

Abstract
Ischemia is known to increase the deleterious effect of β-amyloid (Aβ), contributing to early cognitive impairment in Alzheimer's disease. Here, we investigated whether transient ischemia may function as a trigger for Aβ-dependent synaptic impairment in the entorhinal cortex (EC), acting through specific cellular signaling. We found that synaptic depression induced by oxygen glucose deprivation (OGD) was enhanced in EC slices either in presence of synthetic oligomeric Aβ or in slices from mutant human amyloid precursor protein transgenic mice (mhAPP J20). OGD-induced synaptic depression was ameliorated by functional suppression of RAGE. In particular, overexpression of the dominant-negative form of RAGE targeted to microglia (DNMSR) protects against OGD-induced synaptic impairment in an amyloid-enriched environment, reducing the activation of stress-related kinases (p38MAPK and JNK) and the release of IL-1β. Our results demonstrate a prominent role for the RAGE-dependent neuroinflammatory pathway in the synaptic failure induced by Aβ and triggered by transient ischemia.
Keywords: entorhinal cortex, interleukin1-beta, jnk, neuroinflammation, p38mapk, synaptic transmission
Introduction
Alzheimer's disease (AD), is increased by vascular pathologies such as stroke or hypertension, suggesting common pathogenic mechanisms closely related to ischemic injury (Girouard and Iadecola, 2006). In particular, β-amyloid (Aβ) overproduction, which is believed to contribute to early impairment of cognitive functions during AD, is triggered by ischemia. Indeed, hypoxia modulates the expression and activity of Aβ-metabolizing enzymes, resulting in increased Aβ peptides, including the deleterious Aβ(1-42) (Sun et al., 2006; Guglielmotto et al., 2009; Pluta et al., 2013). Moreover, an increased susceptibility to ischemia has been shown in the brains of amyloid precursor protein (APP)-overexpressing mice (Zhang et al., 1997). However, a causal relationship between AD and brain ischemia at the molecular and functional level has not been established.
Some evidence shows a progressive impairment of synaptic function with increasing extracellular Aβ. In particular, synthetic oligomeric Aβ(1-42) in the nanomolar range was shown to specifically inhibit LTP in the hippocampus (Cullen et al., 1997; Lambert et al., 1998; Vitolo et al., 2002; Walsh et al., 2002; Wang et al., 2004; Shankar et al., 2008) and cortical areas (Origlia et al., 2008, 2009a), whereas higher micromolar concentrations caused synaptic depression and LTD impairment (Snyder et al., 2005; Hsieh et al., 2006; Parameshwaran et al., 2007; Origlia et al., 2010).
Interestingly, RAGE (Receptor for Advanced Glycation Endproducts) expression changes in neuronal and non-neuronal cells under ischemic conditions (Pichiule et al., 2007; Zhai et al., 2008) and AD (Yan et al., 1996). RAGE is a receptor mediating and focusing Aβ effects on neuronal and non-neuronal cells (Yan et al., 1995; Lue et al., 2001; Schmidt et al., 2001; Deane et al., 2003), contributing to Aβ-dependent functional impairment in hippocampus (Arancio et al., 2004) and entorhinal cortex (EC; Origlia et al., 2008, 2010). Moreover, inhibition of RAGE-mediated transduction was shown to be effective in improving cognition in APP/PS1 mice (Liu et al., 2014). However, whether RAGE has a role in the development of neuronal impairment following transient ischemia in AD is still unknown. We have previously investigated the role of RAGE using transgenic mice expressing a dominant-negative form of RAGE targeted to microglia (DNMSR) or neurons (DN-RAGE). We found that the effect of nanomolar Aβ on LTP is mediated by neuronal RAGE activation of p38MAPK (Arancio et al., 2004; Origlia et al., 2008), whereas LTD impairment and synaptic depression by micromolar Aβ involve the phosphorylation of p38MAPK and JNK, which depend on microglial RAGE activation, along with the induction of the proinflammatory cytokine IL-1β (Origlia et al., 2010). Here, we investigated how RAGE expressed in different cell types might contribute to Aβ-dependent synaptic dysfunction occurring under transient ischemia. Using in vitro oxygen/glucose deprivation (OGD; Tozzi et al., 2007; Medvedeva et al., 2009), we found that increased extracellular amyloid resulting from either exogenous supply of synthetic Aβ(1-42) (Origlia et al., 2008, 2009a, 2010) or from transgenic expression of APP (Mucke et al., 2000) facilitates synaptic depression induced by OGD in EC slices. More importantly, overexpression of the dominant-negative form of RAGE in microglia (DNMSR) protects against OGD-induced synaptic impairment in an Aβ-enriched environment by reducing the activation of p38MAPK and JNK and the release of IL-1β. Our results demonstrate a role for the RAGE-dependent neuroinflammatory pathway in the synaptic failure induced by Aβ and triggered by ischemia.
Materials and Methods
Animals.
Homozygous RAGE-null mice were generated and characterized as described previously (Origlia et al., 2008, 2010). Transgenic mice with signal-transduction-deficient mutants of RAGE in which the cytosolic domain of the receptor has been deleted, thereby imparting a dominant-negative RAGE effect, targeted to neurons (DN-RAGE, driven by the platelet derived growth factor-B chain promoter) or to microglia (DNMSR, driven by the macrophage scavenger receptor promoter), have been used previously by our group (Arancio et al., 2004; Origlia et al., 2008, 2010; Fang et al., 2010). Furthermore, mhAPP-transgenic mice overexpressing an alternatively spliced human APP (hAPP) minigene that encodes hAPP695, hAPP751, and hAPP770 bearing mutations linked to familial AD (V717F, K670N/M671L) have been used (APPsweInd, line J20; Mucke et al., 2000). Double-transgenic mice expressing DN-RAGE in microglia and mhAPP were obtained by crossing DN-RAGE mice to mhAPP mice as described previously (Fang et al., 2010). Male transgenic mice and their littermate controls were used for the in vitro electrophysiology (age range, 2–3 months).
Pharmacological agents.
Aβ(1-42) and the reverse peptide, Aβ(42-1), were purchased from Abbiotec. Oligomeric Aβ(1-42) peptide was prepared as described previously and characterized by atomic force microscopy (Yan et al., 2007) and mass spectrometry as reported by Origlia et al. (2009a). Aliquots were stored at −20°C in DMSO as a 200 mm stock solution and diluted, immediately before application, to the desired final concentration in artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 2.5 KCl, 2 CaCl2, 1.2 MgSO4 1 NaH2PO4, 6.2 NaHCO3, 10 HEPES, and 11 glucose. 4-(4-Fluorophenyl)-2-(4-metylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole (SB203580) and anthra(1-9-cd)pyrazol-6(2H)-one (SP600125) were purchased from Alexis; (5S)-(tert-butoxycarbonylamino)-6-phenyl-(4R)-hydroxy-(2R)-benzylhexanoyl)-l-leucy-l-phenylalaninamide (L-685,458) was purchased from Tocris Bioscience; and 2-amino-3-methoxyflavone (PD 98059) was purchased from Calbiochem. All drugs were prepared in DMSO stock solutions (final concentration of DMSO in perfusate was 0.1%). MW108 compound was obtained from the laboratory of Dr. Watterson and synthesized as reported in Watterson et al. (2013). The cell-permeable JNK inhibitor I (L)-form was purchased from Millipore and Interleukin-1 receptor antagonist (IL-1Ra) was purchased from PeproTech. These drugs were dissolved in sterile saline solution and then diluted to the desired final concentration in ACSF. Specific antibodies to RAGE have been described in our previous studies (Yan et al., 1996). Purified rabbit nonimmune IgG was used as a control (Bethyl).
Slice preparation.
Animals were deeply anesthetized using urethane (20% solution, 1 ml/100 g of body weight) via intraperitoneal injection and then decapitated after disappearance of the tail pinch reflex. The brain was rapidly removed and thick horizontal sections (400 μm) containing the entorhinal area were made on a Vibratome. All steps were performed in ice-cold oxygenated ACSF solution. Before recording, slices were stored for at least 1 h in a recovery chamber containing oxygenated ACSF solution at room temperature. During electrophysiological recordings, slices were perfused at a rate of 2.5–3 ml/min with oxygenated ACSF at 33 ± 1°C.
Electrophysiological recordings.
Extracellular field potentials (FPs) were evoked in layer II/III in response to electrical stimulation of layer II (Origlia et al., 2008, 2010). The amplitude of the FPs was used as a measure of the evoked population excitatory current. All FPs had peak latency between 4.5 and 6 ms. Baseline responses were obtained with a stimulation intensity that yielded 50–60% of maximal amplitude. After 15 min of stable baseline recordings, slices were perfused for 10 min with deoxygenated glucose-free ACSF (glucose was substituted with d-mannitol at equimolar concentration) to obtain a transient OGD. The amplitude of FP was monitored every 20 s and averaged every three responses by online data acquisition software. Effects on synaptic function were evaluated either during ischemia (as the average of FP amplitude during the last 3 min of 10 min OGD application) or as the recovery of FPs calculated as the averaged relative amplitude of FPs respect to the baseline values after reintroduction of regular ACSF (from minutes 41–50 after the end of OGD).
ELISAs.
Invitrogen human Aβ ELISA kits were used for the quantification of Aβ(1-42) and Aβ(1-40) in transgenic mice tissue homogenate following the procedure described in Chishti et al. (2001). Quantification of phosphorylated [pTpY180/182]p38 MAPK or phosphorylated [pTpY183/185]JNK were detected in protein extracts from slices collected after electrophysiology using two different ELISA kits (Millipore). Levels of phosphorylated forms were normalized with respect to the total p38 MAPK and total JNK protein content that was assessed using two ELISA kits purchased from the same company (Millipore). Quantification of IL-1β was performed using an ELISA kit purchased from Bender Medsystem. After electrophysiology, slices were collected, immediately frozen at −80°C, and subsequently lysed in cell extraction buffer containing the following (in mm): 10 Tris, pH 7.4, 100 NaCl, 1 EDTA, 1 EGTA, 1 NaF, 20 Na4P2O7, 2 Na3VO4, along with 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% deoxycholate, and 1 mm PMSF (protease mixture inhibitor; Sigma-Aldrich). The extract was then centrifuged (13,000 rpm, 10 min at 4°C) to obtain a clear lysate that was used for the assay.
