1's interaction with Homer is required for its effect on LTP.
OPHN1–Homer interaction is essential for positioning the EZ near the PSD
Next, we sought to define the mechanism(s) by which the OPHN1–Homer complex mediates synaptic potentiation. We reasoned that a plausible mechanism could involve the positioning of the EZ near the PSD, since Homer as a tetrameric adaptor protein participates in coupling the EZ to the PSD scaffold, and the latter is important for local AMPAR recycling and expression of synaptic potentiation (Lu et al., 2007; Petrini et al., 2009). To test this hypothesis, we first examined whether endogenous OPHN1 is required for spine EZ positioning. CA1 neurons in hippocampal brain slices were biolistically transfected with a plasmid expressing OPHN1#2 or control scrambled (scr#1) shRNA, together with plasmids expressing clathrin-DsRed and PSD-95-GFP to visualize the EZ and PSD, respectively. Three to four days post transfection, clathrin-DsRed and PSD-95-GFP puncta were monitored. Consistent with previous findings (Lu et al., 2007), we observed that in scr#1 shRNA-expressing neurons PSD-95-GFP puncta were nearly always associated with clathrin-DsRed (Fig. 2A). In OPHN1#2 shRNA-expressing neurons, however, we noted a considerable loss of PSD-associated clathrin puncta (Fig. 2A). For quantitative analysis, we measured the percentage of PSD-95-GFP puncta that lacked adjacent clathrin-DsRed (i.e., EZ-negative synapses). Compared with scr#1 shRNA, OPHN1#2 shRNA caused an ∼5-fold increase in the fraction of EZ-negative synapses (Fig. 2B). Notably, OPHN1 knockdown did not change the total number of clathrin-positive puncta over dendrites (Fig. 2E), supporting a defect in clathrin localization rather than clathrin coat formation. Also, OPHN1 knockdown over a period of 3–4 d did not affect spine/synapse density (Nadif Kasri et al., 2009). These data indicate that OPHN1 is required for the positioning of EZs near the PSD.
We then examined whether disruption of the OPHN1–Homer interaction is sufficient to displace the EZ from the PSD. Clathrin-DsRed and PSD-95-GFP were coexpressed in CA1 neurons in hippocampal slices via biolistic delivery, and slices were incubated with pep-OPHN1Hom, pep-contHom, or no peptide for 24 h. As expected, in control-treated slices (pep-contHom or no pep) most PSD-95-GFP puncta were associated with clathrin-DsRed. In contrast, treatment of slices with pep-OPHN1Hom resulted in a marked reduction in PSD-associated clathrin puncta and a corresponding increase (∼5-fold) in EZ-negative synapses (Fig. 2A,B). Of note, treatment of slices with pep-OPHN1Hom for 24 h did not affect the total number of clathrin-positive puncta over dendrites (Fig. 2E) or synapse density (Nadif Kasri et al., 2011). Given that OPHN1 was also found to interact with Endophilin-A2/3 (Endo2/3), we also assessed the impact of a peptide that specifically disrupts the OPHN1–Endo2/3 interaction (pep-OPHN1Endo; Nadif Kasri et al., 2011) on spine EZ positioning. Contrary to pep-OPHN1Hom, pep-OPHN1Endo had no effect on EZ positioning (Fig. 2B), indicating that OPHN1's interaction with Homer, but not Endo2/3, is required for EZ localization. To corroborate these findings and exclude potential effects of ectopically expressed PSD-95-GFP, an analogous set of experiments as above was performed, but this time Homer-GFP, instead of PSD-95-GFP, was coexpressed with clathrin-DsRed to label/visualize excitatory synapses. In essence, we observed no significant difference between this set (Fig. 2C,D) and the above set (Fig. 2A,B) of experiments. Moreover, similar results were obtained when sparsely plated hippocampal neurons treated or not with above peptides were immunostained for endogenous PSD-95 and clathrin heavy chain to visualize the PSD and EZ, respectively (Fig. 2F,G). Thus, these data indicate that OPHN1's interaction with Homer is essential for the positioning of EZs near the PSD.
