

Abstract
It is now clear that many neuronal populations release more than one classical neurotransmitter, yet in most cases the functional role of corelease is unknown. Striatal cholinergic interneurons release both glutamate and acetylcholine, and vesicular loading of glutamate has been shown to enhance acetylcholine content. Using a combination of optogenetics and whole-cell recordings in mice, we now provide physiological evidence that optogenetic stimulation of cholinergic interneurons triggers monosynaptic glutamate- and acetylcholine-mediated currents in striatal fast-spiking interneurons (FSIs), both of which depend on the expression of the vesicular glutamate transporter 3 (VGLUT3). In contrast to corticostriatal glutamatergic inputs onto FSIs, which are mediated primarily by AMPA-type glutamate receptors, glutamate release by cholinergic interneurons activates both AMPA- and NMDA-type glutamate receptors, suggesting a unique role for these inputs in the modulation of FSI activity. Importantly, we find that the loss of VGLUT3 not only markedly attenuates glutamatergic and cholinergic inputs on FSIs, but also significantly decreases disynaptic GABAergic input onto medium spiny neurons (MSNs), the major output neurons of the striatum. Our data demonstrate that VGLUT3 is required for normal cholinergic signaling onto FSIs, as well as for acetylcholine-dependent disynaptic inhibition of MSNs. Thus, by supporting fast glutamatergic transmission as well as by modulating the strength of cholinergic signaling, VGLUT3 has the capacity to exert widespread influence on the striatal network.
Keywords: acetylcholine, corelease, fast-spiking interneuron, glutamate, striatum, VGLUT3
Introduction
Many neuronal populations release multiple fast neurotransmitters (for review, see Hnasko and Edwards, 2011), which has the potential to increase the temporal complexity of neural coding (Lee et al., 2010; Ren et al., 2011) or vesicular neurotransmitter content (Gras et al., 2008; Hnasko et al., 2010). Nevertheless, the functional role of multitransmitter signaling in most cases is still unknown.
Identification of the vesicular glutamate transporters (VGLUTs) has greatly enhanced our ability to identify glutamatergic neurons, including many unexpected populations (El Mestikawy et al., 2011; Hnasko and Edwards, 2012). The three VGLUT isoforms have a largely mutually exclusive distribution. VGLUT1 and 2 are the predominant isoforms (Fremeau et al., 2001; Herzog et al., 2001), while VGLUT3 is expressed sparsely, often in neurons better known for releasing a different classical transmitter. These include rostral raphe serotonergic neurons and striatal cholinergic interneurons (Fremeau et al., 2002; Gras et al., 2002). In cholinergic interneurons, VGLUT3 and the vesicular acetylcholine transporter (VAChT) are localized to the same synaptic vesicles, consistent with the idea that cholinergic interneurons release both transmitters in vivo (Gras et al., 2008). Indeed, optogenetic activation of striatal cholinergic interneurons triggers glutamatergic currents in striatal medium spiny neurons (MSNs; Higley et al., 2011). In addition, VGLUT3 may enhance vesicular loading of acetylcholine. A reduction in radiolabeled acetylcholine loading was observed in VGLUT3 knock-out (KO) striatum in brain slices, indicating a potential role for glutamate transport in vesicular acetylcholine content (Gras et al., 2008). However, physiological measurements of the impact of glutamate copackaging on the striatal microcircuit are lacking.
Electron microscopy studies demonstrated that cholinergic interneurons contact MSNs as well as parvalbumin-positive interneurons (Chang and Kita, 1992), which correspond physiologically to fast-spiking interneurons (FSIs). Though small in number, FSIs provide potent inhibitory input to numerous MSNs (Koós and Tepper, 1999; Gittis et al., 2010). A pharmacological study in striatal slices further suggested that cholinergic signaling regulates FSIs and their synaptic output (Koós and Tepper, 2002). Here, we take advantage of optogenetics and patch-clamp electrophysiology to examine the synaptic relationship between these two sparse populations in a more physiological context.
Materials and Methods
Animals.
Lhx6-GFP and ChAT-Cre mice (both from GENSAT; www.gensat.org) were crossed to yield ChAT-Cre::Lhx6-GFP mice. VGLUT3 KO mice (Seal et al., 2008) were bred separately to generate ChAT-Cre::Lhx6-GFP::VGLUT3 KO mice. We used mice of either sex on a C57BL/6 background.
Surgery.