Statistical analysis.
All data are reported as mean ± SEM. Statistical comparisons between experimental groups or between FP amplitudes measured during baseline and after OGD protocol were performed by applying two-way repeated-measures ANOVA with pairwise multiple-comparison procedures (Holm–Sidak method). Comparisons between different groups in ELISA experiments were performed using one-way ANOVA. Differences were considered significant when p < 0.05.
Results
OGD-dependent synaptic depression is enhanced in the presence of Aβ
Synaptic failure is the earliest consequence of ischemic brain damage, resulting in acute and reversible or long-lasting changes in synaptic function (Hofmeijer and van Putten, 2012).
Previous data in hippocampus and corticostriatal slices (Tozzi et al., 2007; Medvedeva et al., 2009) showed that OGD induces fast depression of FPs with a duration and persistence that are a function of OGD length. As in previous work (Origlia et al., 2008, 2010), we focused our studies on the EC, an area of the brain crucially involved in cognitive functions (Witter et al., 1989) and affected at an early stage of AD (Braak and Braak, 1991). We recorded FPs from superficial cortical layer II/III of EC slices after stimulation of layer II, as described in Origlia et al. (2008). Our results show a rapid fall of FP amplitude during 10 min OGD (mean amplitude of the last 3 min of OGD was 32 ± 3% of baseline, n = 11, 6 mice; Fig. 1A, vehicle-treated slices). FP amplitude partially recovered at the end of the OGD protocol after reintroduction of oxygenated ACSF, reaching a stable level of depression after 50 min (mean relative amplitude of FPs was 80 ± 4% of baseline, n = 11 slices, 6 mice p < 0.05 vs baseline; Fig. 1A, vehicle-treated slices).
Figure 1.

OGD-dependent synaptic depression is enhanced in the presence of Aβ. A, OGD for 10 min (corresponding to dark bar) induced a decrease of FP amplitude in control vehicle-treated slices (open circles), reaching a stable level of depression after 50 min. OGD-induced depression was enhanced in slices treated with Aβ(1-42) at 1 μm (gray circles) for 10 min (corresponding to dark bar). Insets show typical traces of FP recordings before during and after OGD exposure in vehicle- and Aβ-treated slices. B, Synaptic depression after 10 min of OGD (dark bar) was significantly increased in mhAPP slices (gray diamonds) with respect to WT (open circles). Insets show typical traces of FP recordings before, during, and after OGD in WT or mhAPP slices. C, OGD (dark bar, 10 min)-induced depression was significantly reduced in mhAPP slices continuously perfused with 0.5 μm l-685,458 (white squares) with respect to vehicle-treated mhAPP slices. D, Bars represent the average Aβ(1-40) (right) and Aβ(1-42) (left) levels measured using an ELISA assay. Both Aβ(1-40) and Aβ(1-42) were significantly elevated (#p < 0.05) in mhAPP slices exposed to OGD with respect to control nonexposed mhAPP slices. Scale bars: A, B, horizontal, 5 ms; vertical, 0.5 mV. Error bars indicate SEM.
To investigate the effect of transient ischemia in an Aβ-enriched condition, we applied exogenously an oligomeric preparation of synthetic Aβ(1-42). When simultaneous OGD application and Aβ perfusion (1 μm for 10 min) were used, we observed a significant increase in long-lasting depression with respect to control vehicle-treated slices exposed to OGD alone (66 ± 4%, n = 6 slices, 4 mice p < 0.05; Fig. 1A); additional control experiments were performed using the reverse peptide Aβ(42-1, data not shown). This synergistic effect of OGD with Aβ has been described previously (Mozes et al., 2012), but to establish a causal relationship between transient ischemia and Aβ-dependent synaptic dysfunction, we repeated the experiment in slices from mice expressing a mutant form of human APP (mhAPP; Mucke et al., 2000). As reported in Figure 1B, OGD-induced depression was significantly enhanced in mhAPP slices (55 ± 10%, n = 10 slices, 6 mice) with respect to OGD in WT slices (80 ± 6.7%, n = 8, 5 mice; p < 0.05). To investigate whether the different vulnerability of mhAPP slices to OGD reflected an increased amyloid production, we used the specific γ-secretase inhibitor l-685,458 (Li et al., 2000; Parent et al., 2005). Perfusion with this compound did not induce significant changes in FP amplitude during baseline recordings in EC slices (data not shown). When mhAPP slices were continuously perfused with 0.5 μm l-685,458 (Fig. 1C), synaptic depression was significantly lower than in mhAPP vehicle-treated slices either during OGD (67 ± 11%, n = 8, 4 mice vs 40 ± 2%, n = 10 slices, 6 mice; p < 0.05) or after reoxygenation (80 ± 2%, n = 8, 4 mice p < 0.05 vs 65 ± 10%, n = 10, 6 mice). The protective effect of l-685,458 was not present in OGD-treated WT slices (data not shown), suggesting that its efficacy was related to inhibition of the amyloidogenic pathway rather than to unspecific effect on other metabolic pathways. Furthermore, the modulation of amyloid production by OGD was confirmed after measurement of Aβ peptides in mhAPP slices collected after electrophysiology. In EC slices that were analyzed after 10 min of OGD, we found an increase of Aβ(1-42) (Fig. 1D). Tissue level of Aβ(1-42) after OGD was significantly higher than that observed in mhAPP slices not exposed to OGD (14.6 ± 0.7 vs 6.5 ± 2.3 pg/mg protein, p < 0.05; n = 6, 6 mice each group). Similar results were obtained measuring Aβ(1–40) levels in mhAPP EC slices tissue extract (n = 6; p < 0.05 vs control; Fig 1D). An OGD-induced increased activity of secretases and Aβ production was shown previously in cultured neuroblastoma cells expressing APP (T. Zhang et al., 2014). Moreover, a modulation of Aβ production was reported in the hippocampus 1 min after electrical stimulation (Puzzo et al., 2011). Therefore, OGD can modulate amyloid-metabolism and an Aβ-enriched environment exacerbates synaptic dysfunction after transient ischemia.
Microglial RAGE inhibition protects against OGD-induced synaptic dysfunction in Aβ-enriched environment
RAGE is a multiligand membrane receptor belonging to the IgG superfamily. Several molecules released during cerebral ischemia may activate RAGE and participate in neuronal damage, including HMGB1 (Muhammad et al., 2008), S100 (Gürsoy and Büyükuysal, 2010). To determine whether RAGE is involved in OGD-induced depression, we first examined the effect of transient ischemia in EC slices from RAGE-null mice (RAGE KO) and in slices from WT mice pretreated with anti-RAGE IgG (2.5 μg/ml in ACSF). Previous data showed that complete RAGE inhibition by either knocking out the RAGE gene or using blocking antibodies did not affect basal synaptic transmission or plasticity in EC slices (Origlia et al., 2008, 2010). The long-lasting synaptic depression induced by OGD was not observed in presence of RAGE inhibition, because a complete recovery of FP amplitude was achieved in slices treated with anti-RAGE IgG (100 ± 4% of baseline, n = 6 slices, 4 mice p > 0.05 vs baseline, p < 0.05 vs 73 ± 8%, n = 6, 4 mice in vehicle-treated slices; Fig. 2A) and in slices from RAGE KO mice (99 ± 5%, n = 6 slices, 4 mice p > 0.05 vs baseline; p < 0.05 vs 81 ± 4.6%, n = 13, 6 mice in WT slices; Fig. 2B). These results confirm that RAGE is involved in the synaptic alteration that follows transient ischemia and prompted us to investigate its cell-specific contribution using slices from mice overexpressing a dominant-negative form of RAGE targeted to neurons (DN-RAGE) and to microglia (DNMSR), as described previously (Origlia et al., 2010). Remarkably, introducing DN-RAGE in neurons did not protect against transient ischemia (Fig. 2C) and the effect of OGD was similar to that observed in control WT slices (mean FP amplitude after OGD was 85 ± 5% of baseline, n = 5, 4 mice; p > 0.05 vs control WT slices after OGD). In contrast, the effect of transient ischemia was reduced when DN-RAGE was targeted to microglia. As shown in Figure 2C, synaptic depression during OGD in DNMSR slices was significantly reduced with respect to control WT (70 ± 5%, n = 9 slices, 5 mice, p < 0.05 vs 33 ± 3%, n = 13 slices, 6 mice) and there was a full recovery of FP amplitude after reintroduction of oxygenated ACSF (100 ± 6%, n = 9 slices, 5 mice; p < 0.05 vs control WT). Therefore, our results indicate a different role for RAGE in ischemia depending on its localization (neuronal vs microglial).
Figure 2.