Disruption of OPHN1–Homer interaction impairs AMPAR recycling and reduces AMPAR abundance at synapses
Previous studies reported a pivotal role for spine-localized EZs in the recapturing and recycling of AMPARs, and thereby the mobility and accumulation of AMPARs at synapses, both during basal activity and synaptic potentiation (Lu et al., 2007; Petrini et al., 2009). Hence, we explored whether disruption of the OPHN1–Homer interaction causes impaired AMPAR recycling, along with a decrease in synaptic AMPARs. Given that a defect in AMPAR recycling consequentially reduces the insertion of AMPARs into the postsynaptic membrane, we began by examining whether pep-OPHN1Hom decreases the number of newly exocytosed AMPARs at synaptic sites during basal synaptic activity. To this end, we implemented a previously established approach based on the pH-sensitive PHluorin-tagged GluA1 receptor (SEP-GluA1; Kopec et al., 2006). SEP-GluA1 displays fluorescence when on the surface membrane and is nonfluorescent when trapped in intracellular compartments, and so intracellular, but not surface, SEP-GluA1s are protected from photobleaching (Makino and Malinow, 2009; Petrini et al., 2009). As such, following bleaching a large portion of the dendrites, receptor exocytosis can be measured as a return of SEP fluorescence due to unbleached intracellular receptors delivered to the surface membrane (exemplified in Fig. 3A,B). We cotransfected hippocampal cultured neurons with plasmids expressing SEP-GluA1 and mCherry-Homer (to identify synapses), and 3–5 d post transfection neurons were treated with pep-OPHN1Hom, pep-contHom, or no peptide for 24 h. We then bleached a large portion of their dendrites and measured SEP fluorescence at synaptic sites in the bleached areas during a 20 min time period. Interestingly, we found that 20 min following photobleaching, GluA1 exocytosis at synaptic sites was significantly reduced in neurons treated with pep-OPHN1Hom, compared with pep-contHom or no peptide-treated neurons (Fig. 3C,D). These findings imply that spontaneous recycling of AMPARs to the surface is impaired upon disruption of the OPHN1–Homer interaction, leading to a reduced amount of surface AMPARs at synapses. To corroborate the latter, we undertook an immunofluorescence approach to measure AMPAR surface levels. The number of GluA1 receptors at synapses (revealed by synapsin-1 staining) was significantly reduced in pep-OPHN1Hom-treated neurons compared with pep-contHom or no peptide-treated neurons (Fig. 3E,F). In accordance, basal AMPAR-mediated transmission was reduced upon blockage of the OPHN1–Homer interaction (Nadif Kasri et al., 2011). Together, these data indicate that disruption of the OPHN1–Homer interaction causes a loss of synaptic AMPARs in basal conditions.
Next, we assessed the impact of pep-OPHN1Hom on AMPAR recycling/exocytosis and synaptic AMPAR surface expression during synaptic potentiation by applying a glycine stimulation protocol commonly used to induce chemical LTP in cultured hippocampal neurons (Park et al., 2004). To ensure the efficacy of this protocol, we first tested the effect of glycine stimulation on SEP-GluA1-expressing neurons pretreated with no peptide or pep-contHom for 24 h. Consistent with previous studies (Petrini et al., 2009), glycine induced a robust increase in the amount of SEP-GluA1 at the spine surface of these neurons (Fig. 4A–C). In contrast, when neurons were pretreated with pep-OPHN1Hom, only a modest increase in synaptic SEP-GluA1 was observed upon glycine stimulation (Fig. 4A–C), indicating that disruption of the OPHN1–Homer interaction impedes glycine-induced increase in synaptic AMPARs. To determine whether this impediment was due to impaired receptor exocytosis (and recycling), we monitored SEP-GluA1 exocytosis during synaptic potentiation. Specifically, glycine stimulation was applied immediately after photobleaching dendritic areas of SEP-GluA1-expressing neurons that were pretreated with pep-OPHN1Hom, pep-contHom, or no peptide, and receptor exocytosis was measured by means of the return of SEP fluorescence at synaptic sites in the bleached areas during a 30 min time period (Fig. 4D,E). Whereas glycine stimulation strongly promoted GluA1 exocytosis in neurons pretreated with pep-contHom or no peptide, it hardly did so in pep-OPHN1Hom-pretreated neurons (Fig. 4D–F), demonstrating that pep-OPHN1Hom impairs glycine-induced AMPAR exocytosis at synaptic sites.
Together, these results support a model in which disruption of the OPHN1–Homer interaction causes a displacement of EZs from the PSD, leading to impaired receptor recycling/exocytosis, along with a decrease in the abundance of AMPARs at synapses, in both basal conditions and conditions that can induce synaptic potentiation.
Discussion
In the present study, we provide novel mechanistic insight into how the X-linked mental retardation protein OPHN1 controls activity-dependent maturation and plasticity of excitatory synapses. In earlier work, we had shown that a key underlying mechanism by which OPHN1 controls these processes involves its Rho-GAP activity; the latter by locally modulating RhoA/Rho-kinase activities and thereby actin dynamics promotes the stabilization of synaptic AMPARs (Nadif Kasri et al., 2009). In follow-up studies, we found that also OPHN1's interaction with Homer is required for its role in regulating basal synaptic strength (Nadif Kasri et al., 2011), and, importantly, we show here that this interaction is essential for its role in controlling functional changes during LTP as well. Thus, activity-dependent strengthening of excitatory synapses requires not only OPHN1's Rho-GAP activity, but also its interaction with Homer.
How does OPHN1 via its interaction with Homer contribute to the regulation of excitatory synaptic strength? Our current study unveils that a physical interaction between OPHN1 and Homer is critical for the positioning of EZs near the PSD. Recent studies indicated that a major function of EZs in spines is to capture and recycle a local pool of AMPARs, thereby, maintaining a mobile population of surface AMPARs required for synaptic potentiation (Lu et al., 2007; Petrini et al., 2009). Consequently, in spines lacking an EZ, local endocytic receptor recycling is disrupted, leading to a loss of synaptic AMPARs and an impairment in synaptic transmission. Similarly, we find that disruption of the OPHN1–Homer interaction results in impaired receptor recycling/exocytosis along with a decrease in synaptic AMPAR expression, in both basal conditions and conditions that can induce synaptic potentiation. Thus, we posit that the OPHN1–Homer complex, by coupling the EZ to the PSD and thereby facilitating receptor recycling, contributes to activity-dependent synapse strengthening. Notably, disruption of OPHN1's interaction with Endo2/3 does not affect the localization of EZs, indicating that OPHN1's function in spine EZ positioning is independent of its role in postsynaptic endocytosis. In line with this, we found that the OPHN1–Endo2/3 interaction is required for its role in mediating mGluR-LTD, but not for its effects on basal synaptic strength and LTP (Nadif Kasri et al., 2011).
While the precise mechanism by which the OPHN1–Homer interaction mediates the coupling of EZs to the PSD requires further investigation, we know that OPHN1 is enriched at the PSD in an activity-dependent manner (Nadif Kasri et al., 2009), and Homer has been linked to the EZ via its interaction with Dynamin-3, a component of the clathrin endocytic machinery in spines (Lu et al., 2007). As such, the OPHN1-Homer-Dynamin-3 complex is well suited to couple the EZ to the PSD. Noteworthy, OPHN1 has been shown to dimerize with itself (Eberth et al., 2009), and thus, like Dynamin-3, which forms oligomers (Lu et al., 2007), probably serves as a scaffold to link the PSD to the EZ. This function of OPHN1 is likely to be independent of its actin remodeling function mediated by its Rho-GAP activity, as disruption of the OPHN1–Homer interaction does not affect OPHN1's Rho-GAP activity. Moreover, the positioning of EZs in spines is unaffected by actin polymerization or depolarization (Blanpied et al., 2002). These data support the notion that OPHN1 exerts its effects on synapse strengthening by serving at least two distinct roles. Via its interaction with Homer, OPHN1 facilitates receptor recycling and thereby maintenance of a mobile pool of surface AMPARs, while via its Rho-GAP activity OPHN1 contributes to the stabilization of synaptic AMPARs.
In summary, our current data unveil a novel function for OPHN1 as an interaction partner of Homer in spine EZ positioning, and provide additional insight into how genetic deficits in OPHN1 can lead to glutamatergic dysfunction.
Footnotes
This work was supported by National Institutes of Health Grants RO1-MH082808 and RO1-NS082266 to L.V.A.
The authors declare no competing financial interests.
References
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22:461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billuart P, Bienvenu T, Ronce N, des Portes V, Vinet MC, Zemni R, Roest Crollius H, Carrié A, Fauchereau F, Cherry M, Briault S, Hamel B, Fryns JP, Beldjord C, Kahn A, Moraine C, Chelly J. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature. 1998;392:923–926. doi: 10.1038/31940. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Scott DB, Ehlers MD. Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron. 2002;36:435–449. doi: 10.1016/S0896-6273(02)00979-0. [DOI] [PubMed] [Google Scholar]
- Choquet D, Triller A. The dynamic synapse. Neuron. 2013;80:691–703. doi: 10.1016/j.neuron.2013.10.013. [DOI] [PubMed] [Google Scholar]
- Eberth A, Lundmark R, Gremer L, Dvorsky R, Koessmeier KT, McMahon HT, Ahmadian MR. A BAR domain-mediated autoinhibitory mechanism for RhoGAPs of the GRAF family. Biochem J. 2009;417:371–377. doi: 10.1042/BJ20081535. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Akerman CJ, Cross JR, Van der Veken L, Van Aelst L. The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat Neurosci. 2004;7:364–372. doi: 10.1038/nn1210. [DOI] [PubMed] [Google Scholar]
- Henley JM, Barker EA, Glebov OO. Routes, destinations and delays: recent advances in AMPA receptor trafficking. Trends Neurosci. 2011;34:258–268. doi: 10.1016/j.tins.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Helton TD, Blanpied TA, Rácz B, Newpher TM, Weinberg RJ, Ehlers MD. Postsynaptic positioning of endocytic zones and AMPA receptor cycling by physical coupling of dynamin-3 to Homer. Neuron. 2007;55:874–889. doi: 10.1016/j.neuron.2007.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadif Kasri N, Nakano-Kobayashi A, Malinow R, Li B, Van Aelst L. The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev. 2009;23:1289–1302. doi: 10.1101/gad.1783809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadif Kasri N, Nakano-Kobayashi A, Van Aelst L. Rapid synthesis of the X-linked mental retardation protein OPHN1 mediates mGluR-dependent LTD through interaction with the endocytic machinery. Neuron. 2011;72:300–315. doi: 10.1016/j.neuron.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D. Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron. 2009;63:92–105. doi: 10.1016/j.neuron.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]