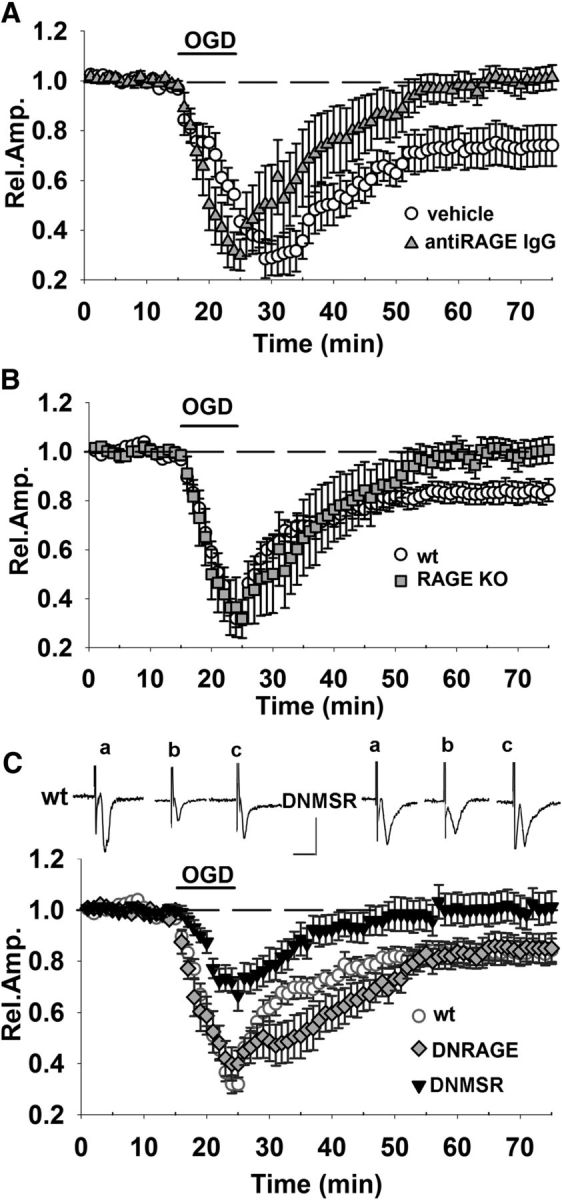
Microglial RAGE contributes to OGD-induced synaptic depression. Blockade of RAGE by either knocking out the RAGE gene or with neutralizing anti-RAGE IgG was able to prevent ischemia-dependent long-lasting depression. FP amplitude in anti-RAGE IgG (A, gray triangles) and RAGE-KO (B, gray squares) slices was completely rescued 50 min after OGD (10 min corresponding to dark bar) and was significantly different from FP amplitude in OGD-exposed WT (white circles). C, RAGE-deficient signaling in neurons (DN-RAGE, gray diamonds) was not sufficient to prevent OGD-induced synaptic depression. In contrast, DNMSR slices (c, black triangles), characterized by RAGE signaling deficiency in microglia, displayed a significant reduction of synaptic depression during OGD (10 min, dark bar) compared with WT (white circles) and a full recovery of FP amplitude was achieved 50 min after. Insets show typical traces of FP recordings before (a), during (b), and after (c) OGD in WT or DNMSR slices. Scale bars, 10 ms (horizontal) and 1 mV (vertical). WT slices recorded in interleaved experiments were pooled together and reported as one group in B and C. In A, vehicle control solution contained rabbit nonimmune IgG.
Because RAGE is an Aβ receptor that mediates synaptic plasticity impairment in the hippocampus (Arancio et al., 2004) and cortical areas (Origlia et al., 2008, 2009a, 2010), it is important to know the role of cell-specific activation of RAGE under Aβ load. The Aβ oligomers at a nanomolar concentration impair LTP-activating RAGE expressed selectively in neurons (Origlia et al., 2008). Interestingly, increasing synthetic Aβ concentration up to a micromolar level induces RAGE activation in microglial cells that progressively affects basal synaptic transmission and LTD in addition to LTP (Origlia et al., 2010). We therefore wanted to confirm the protective role of specific RAGE inhibition in an amyloid-enriched environment. First, we investigated the effect of OGD coupled to an exogenous supply of Aβ(1-42) (1 μm for 10 min) peptide in slices from DNMSR mice. As reported in Figure 3A, synaptic depression was reduced in DNMSR after OGD and Aβ exposure (mean FP amplitude = 93 ± 9%, n = 6, 4 mice; p < 0.05 vs 65 ± 4%, n = 8, 5 mice in WT Aβ-treated slices after OGD). Similarly, deficiency of RAGE in microglia was able to contrast the effect of OGD in presence of mutant APP overexpression. The synaptic depression was significantly lower in slices from double-transgenic mice (mhAPPxDNMSR) with respect to single mhAPP-transgenic slices (Fig. 3B) either during OGD (67 ± 11%, n = 8, 4 mice vs 35 ± 7%, n = 10 slices, 6 mice; p < 0.05) or after reoxygenation (85 ± 8%, n = 8, 4 mice vs 63 ± 7%, n = 10 slices, 6 mice; p < 0.05). To investigate whether the protective effect of DNMSR expression in mhAPP mice was due to a modulation of Aβ production, we repeated the measurement of Aβ(1-42) and Aβ(1-40) in slices from mhAPPx DNMSR mice either under basal condition or after 10 min of OGD. The results reported in Figure 3C show that OGD induced an increase of Aβ(1-42) in mhAPPxDNMSR with respect to nonstimulated slices (14.6 ± 0.1 vs 6.48 ± 2.6 pg/mg protein, n = 6 slices, 3 mice each group; p < 0.05). A similar result was obtained for Aβ(1-40) levels (Fig. 3C), suggesting that amelioration of synaptic function in mhAPPx DNMSR is due to an inhibition of RAGE/Aβ interaction rather than to a reduction of Aβ production. Therefore, our results suggest a prominent role for RAGE expressed in microglia in the synaptic alterations that follow transient ischemia and also that specific molecular pathways driven by the RAGE/Aβ interaction contribute to exacerbate these effects.
Figure 3.
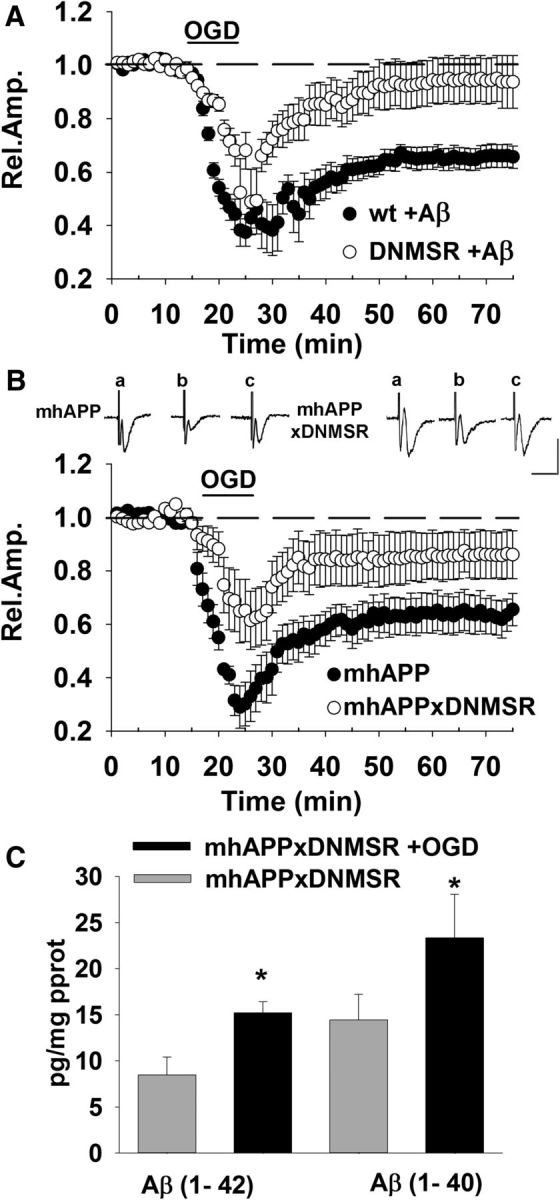
Microglial RAGE inhibition protects against OGD-induced synaptic dysfunction in an Aβ-enriched environment. A, Effect of OGD (dark bar, 10 min) coupled to exogenous supply of Aβ(1-42) (1 μm perfused for 10 min) in slices from DNMSR mice (gray diamonds) was significantly reduced with respect to Aβ-treated WT slices exposed to OGD (black circles). B, A deficiency of RAGE in microglia was able to counteract the effect of OGD in the presence of mutant APP overexpression. The synaptic depression was significantly lower in slices from double-transgenic mice (DNMSR × mhAPP, white circles) with respect to single mhAPP transgenic slices (black circles). Insets show typical traces of FP recordings before (a), during (b), and after (c) OGD in mhAPP or mhAPPxDNMSR slices. Scale bars, 10 ms (horizontal) and 1 mV (vertical). C, Bars represent the average Aβ(1-40) (right) and Aβ(1-42) (left) levels measured using an ELISA assay. Both Aβ(1-40) and Aβ(1-42) were significantly elevated (*p < 0.05) in mhAPPxDNMSR slices exposed to OGD with respect to control nonexposed mhAPPxDNMSR slices.
IL1β is increased during OGD and its blockade by IL1Ra reduces synaptic dysfunction in the presence of elevated Aβ
The results reported so far indicate a clear role for microglial activation in the synaptic failure induced by OGD. This led us to investigate in more detail the possible mediators that could be released by microglia and modulate synaptic function. We and others have demonstrated previously that IL-1β is a key molecule that can modulate synaptic function and plasticity favoring synaptic depression (Coogan et al., 1999; Ikegaya et al., 2003; Kelly et al., 2003; Origlia et al., 2010). In particular, Aβ mediated the induction of IL-1β in EC slices at a concentration that is capable of inducing synaptic dysfunction, and this effect was suppressed in DNMSR mice (Origlia et al., 2010). To determine whether IL1-β is involved in OGD-induced depression, we investigated the effect of transient ischemia in EC slices perfused with IL1Ra (Fig. 4A). When slices were continuously perfused with IL1Ra (20 ng/ml), we observed a significant rescue of synaptic function during OGD (mean amplitude = 80 ± 4%, n = 6 slices, 4 mice; p < 0.05 vs 37 ± 5%, n = 8 slices, 4 mice in vehicle-treated slices during OGD; Fig. 4A). The protective effect of the IL-1β signaling blockade was also present after reoxygenation because FP amplitude returned to the baseline values 50 min after OGD (96 ± 5%, n = 6 slices, 4 mice p > 0.05 vs baseline; p < 0.05 vs 80 ± 6%, n = 8 slices, 4 mice in vehicle-treated slices after OGD; Fig. 4A). These results prompted us to investigate the possible protective effect of IL1Ra in mhAPP slices exposed to transient ischemia. As shown in Figure 4B, synaptic depression caused by OGD was significantly reduced by IL1Ra pretreatment in mhAPP slices (during OGD mean amplitude = 67 ± 11%, n = 6 slices, 4 mice vs 39 ± 3%, n = 9, 5 mice, and 50 min after OGD mean amplitude = 77 ± 8%, n = 6, 4 mice vs 61 ± 7%, n = 9, 5 mice in IL1Ra mhAPP and vehicle mhAPP slices, respectively; p < 0.05). Remarkably, the involvement of IL1β was confirmed by measurement of the protein levels in slices exposed to ischemia (Fig. 4C). Using an ELISA assay, we found that IL1-β tissue levels were not significantly different between WT and 2- to 3-month-old mhAPP slices (12.23 ± 1.5 and 15.36 ± 3.1 pg/ml, respectively, n = 6 slices, 4 mice each; p > 0.05), in agreement with previously reported findings (Fang et al., 2010). However, IL1β was elevated in WT slices after 10 min of OGD with respect to nonexposed WT slices (24.75 ± 2.3 pg/ml, n = 6, 4 mice; p < 0.05). The effect of OGD on IL-1β levels was also present in mhAPP slices (39.7 ± 2.9 vs 15.36 ± 3.1 pg/ml in mhAPP slices not exposed to OGD; n = 6 slices, 4 mice, p < 0.001) and was significantly enhanced respect to that observed in OGD-exposed WT slices (24.75 ± 2.3 pg/ml, n = 6, 4 mice; p < 0.001). Deficiency of RAGE signaling in microglia (DNMSR slices) and RAGE blockade by anti-RAGE IgG were not able to modify basal IL-1β levels (as in Origlia et al., 2010). More importantly, RAGE blockade by anti-RAGE IgG and deficiency of RAGE signaling in microglia (DNMSRxmhAPP) completely prevented OGD effect, maintaining IL-1β levels comparable to basal values (18 ± 3.91 and 21.7 ± 4.8 pg/ml, respectively; n = 6, 4 mice; p > 0.05 vs WT, p > 0.05 vs mhAPP). These results confirm that IL-1β is an inflammatory mediator that links RAGE activation in microglia to Aβ-induced synaptic dysfunction under ischemic conditions.
Figure 4.
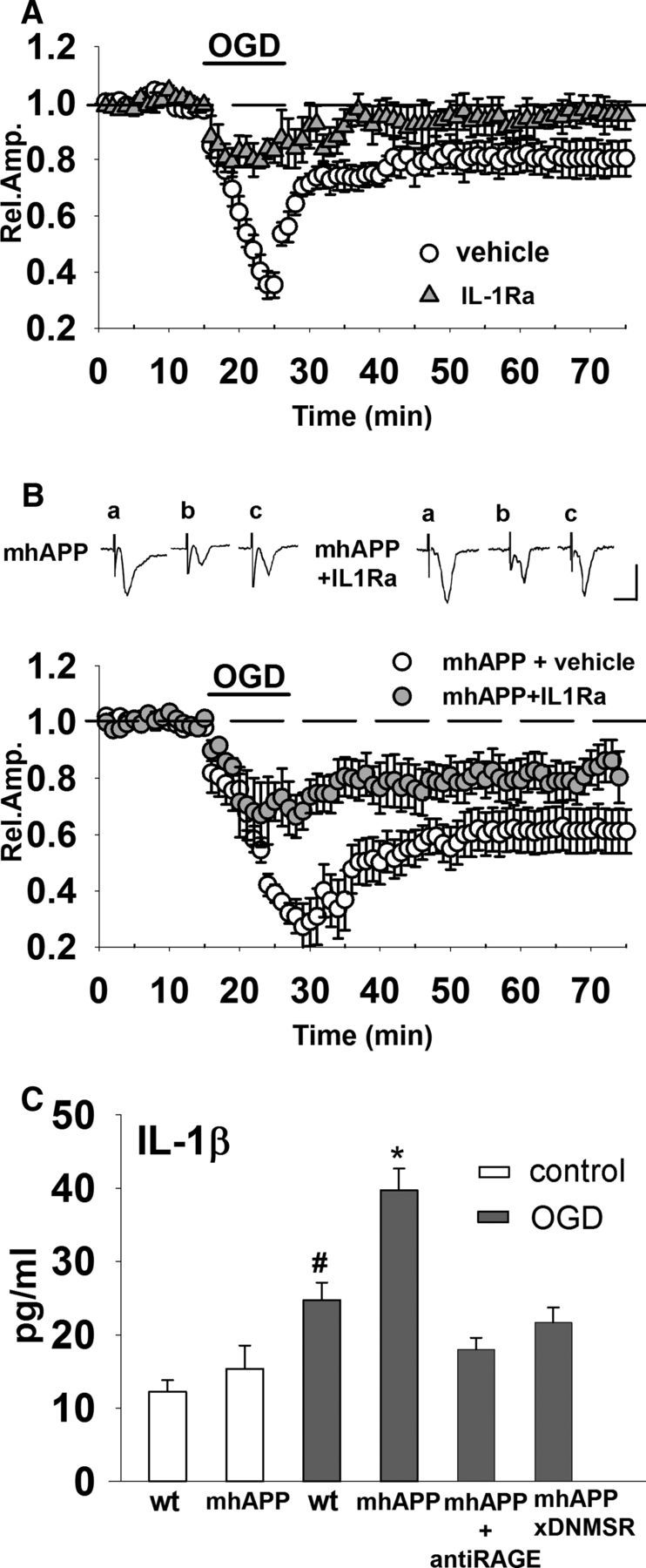
IL1β is increased during OGD and its blockade by IL1Ra reduces synaptic dysfunction in mhAPP slices. A, When slices were treated with 20 ng/ml IL-1Ra (gray triangles), synaptic depression during OGD (10 min, dark bar) was significantly decreased with respect to OGD-exposed vehicle-treated slices. In the presence of IL-1Ra (gray triangles), OGD failed to induce long-lasting synaptic depression of FP amplitude that returned to baseline values 50 min after transient ischemia. B, Synaptic depression caused by OGD (dark bar) was significantly reduced by IL1Ra pretreatment in mhAPP slices (gray circles) compared with vehicle-treated mhAPP slices (white circles). Insets show typical traces of FP recordings before (a), during (b), and after (c) OGD in mhAPP vehicle-treated or mhAPP IL1Ra-treated slices. Scale bars, 10 ms (horizontal) and 1 mV (vertical). C, Plot representing IL-1β levels in EC slices exposed to OGD. The basal levels of IL-1β did not differ between WT and mhAPP mice (p < 0.05), but were significantly elevated in WT slices exposed to OGD compared with nonexposed WT and mhAPP slices (#p < 0.01). Ten minutes of OGD induced a significantly higher level of IL-1β in mhAPP slices compared with nonexposed mhAPP (*p < 0.001) and this increase was significantly higher than that found in WT slices exposed to OGD (*p < 0.001). Error bars indicate SEM.
Inhibition of the stress-related kinases p38MAPK and JNK protects against OGD-induced synaptic dysfunction in WT and mhAPP slices
So far, our results suggested that RAGE in microglia activates an inflammatory pathway involving IL-1β that contributes in controlling synaptic responses under transient ischemia. The next step was to identify intracellular pathways associated with synaptic dysfunction induced by transient ischemia in an Aβ-enriched environment. Interestingly, the RAGE-dependent inflammatory signaling is linked to the activation of intracellular transduction pathways related to stress, such as p38MAPK and JNK (for review, see Simm et al., 2004; Origlia et al., 2009b). Activation of these intracellular pathways is also triggered by OGD in neurons (Kikuchi et al., 2009; Benakis et al., 2010). In particular, a recent study by Chen et al. (2014) demonstrated that RAGE inhibition prevented p38MAPK phosphorylation and rescued cognitive and synaptic plasticity deficits induced by glycated Aβ (Aβ-AGE). Moreover, our previous work demonstrated that p38MAPK and JNK activation are important key events in Aβ-dependent synaptic depression and LTD impairment (Origlia et al., 2010). Given this evidence, we wanted to verify whether the phosphorylation of p38MAPK and JNK is relevant for synaptic depression under transient ischemia either alone or in combination with Aβ toxicity. At first, we used inhibitors of p38MAPK (SB203580, 1 μm) and JNK (SP600125, 20 μm) to prevent synaptic depression in WT slices exposed to transient ischemia. Perfusion with either compound did not induce significant changes in extracellular FP amplitude during baseline recording in EC slices, in agreement with what has been found previously (Origlia et al., 2008). As shown in Figure 5A, continuous perfusion with SB203580 reduced synaptic depression during OGD (mean amplitude = 72 ± 8% of baseline, n = 6, 4 mice p < 0.05 vs 34 ± 2%, n = 12, 6 mice in vehicle-treated slices) and allowed a full recovery of FP amplitude after reoxygenation (98 ± 3%, n = 6, 4 mice p > 0.05 vs baseline, p < 0.05 vs 82 ± 4% in vehicle-treated slices, n = 12, 6 mice). This result was confirmed using 10 μm MW-108, a novel selective inhibitor of p38αMAPK isoform (Watterson et al., 2013) that, when perfused on slices, had a protective effect that was comparable to that of SB203580 (mean amplitude 84 ± 4% of baseline during OGD and 102 ± 2% after recovery, n = 6, 4 mice; p > 0.05 vs SB203580-treated slices; Fig. 5A). Synaptic function was also ameliorated in slices continuously perfused with SP600125 (Fig. 5B) considering both the short-term effect of OGD (73 ± 8% of baseline, n = 7, 4 mice p < 0.05 vs vehicle-treated slices) and long-lasting depression (94 ± 11%, n = 7, 4 mice p > 0.05 vs baseline, p < 0.05 vs vehicle-treated slices). A similar protection was obtained using the cell-permeable inhibitor of JNK (JNKI) at 2 μm (mean amplitude was 71 ± 8% of baseline during OGD and 101 ± 8% after reoxygenation, n = 6, 4 mice; p > 0.05 vs SP600125; Fig. 5B). In contrast, when inhibition of p42-p44 MAP kinase was investigated in OGD-exposed slices, using the MAP kinase kinase (MEK) inhibitor PD98059 (50 μm), we did not observe any significant difference respect to control vehicle OGD-exposed slices (mean amplitude was 26 ± 10% of baseline during OGD and 82 ± 5% after reoxygenation, n = 6, 4 mice; Fig. 5A). These results suggest that synaptic depression elicited by transient ischemia is dependent on the activation of both JNK and p38MAPK. We then investigated the effect of p38MAPK or JNK inhibition in slices from mhAPP mice. As reported in Figure 5, C and D, perfusion of slices throughout the experiment with either SB203580 or SP600125 mitigated the effect of transient ischemia in mhAPP slices, significantly reducing OGD-dependent depression (85 ± 11%, n = 7 slices, and 85 ± 9%, n = 7 slices, p < 0.05 vs 53 ± 7%, n = 7 in mhAPP vehicle-treated slices). Similar to WT OGD-exposed slices, we confirmed the specific involvement of these kinases using different inhibitors (MW-108 and JNKI). As shown in Figure 5C, 10 μm MW-108 perfusion in mhAPP slices partially rescued OGD depression in a manner comparable to the SB203580 compound (82 ± 7%, n = 6 slices, 4 mice, p > 0.05 vs SB203580 in mhAPP OGD-exposed slices). Moreover, JNKI perfusion in mhAPP OGD-treated slices revealed a comparable effect with respect to SP600125 (85 ± 7%, n = 6 slices, 4 mice, p > 0.05 vs SP600125 in mhAPP OGD-exposed slices; Fig. 5D). Therefore, both p38MAPK (in particular the α isoform) and JNK activation are involved in synaptic changes induced by OGD and they contribute to the synaptic dysfunction induced by Aβ and triggered by transient ischemia.
Figure 5.

OGD-induced synaptic dysfunction in WT and mhAPP slices is prevented by inhibition of the stress-related kinases p38MAPK and JNK. A, Treatment of slices with either 1 μm SB203580 (gray squares) or 10 μm MW-108 (filled triangles) was able to reduce synaptic depression during OGD (dark bar) and allowed a full recovery of FPs 50 min after transient ischemia. In contrast, the MEK inhibitor PD 98059 (50 μm, gray diamonds) did not affect OGD-dependent depression. B, A similar protective effect was achieved in slices after treatment with either 20 μm SP600125 (black triangles) or JNKI (filled triangles). C, In mhAPP slices, the OGD-induced depression was significantly diminished by SB203580 (gray squares) or MW-108 (black circles) compared with vehicle-treated mhAPP slices (white circles). D, SP600125 (black triangles) and JNKI (black circles) were capable of reducing OGD-induced depression in mhAPP slices compared with vehicle-treated mhAPP slices (white circles). The insets in A–D represent typical traces of FP recordings before (a), during (b), and after (c) OGD. Scale bars, 5 ms (horizontal) and 1 mV (vertical).
OGD modulates p38MAPK and JNK phosphorylation differently in WT and mhAPP cortical slices
To better clarify whether the phosphorylation of stress-related kinases follows cell-specific RAGE activation after transient ischemia, we measured tissue levels of phosphorylated p38MAPK and phosphorylated JNK in EC slices that were collected after the end of electrophysiology. WT slices that were exposed to 10 min of OGD showed a significant increase in phospho-p38MAPK tissue levels with respect to nonexposed WT slices (141.75 ± 27 U/ng vs 32.33 ± 19.8 U/ng, n = 6; p < 0.01; Fig. 6A). As reported previously (Origlia et al., 2010), selective deficiency of RAGE signaling in microglia (DNMSR) or complete inactivation of RAGE (anti-RAGE IgG) did not modify the basal level of phospho-p38MAPK (data not shown). However, slices either from DNMSR mice or WT mice treated with anti-RAGE IgG displayed complete suppression of phospho-p38MAPK after OGD (20.86 ± 5 U/ng and 51.6 ± 14 U/ng, respectively, n = 6; p < 0.05 vs WT exposed to OGD and p > 0.05 vs control WT slices; Fig. 6A). The p38MAPK activation that follows OGD was even more robust in mhAPP slices. After OGD, tissue phospho-p38 levels were enhanced in mhAPP with respect to nonexposed mhAPP slices (418.22 ± 65 U/ng and 55.16 ± 7.7 U/ng respectively, n = 6; p < 0.001; Fig. 6A) and significantly higher with respect to those found in OGD-exposed WT slices (p < 0.001). Like WT, in mhAPP slices, selective deficiency of RAGE signaling in microglia or RAGE inhibition with specific IgG were capable of preventing p38MAPK phosphorylation after OGD (30.39 ± 11.7 U/ng and 58.2 ± 25.9 U/ng in DNMSR × mhAPP and anti-RAGE IgG treated slices, respectively, n = 6; p < 0.001 vs mhAPP OGD-exposed slices; p > 0.05 vs mhAPP nonexposed slices). Interestingly, the pharmacological inhibition of the other stress-related kinase (JNK) abolished the effect of OGD in either WT or mhAPP slices. Indeed, in presence of SP600125 phospho-p38MAPK after OGD returned to baseline levels both in WT (22.43 ± 2.8 U/ng, n = 6; p < 0.01 vs WT OGD exposed slices; p > 0.05 vs WT nonexposed slices) and mhAPP slices (29.51 ± 9.18, n = 6; p < 0.001 vs mhAPP after OGD; p > 0.05 vs mhAPP without OGD exposure). To further explore the molecular link between kinase activation and IL-1β release, we measured phospho-p38MAPK levels in slices treated with IL-1Ra. As shown in Figure 6A, IL1Ra was not capable of preventing the increase of phospho-p38MAPK after OGD either in WT (110 ± 35 U/ng; n = 6; p < 0.05 vs WT nonexposed slices; p > 0.05 vs WT OGD exposed slices) or in mhAPP slices (370 ± 63.4 U/ng; n = 6; p < 0.001 vs mhAPP nonexposed slices; p < 0.05 vs mhAPP slices after OGD), suggesting that p38MAPK activation represents an upstream event with respect to IL-1β release. These data confirm that p38MAPK is strongly activated during transient ischemia in cortical slices overexpressing mutant human APP, and this phenomenon is specifically driven by RAGE expressed in microglia and requiring JNK phosphorylation.
Figure 6.

OGD increases p38MAPK/JNK phosphorylation in WT and mhAPP cortical slices. A, Plot representing averaged phospho-p38MAPK levels measured using ELISA and expressed as units per total content of p38MAPK protein. In WT slices, tissue levels of phospho-p38MAPK after 10 min of OGD were significantly higher with respect to control vehicle-treated slices (left, *p < 0.01). Selective deficiency of RAGE signaling in microglia (DNMSR) and complete inactivation of RAGE with anti-RAGE IgG was able to prevent the OGD-induced increase of phospho-p38MAPK (left, p < 0.05 vs WT OGD-exposed slices). p38MAPK activation by OGD was also reduced in presence of JNK inhibition (left, SP600125, p < 0.05 vs OGD-exposed WT slices), but not by treatment with IL-1Ra (left, *p < 0.05 vs WT control slices). A robust increase in phospho-p38MAPK was also observed in mhAPP slices after OGD (right, °p < 0.001 vs non-OGD mhAPP) and this increase was prevented in double-transgenic DNMSRxmhAPP or in anti-RAGE IgG-treated mhAPP slices (right, p > 0.05 vs non-OGD mhAPP slices). In mhAPP slices, JNK inhibition (right, SP600125) was effective in reducing p38MAPK activation, whereas phosphor-p38 was still elevated after IL-1Ra treatment (right, °p < 0.001 vs non-OGD mhAPP slices). B, Plot representing averaged phospho-JNK levels measured using ELISA and expressed as units/total content of JNK protein. Tissue levels of phospho-JNK were elevated after 10 min of OGD in WT cortical slices (left, *p < 0.05 vs WT nonexposed slices). Inhibition of RAGE (in DNMSR or anti-RAGE IgG-treated slices) or blockade of IL-1β signaling with IL-1Ra did not reduce the OGD effect (left, *p < 0.05 vs WT nonexposed slices), whereas inhibition of p38MAPK (SB203580) significantly reduced JNK activation by OGD (left, p > 0.05 vs WT nonexposed slices). A similar increase of JNK activation was observed in mhAPP slices exposed to OGD (right, °p < 0.05 vs mhAPP nonexposed slices). Blockade of RAGE by IgG or inhibition of RAGE in microglia in double-transgenic DNMSRxmhAPP mice suppressed OGD-induced increase of phospho-JNK (right, p > 0.05 vs nonexposed mhAPP slices). Basal levels of phospho-JNK were also maintained in mhAPP slices exposed to OGD that were treated with p38MAPK inhibitor (right, SB203580, p > 0.05 vs nonexposed mhAPP slices), whereas phospho-JNK was still activated in mhAPP slices treated with IL-1Ra and exposed to OGD (°p < 0.05 vs mhAPP nonexposed slices). Error bars indicate SEM.
Similarly, we measured JNK phosphorylation levels after OGD exposure in EC slices from either WT or mhAPP mice (Fig. 6B). Slice perfusion with ischemic solution for 10 min significantly increased the JNK phosphorylation compared with the vehicle-treated slices (15.47 ± 3.8 vs 6.53 ± 0.4 U/ng, n = 6; p < 0.05). However, different from p38MAPK, the activation of JNK was not prevented in DNMSR slices and in slices pretreated with anti-RAGE IgG (15.04 ± 1.45 U/ng and 16.38 ± 2.2, n = 6; p < 0.05 vs control WT nonexposed slices; p > 0.05 vs Wt OGD-exposed slices). Therefore, JNK phosphorylation in cortical slices after OGD is independent from RAGE signaling. A different result emerged when we examined the effect of OGD on JNK phosphorylation in mhAPP slices. Levels of phospho-JNK were elevated after OGD with respect to nonexposed mhAPP slices (11.82 ± 1.45 U/ng vs 6.02 ± 1.6 U/ng, n = 6, p < 0.05), but in this case, genetic inhibition of RAGE in microglia (DNMSR × mhAPP) or anti-RAGE blocking IgG were able to maintain basal values of phospho JNK after OGD (5.17 ± 0.9 and 7 ± 1.56 U/ng, respectively, n = 6; p < 0.05 vs mhAPP exposed to OGD; p > 0.05 vs mhAPP not exposed to OGD). Therefore, in the presence of mhAPP overexpression, JNK activation under transient ischemia is specifically controlled by microglial RAGE. Finally, to complete the analysis of stress-related signaling pathways involved in OGD-induced synaptic dysfunction, we measured JNK activation in WT and mhAPP slices treated with SB203580 and IL1Ra. Consistently with the results for p38MAPK, IL1Ra was not able to prevent JNK activation after OGD either in WT (14.79 ± 0.9 U/ng, n = 6; p < 0.05 vs WT not exposed to OGD) or in mhAPP cortical slices (14.53 ± 2 U/ng, n = 6; p < 0.05 vs mhAPP not exposed to OGD), confirming that the JNK pathway is upstream with respect to IL-1β signaling. In contrast, pharmacological inhibition of p38MAPK with SB203580 completely prevented OGD-induced activation of JNK in WT (4.86 ± 1.89 U/ng, n = 6; p < 0.05 vs WT after OGD) and in mhAPP (6.39 ± 0.1 U/ng, n = 6; p < 0.05 vs mhAPP exposed to OGD). These results confirm that, after transient ischemia and in the presence of mhAPP overexpression, a cell-specific RAGE signaling involves the phosphorylation of JNK and p38MAPK that are probably coactivated and reciprocally required to induce synaptic depression.
Discussion
The present study focused on RAGE as a key molecule that mediates the effect of transient ischemia on synaptic function. Previous results have linked RAGE to brain ischemia. In particular, it has been shown that RAGE mediates ischemic damage in an in vivo model of medial cerebral artery occlusion (MCAO), contributing to neuroinflammation (Muhammad et al., 2008) and modulating the severity of injury (Hassid et al., 2009). In addition, RAGE expression is modulated by the Hypoxia-inducible Factor-1-α (HIF-1α), a transcription factor activated during ischemia (Pichiule et al., 2007). Here, using an in vitro model of glucose/oxygen deprivation and genetic inhibition of RAGE, we provide evidence that this receptor, and particularly its expression on microglial cells, can modulate an inflammatory pathway controlling synaptic function during transient ischemia. However, activation of RAGE signaling during ischemia can lead to deleterious or beneficial consequences. In particular, overexpression of neuronal RAGE renders the brain more vulnerable to ischemia/reperfusion injury (Hassid et al., 2009), whereas another study demonstrated that inhibition of RAGE signaling resulted in neuroprotection (Pichiule et al., 2007). Our present work partially confirmed this dual role for RAGE because brain slices were protected against an OGD effect on synaptic function either by complete inhibition (RAGE KO or anti-RAGE IgG) or selective inhibition of RAGE in microglia (DNMSR), but not in slices with deficient RAGE signaling in neurons. One possible explanation for this difference is the diversity of ligands that may interact with RAGE, triggering specific intracellular pathways and the localization of RAGE expression in different cell types. Indeed, a search for ligands capable of binding RAGE resulted in an array of molecules; for example, other than the AGEs (advanced glycation end-products), RAGE binds high mobility group (HMG) proteins, including HMGB1 (also known as amphoterin), S100/calgranulin, and β-amyloid. The protective anti-inflammatory effect of HMGB1 antagonism at the receptor RAGE has been demonstrated in an in vivo model of brain ischemia and confirmed in vitro, when RAGE expression in microglial cells mediated the toxic effect of HMGB1 (Muhammad et al., 2008). Conversely, treatment of neurons with low concentrations of S100B decreased neuronal death after OGD and this effect was abolished by a neutralizing antibody against RAGE. Conversely, high concentrations of exogenous S100B had a cytotoxic effect that was RAGE independent (Pichiule et al., 2007). Furthermore, some experimental evidence has demonstrated a clear role for the Aβ protein in the development of neuronal injury under ischemic conditions (for review, see Pluta et al., 2013). It is important to note that hypoxia upregulates APP cleaving enzymes such as BACE1 (β-site APP cleaving enzyme) and γ-secretase, resulting in Aβ overproduction (Tanimukai et al., 1998; Pennypacker et al., 1999; Sun et al., 2006; Zhang et al., 2007; Guglielmotto et al., 2009; Li et al., 2010; Zhiyou et al., 2009) and an increased risk of AD development. The link between Aβ protein accumulation and ischemia has been strengthened by observations in AD mouse models carrying either single (Zhang et al., 1997) or double (Kitaguchi et al., 2009) APP mutations. Conversely, suppression of the WT protein in APP knock-out mice increased the risk of mortality as a result of cerebral ischemia (Koike et al., 2012); accordingly, overexpression of WT APP provides neuroprotection after MCAO in rats (Clarke et al., 2007). This evidence suggests that, when expressed normally, APP might be a stress response protein that is beneficial in a hypoxic/ischemic environment; however, it becomes detrimental when it is abnormally processed to form toxic Aβ peptides. In agreement with this hypothesis, our experiments demonstrate that the OGD effect on synaptic function is worsened in the presence of elevated Aβ or in an environment that is prone to overproducing Aβ, such as in slices from mhAPP mice. An additional important finding in mhAPP mice was that Aβ levels were increased after OGD and that by inhibiting γ-secretase, we were able to ameliorate synaptic function.
Previous reports showed that RAGE is implicated in disruption of synaptic function by Aβ (Arancio et al., 2004; Origlia et al., 2008). In the EC, nanomolar oligomeric Aβ inhibits LTP through activation of RAGE in neurons (Origlia et al., 2008), whereas microglial RAGE contributes to synaptic depression and LTD impairment induced by higher concentrations of Aβ (Origlia et al., 2010). In the present study, we investigated the cell-specific Aβ/RAGE signaling that is triggered by ischemia and lead to synaptic depression. The important finding was that selective inhibition of RAGE in microglial cells was able to mitigate the effect of OGD, ameliorating synaptic function. This result is in agreement with previous observations that microglial RAGE plays a key role in the development of inflammatory response and cellular dysfunction in an Aβ milieu (Du Yan et al., 1997; Lue et al., 2001; Fang et al., 2010) and raise the important issue of identifying the mediators released by stimulated microglial cells on exposure to OGD that are capable of interacting with neurons. The activation of RAGE in microglia enhances production of proinflammatory cytokines such as IL-1β and TNF-α (Lue et al., 2001; for review, see Simm et al., 2004; Chen et al., 2007; Fang et al., 2010). Importantly, IL-1β is implicated in disruption of synaptic efficacy induced by proinflammatory agents (Curran et al., 2003; Kelly et al., 2003) and is capable of inducing synaptic depression in the hippocampus (Ikegaya et al., 2003). In our previous work, we confirmed that IL-1β perfusion on EC slices causes a depression of FP amplitude that can persist after the washout. Moreover, we demonstrated that Aβ/RAGE signaling in microglia mediated the induction of IL-1β in EC slices (Origlia et al., 2010). Consistent with a previous work in the hippocampus (Yoshioka et al., 1999), our present data demonstrated that IL-1β is involved in synaptic impairment induced by OGD that can be prevented by IL-1Ra. More importantly, IL-1 antagonism was effective in ameliorating synaptic function in OGD-exposed mhAPP slices, in which IL-1β elevation was particularly evident. This effect was RAGE specific, because functional inactivation of the receptor or deficiency of RAGE signaling in microglia significantly suppressed OGD-induced IL-1β production. Therefore, Aβ/RAGE signaling in microglia under an ischemic insult controls the release of IL-1β that can exert its effect on neurons, causing synaptic depression.
To gain further insight in to the intracellular signaling cascade(s) that might be activated by Aβ-RAGE interaction during OGD, leading to IL-1β release, we investigated the role of p38MAPK and JNK. We focused our attention to these kinases because they are involved in the cellular response to OGD (Shinozaki et al., 2007; Guglielmotto et al., 2009; Liu et al., 2011) and because they are strongly activated in EC slices exposed to high levels of Aβ (Takuma et al., 2005; Origlia et al., 2008, 2010). We found that OGD induced the phosphorylation of p38MAPK and that this effect was enhanced in mhAPP slices. In agreement with our hypothesis, p38MAPK activation was reduced by RAGE inhibition in microglia, but not in the presence of IL-1β antagonism. Interestingly, when ischemia was applied to WT slices, the phosphorylation of the other kinase, JNK, was independent from RAGE because it was not prevented by functional or genetic inhibition of the receptor. This result could be the consequence of an early direct activation of JNK induced by hypoxia via oxidative stress (Tamagno et al., 2008, 2009; Guglielmotto et al., 2009). In contrast, the increase of phospho-JNK was predominantly controlled by microglial RAGE in mhAPP mice, confirming that in presence of elevated Aβ, a specific inflammatory pathway, is stimulated during OGD, contributing to synaptic dysfunction. This result is consistent with the robust activation of JNK induced by Aβ that has been described previously (Shoji et al., 2000; Morishima et al., 2001; Troy et al., 2001; Jang and Surh, 2002; Wei et al., 2002; Fogarty et al., 2003; Minogue et al., 2003, Origlia et al., 2010). However, regardless of Aβ, the JNK activation was upstream of IL-1β release because it was not inhibited by IL-1Ra. Finally, we demonstrated that specific inhibition of only one of the two kinases using SB203580 or SP600125 was sufficient to prevent the activation of the other and to prevent synaptic depression. Therefore, p38MAPK and JNK coactivation is an important key event required for Aβ-mediated synaptic depression that is triggered by OGD. Together, our results suggest that in Aβ-enriched conditions, the stimulation of RAGE in microglial cells leads to the simultaneous activation of JNK and p38MAPK. These kinases are capable of modulating IL-1β release, which affects neuronal function, inducing synaptic depression (Fig. 7). Microglial cells were shown to be critically involved in plastic changes associated with either physiological (Wake et al., 2009) or pathological (for review, see Kettenmann et al., 2013) conditions. In particular, the amyloid-dependent LTP blockade in hippocampus was prevented by microglia inhibition (Wang et al., 2004b). More recently, a specific role for microglia in synaptic depression induced by hypoxia has emerged (J. Zhang et al., 2014). In that study, the combination of hypoxia and an inflammatory stimulus (LPS) was shown to trigger a long-term form of synaptic depression with a mechanism that requires microglial CR3 receptor. In addition, LPS + hypoxia LTD was caused by AMPA receptor endocytosis (J. Zhang et al., 2014). Furthermore, elevated extracellular Aβ causes synaptic depression, negatively modulating AMPA-mediated currents and inducing AMPA receptor endocytosis with a mechanism dependent on p38MAPK activation (Hsieh et al., 2006; Origlia et al., 2010). This is somewhat consistent with our present findings, which demonstrate a synergistic effect of Aβ and OGD capable of inducing a similar form of LTD that is dependent on RAGE signaling in microglial cells and on activation of cell-stress kinases.
Figure 7.

Schematic drawing representing the interaction of neurons and microglia that is triggered by OGD in an amyloid-enriched environment. An ischemic insult enhances the deleterious effect of amyloid on synaptic function. Amyloid overproduction induced by OGD leads to RAGE engagement in microglia and activation of p38mapk and JNK. The kinases, in turn, control the release of proinflammatory cytokines such as IL-1β, which may affect synaptic function. This is a self-maintained loop that amplifies the effect of Aβ and shift synaptic activity toward depression.
Some clinical and experimental evidence supports the hypothesis that transient ischemic episodes may accelerate Aβ-dependent synaptic dysfunction, leading to cognitive impairment and development of AD pathology (De la Torre, 2005, 2008; Gorelick et al., 2011; Pimentel-Coelho et al., 2013; for review, see Pluta et al., 2013). In view of this, our present study suggests that the inflammatory pathway driven by the RAGE/Aβ interaction may represent a basic mechanism that links vascular pathology to AD.
Footnotes
This work was supported by the Alzheimer's Association (Grant NIRG-173407 to N.O.), the Ministry of Education, Universities and Research Ageing Project “Research on the Molecular Mechanisms Involved in Ageing,” Regione Toscana (Health Research Program) and Research Projects of National Interest (2010 Italian Ministry of Research to L.D.), and the National Institutes of Health (Grant R37AG037319 to S.S.D.Y.). We thank R. Di Renzo and C. Orsini for technical assistance and Marty Watterson for the gift of MW108 compound.
The authors declare no competing financial interests.
References
- Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, et al. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004;23:4096–4105. doi: 10.1038/sj.emboj.7600415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benakis C, Bonny C, Hirt L. JNK inhibition and inflammation after cerebral ischemia. Brain Behav Immun. 2010;24:800–811. doi: 10.1016/j.bbi.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- Chen C, Li XH, Tu Y, Sun HT, Liang HQ, Cheng SX, Zhang S. Aβ-AGE aggravates cognitive deficit in rats via RAGE pathway. Neuroscience. 2014;257:1–10. doi: 10.1016/j.neuroscience.2013.10.056. [DOI] [PubMed] [Google Scholar]
- Chen X, Walker DG, Schmidt AM, Arancio O, Lue LF, Yan SD. RAGE: a potential target for Abeta-mediated cellular perturbation in Alzheimer's disease. Curr Mol Med. 2007;7:735–742. doi: 10.2174/156652407783220741. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Clarke J, Thornell A, Corbett D, Soininen H, Hiltunen M, Jolkkonen J. Overexpression of APP provides neuroprotection in the absence of functional benefit following middle cerebral artery occlusion in rats. Eur J Neurosci. 2007;26:1845–1852. doi: 10.1111/j.1460-9568.2007.05807.x. [DOI] [PubMed] [Google Scholar]
- Coogan AN, O'Neill LA, O'Connor JJ. The P38 mitogen-activated protein kinase inhibitor SB203580 antagonizes the inhibitory effects of interleukin-1beta on long-term potentiation in the rat dentate gyrus in vitro. Neuroscience. 1999;93:57–69. doi: 10.1016/S0306-4522(99)00100-1. [DOI] [PubMed] [Google Scholar]
- Cullen WK, Suh YH, Anwyl R, Rowan MJ. Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport. 1997;8:3213–3217. doi: 10.1097/00001756-199710200-00006. [DOI] [PubMed] [Google Scholar]
- Curran BP, Murray HJ, O'Connor JJ. A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1beta and long-term depression in the rat dentate gyrus in vitro. Neuroscience. 2003;118:347–357. doi: 10.1016/S0306-4522(02)00941-7. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. Is Alzheimer's disease preceded by neurodegeneration or cerebral hypoperfusion? Ann Neurol. 2005;57:783–784. doi: 10.1002/ana.20516. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. Alzheimer's disease prevalence can be lowered with non-invasive testing. J Alzheimers Dis. 2008;14:353–359. doi: 10.3233/jad-2008-14310. [DOI] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, Rajavashisth T, Chen X, Godman GC, Stern D, Schmidt AM. Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:5296–5301. doi: 10.1073/pnas.94.10.5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Yan S, Schmidt AM, Chen JX, Yan SS. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer's disease. FASEB J. 2010;24:1043–1055. doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty MP, Downer EJ, Campbell V. A role for c-Jun N-terminal kinase 1 (JNK1), but not JNK2, in the beta-amyloid-mediated stabilization of protein p53 and induction of the apoptotic cascade in cultured cortical neurons. Biochem J. 2003;371:789–798. doi: 10.1042/BJ20021660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, et al. American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmotto M, Aragno M, Autelli R, Giliberto L, Novo E, Colombatto S, Danni O, Parola M, Smith MA, Perry G, Tamagno E, Tabaton M. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J Neurochem. 2009;108:1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x. [DOI] [PubMed] [Google Scholar]
- Gürsoy M, Büyükuysal RL. Mechanism of S100b release from rat cortical slices determined under basal and stimulated conditions. Neurochem Res. 2010;35:429–436. doi: 10.1007/s11064-009-0075-9. [DOI] [PubMed] [Google Scholar]
- Hassid BG, Nair MN, Ducruet AF, Otten ML, Komotar RJ, Pinsky DJ, Schmidt AM, Yan SF, Connolly ES. Neuronal RAGE expression modulates severity of injury following transient focal cerebral ischemia. J Clin Neurosci. 2009;16:302–306. doi: 10.1016/j.jocn.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Hofmeijer J, van Putten MJ. Ischemic cerebral damage: an appraisal of synaptic failure. Stroke. 2012;43:607–615. doi: 10.1161/STROKEAHA.111.632943. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegaya Y, Delcroix I, Iwakura Y, Matsuki N, Nishiyama N. Interleukin-1beta abrogates long-term depression of hippocampal CA1 synaptic transmission. Synapse. 2003;47:54–57. doi: 10.1002/syn.10154. [DOI] [PubMed] [Google Scholar]
- Jang JH, Surh YJ. beta-Amyloid induces oxidative DNA damage and cell death through activation of c-Jun N terminal kinase. Ann N Y Acad Sci. 2002;973:228–236. doi: 10.1111/j.1749-6632.2002.tb04639.x. [DOI] [PubMed] [Google Scholar]
- Kelly A, Vereker E, Nolan Y, Brady M, Barry C, Loscher CE, Mills KH, Lynch MA. Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1 beta on long term potentiation in rat dentate gyrus. J Biol Chem. 2003;278:19453–19462. doi: 10.1074/jbc.M301938200. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77:10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Kawahara K, Biswas KK, Ito T, Tancharoen S, Morimoto Y, Matsuda F, Oyama Y, Takenouchi K, Miura N, Arimura N, Nawa Y, Meng X, Shrestha B, Arimura S, Iwata M, Mera K, Sameshima H, Ohno Y, Maenosono R, et al. Minocycline attenuates both OGD-induced HMGB1 release and HMGB1-induced cell death in ischemic neuronal injury in PC12 cells. Biochem Biophys Res Commun. 2009;385:132–136. doi: 10.1016/j.bbrc.2009.04.041. [DOI] [PubMed] [Google Scholar]
- Kitaguchi H, Tomimoto H, Ihara M, Shibata M, Uemura K, Kalaria RN, Kihara T, Asada-Utsugi M, Kinoshita A, Takahashi R. Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res. 2009;1294:202–210. doi: 10.1016/j.brainres.2009.07.078. [DOI] [PubMed] [Google Scholar]
- Koike MA, Lin AJ, Pham J, Nguyen E, Yeh JJ, Rahimian R, Tromberg BJ, Choi B, Green KN, LaFerla FM. APP knockout mice experience acute mortality as the result of ischemia. PLoS One. 2012;7:e42665. doi: 10.1371/journal.pone.0042665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wang W, Wang C, Tang YY. Possible involvement of NO/NOS signaling in hippocampal amyloid-beta production induced by transient focal cerebral ischemia in aged rats. Neurosci Lett. 2010;470:106–110. doi: 10.1016/j.neulet.2009.12.064. [DOI] [PubMed] [Google Scholar]
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- Liu R, Zhang L, Lan X, Li L, Zhang TT, Sun JH, Du GH. Protection by borneol on cortical neurons against oxygen-glucose deprivation/reperfusion: involvement of anti-oxidation and anti-inflammation through nuclear transcription factor kappaappaB signaling pathway. Neuroscience. 2011;176:408–419. doi: 10.1016/j.neuroscience.2010.11.029. [DOI] [PubMed] [Google Scholar]
- Liu R, Li JZ, Song JK, Zhou D, Huang C, Bai XY, Xie T, Zhang X, Li YJ, Wu CX, Zhang L, Li L, Zhang TT, Du GH. Pinocembrin improves cognition and protects the neurovascular unit in Alzheimer related deficits. Neurobiol Aging. 2014;35:1275–1285. doi: 10.1016/j.neurobiolaging.2013.12.031. [DOI] [PubMed] [Google Scholar]
- Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol. 2001;171:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- Medvedeva YV, Lin B, Shuttleworth CW, Weiss JH. Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J Neurosci. 2009;29:1105–1114. doi: 10.1523/JNEUROSCI.4604-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minogue AM, Schmid AW, Fogarty MP, Moore AC, Campbell VA, Herron CE, Lynch MA. Activation of the c-Jun N-terminal kinase signaling cascade mediates the effect of amyloid-beta on long term potentiation and cell death in hippocampus: a role for interleukin-1beta? J Biol Chem. 2003;278:27971–27980. doi: 10.1074/jbc.M302530200. [DOI] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozes E, Hunya A, Posa A, Penke B, Datki Z. A novel method for the rapid determination of beta-amyloid toxicity on acute hippocampal slices using MTT and LDH assays. Brain Res Bull. 2012;87:521–525. doi: 10.1016/j.brainresbull.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, Schwaninger M. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28:12023–12031. doi: 10.1523/JNEUROSCI.2435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, Domenici L. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci. 2008;28:3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Capsoni S, Cattaneo A, Fang F, Arancio O, Yan SD, Domenici L. Abeta-dependent Inhibition of LTP in different intracortical circuits of the visual cortex: the role of RAGE. J Alzheimers Dis. 2009a;17:59–68. doi: 10.3233/JAD-2009-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Arancio O, Domenici L, Yan SS. MAPK, beta-amyloid and synaptic dysfunction: the role of RAGE. Expert Rev Neurother. 2009b;9:1635–1645. doi: 10.1586/ern.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Bonadonna C, Rosellini A, Leznik E, Arancio O, Yan SS, Domenici L. Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J Neurosci. 2010;30:11414–11425. doi: 10.1523/JNEUROSCI.2127-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameshwaran K, Sims C, Kanju P, Vaithianathan T, Shonesy BC, Dhanasekaran M, Bahr BA, Suppiramaniam V. Amyloid beta-peptide Abeta(1-42) but not Abeta(1–40) attenuates synaptic AMPA receptor function. Synapse. 2007;61:367–374. doi: 10.1002/syn.20386. [DOI] [PubMed] [Google Scholar]
- Parent AT, Barnes NY, Taniguchi Y, Thinakaran G, Sisodia SS. Presenilin attenuates receptor-mediated signaling and synaptic function. J Neurosci. 2005;25:1540–1549. doi: 10.1523/JNEUROSCI.3850-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennypacker KR, Hernandez H, Benkovic S, Morgan DG, Willing AE, Sanberg PR. Induction of presenilins in the rat brain after middle cerebral arterial occlusion. Brain Res Bull. 1999;48:539–543. doi: 10.1016/S0361-9230(99)00031-3. [DOI] [PubMed] [Google Scholar]
- Pichiule P, Chavez JC, Schmidt AM, Vannucci SJ. Hypoxia-inducible factor-1 mediates neuronal expression of the receptor for advanced glycation end products following hypoxia/ischemia. J Biol Chem. 2007;282:36330–36340. doi: 10.1074/jbc.M706407200. [DOI] [PubMed] [Google Scholar]
- Pimentel-Coelho PM, Michaud JP, Rivest S. Effects of mild chronic cerebral hypoperfusion and early amyloid pathology on spatial learning and the cellular innate immune response in mice. Neurobiol Aging. 2013;34:679–693. doi: 10.1016/j.neurobiolaging.2012.06.025. [DOI] [PubMed] [Google Scholar]
- Pluta R, Jabłoński M, Ułamek-Kozioł M, Kocki J, Brzozowska J, Januszewski S, Furmaga-Jabłońska W, Bogucka-Kocka A, Maciejewski R, Czuczwar SJ. Sporadic Alzheimer's disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer's disease genes. Mol Neurobiol. 2013;48:500–515. doi: 10.1007/s12035-013-8439-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Fa' M, Staniszewski A, Hashimoto G, Aziz F, Sakurai M, Ribe EM, Troy CM, Mercken M, Jung SS, Palmeri A, Arancio O. Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann Neurol. 2011;69:819–830. doi: 10.1002/ana.22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki Y, Sato Y, Koizumi S, Ohno Y, Nagao T, Inoue K. Retinoic acids acting through retinoid receptors protect hippocampal neurons from oxygen-glucose deprivation-mediated cell death by inhibition of c-jun-N-terminal kinase and p38 mitogen-activated protein kinase. Neuroscience. 2007;147:153–163. doi: 10.1016/j.neuroscience.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Shoji M, Iwakami N, Takeuchi S, Waragai M, Suzuki M, Kanazawa I, Lippa CF, Ono S, Okazawa H. JNK activation is associated with intracellular beta-amyloid accumulation. Brain Res Mol Brain Res. 2000;85:221–233. doi: 10.1016/S0169-328X(00)00245-X. [DOI] [PubMed] [Google Scholar]
- Simm A, Casselmann C, Schubert A, Hofmann S, Reimann A, Silber RE. Age associated changes of AGE-receptor expression: RAGE upregulation is associated with human heart dysfunction. Exp Gerontol. 2004;39:407–413. doi: 10.1016/j.exger.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang LE, Song W. Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A. 2006;103:18727–18732. doi: 10.1073/pnas.0606298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma K, Yao J, Huang J, Xu H, Chen X, Luddy J, Trillat AC, Stern DM, Arancio O, Yan SS. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 2005;19:597–598. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo DG, Mattson MP, Tabaton M. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem. 2008;104:683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagno E, Guglielmotto M, Giliberto L, Vitali A, Borghi R, Autelli R, Danni O, Tabaton M. JNK and ERK1/2 pathways have a dual opposite effect on the expression of BACE1. Neurobiol Aging. 2009;30:1563–1573. doi: 10.1016/j.neurobiolaging.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Tanimukai H, Imaizumi K, Kudo T, Katayama T, Tsuda M, Takagi T, Tohyama M, Takeda M. Alzheimer-associated presenilin-1 gene is induced in gerbil hippocampus after transient ischemia. Brain Res Mol Brain Res. 1998;54:212–218. doi: 10.1016/S0169-328X(97)00337-9. [DOI] [PubMed] [Google Scholar]
- Tozzi A, Costa C, Di Filippo M, Tantucci M, Siliquini S, Belcastro V, Parnetti L, Picconi B, Calabresi P. Memantine reduces neuronal dysfunctions triggered by in vitro ischemia and 3-nitropropionic acid. Exp Neurol. 2007;207:218–226. doi: 10.1016/j.expneurol.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–164. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid beta-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Rowan MJ, Anwyl R. Beta-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci. 2004b;24:6049–6056. doi: 10.1523/JNEUROSCI.0233-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson DM, Grum-Tokars VL, Roy SM, Schavocky JP, Bradaric BD, Bachstetter AD, Xing B, Dimayuga E, Saeed F, Zhang H, Staniszewski A, Pelletier JC, Minasov G, Anderson WF, Arancio O, Van Eldik LJ. Development of novel in vivo chemical probes to address CNS protein kinase involvement in synaptic dysfunction. PLoS One. 2013;26:e66226. doi: 10.1371/journal.pone.0066226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Norton DD, Wang X, Kusiak JW. Abeta 17–42 in Alzheimer's disease activates JNK and caspase-8 leading to neuronal apoptosis. Brain. 2002;125:2036–2043. doi: 10.1093/brain/awf205. [DOI] [PubMed] [Google Scholar]
- Witter MP, Groenewegen HJ, Lopes da Silva FH, Lohman AH. Functional organization of the extrinsic and intrinsic circuitry of the parahippocampal region. Prog Neurobiol. 1989;33:161–253. doi: 10.1016/0301-0082(89)90009-9. [DOI] [PubMed] [Google Scholar]
- Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P, et al. Non-enzymatically glycated tau in Alzheimer's disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat Med. 1995;1:693–699. doi: 10.1038/nm0795-693. [DOI] [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- Yan Y, Liu Y, Sorci M, Belfort G, Lustbader JW, Yan SS, Wang C. Surface plasmon resonance and nuclear magnetic resonance studies of ABAD-Abeta interaction. Biochemistry. 2007;46:1724–1731. doi: 10.1021/bi061314n. [DOI] [PubMed] [Google Scholar]
- Yoshioka M, Itoh Y, Mori K, Ueno K, Matsumoto M, Togashi H. Effects of an interleukin-1beta analogue [Lys-D-Pro-Thr], on incomplete cerebral ischemia-induced inhibition of long-term potentiation in rat hippocampal neurons in vivo. Neurosci Lett. 1999;261:171–174. doi: 10.1016/S0304-3940(99)00004-X. [DOI] [PubMed] [Google Scholar]
- Zhai DX, Kong QF, Xu WS, Bai SS, Peng HS, Zhao K, Li GZ, Wang DD, Sun B, Wang JH, Wang GY, Li HL. RAGE expression is up-regulated in human cerebral ischemia and pMCAO rats. Neurosci Lett. 2008;445:117–121. doi: 10.1016/j.neulet.2008.08.077. [DOI] [PubMed] [Google Scholar]
- Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci. 1997;17:7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Malik A, Choi HB, Ko RW, Dissing-Olesen L, MacVicar BA. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron. 2014;82:195–207. doi: 10.1016/j.neuron.2014.01.043. [DOI] [PubMed] [Google Scholar]
- Zhang T, Wang H, Li Q, Huang J, Sun X. Modulating autophagy affects neuroamyloidogenesis in an in vitro ischemic stroke model. Neuroscience. 2014;263:130–137. doi: 10.1016/j.neuroscience.2014.01.012. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF, Xu H, Zhang YW. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem. 2007;282:10873–10880. doi: 10.1074/jbc.M608856200. [DOI] [PubMed] [Google Scholar]
- Zhiyou C, Yong Y, Shanquan S, Jun Z, Liangguo H, Ling Y, Jieying L. Upregulation of BACE1 and beta-amyloid protein mediated by chronic cerebral hypoperfusion contributes to cognitive impairment and pathogenesis of Alzheimer's disease. Neurochem Res. 2009;34:1226–1235. doi: 10.1007/s11064-008-9899-y. [DOI] [PubMed] [Google Scholar]