Stereotaxic surgeries were performed at 4–6 weeks of age. Anesthesia was induced with intraperitoneal ketamine/xylazine and maintained with inhaled isoflurane (1%). ChAT-Cre::Lhx6-GFP mice were injected with AAV2/1-DIO-ChR2-mCherry (1–2 μl) in the dorsal striatum (+0.8 mm anterior, − 1.5 mm lateral, and −2.3 mm inferior from bregma). Lhx6-GFP mice were injected with AAV5-hSyn-ChR2-YFP (1 μl) in the bilateral primary motor and sensory cortices (coordinates +0.5 and +1.5 anterior, ± 2 lateral, 0.7 mm inferior from bregma).
Histology.
Two to three weeks following injections, mice were deeply anesthetized with intraperitoneal ketamine-xylazine and transcardially perfused with PBS, followed by 4% paraformaldehyde in PBS. Brains were postfixed overnight and placed in 30% sucrose at 4°C for 2–3 d to cryoprotect. Coronal slices were cut (30 μm) and blocked in 10% normal donkey serum (NDS) and 1% Triton X-100 (Triton) in PBS for 1 h. Primary antibody (Millipore AB144P Anti-ChAT) was added 1:500 in 5% NDS and 1% Triton in PBS overnight at 4°C. After washing with 0.5% Triton-PBS, slices were incubated with DyLight 649-conjugated donkey anti-goat secondary antibody (Jackson ImmunoResearch) for 2 h at room temperature (RT), washed, and mounted with Prolong Antifade Gold (Invitrogen).
Physiology.
Two to four weeks following stereotaxic virus injection, mice were deeply anesthetized with isoflurane, decapitated, and the brain removed in ice-cold sucrose solution containing the following (in mm): 79 NaCl, 23 NaHCO3, 68 sucrose, 12 glucose, 2.3 KCl, 1.1 NaH2PO4, 6 MgCl2, and 0.5 CaCl2. Mounted on a submerged chuck, sequential coronal slices (300 μm) were cut on a vibratome (Leica) and transferred to a chamber of warm (33°C) carbogenated ACSF containing the following (in mm) 125 NaCl, 26 NaHCO3, 2.5 KCl, 1 MgCl2, 2 CaCl2, 1.25 NaH2PO4, and 12.5 glucose for 30 min. Slices were stored in carbogenated ACSF at RT and submerged in a chamber superfused with carbogenated ACSF at 31−33°C for recordings.
For recordings, the zone of maximal mCherry fluorescence within the dorsal striatum was chosen. Cholinergic interneurons showed mCherry fluorescence and large somata under DIC optics. FSIs showed Lhx6-GFP fluorescence, and typical physiological properties (Gittis et al., 2010). MSNs had nonfluorescent cell bodies. Neurons were patched in whole-cell current-clamp or voltage-clamp configuration using glass electrodes (3–5 MΩ) filled with potassium methanesulfonate-based (current-clamp) or cesium methanesulfonate-based (voltage-clamp) internal solution containing the following (in mm), respectively: 130 KMeSO3 or CsMeSO3, 10 NaCl, 2 MgCl2, 0.16 CaCl2, 0.5 EGTA, 10 HEPES, 2 Mg-ATP, and 0.3 Na-GTP or 120 CsMeS03, 15 CsCl, 8 NaCl, 0.5 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, and 5 QX-314. In experiments assessing the voltage dependence of EPSCs, spermine (0.1 mm) was included. For experiments measuring disynaptic IPSCs, the high-chloride, cesium-based internal solution contained the following (in mm): 120 CsCl, 15 CsMeSO3, 8 NaCl, 0.5 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, and 5 QX-314. Membrane potential was not corrected for the liquid junction potential.
Channelrhodopsin-2 (ChR2) was activated by 5 ms field illuminations through the 40× microscope objective using a mercury arc lamp (100 W) gated with a TTL-controlled shutter (Uniblitz). The filter cube contained a D470/40× excitation filter and cold mirror. Illumination intensity was adjusted to 1–3 mW, measured at the brain slice by adjustment of the aperture stop. Light pulses were delivered every 20 s.
Drugs.
All drugs were prepared as concentrated stock solutions and diluted in ACSF for each experiment. NBQX (Tocris Bioscience) was prepared in DMSO (Sigma), while APV (Tocris Bioscience), scopolamine (Sigma), mecamylamine (Tocris Bioscience), methyllycaconitine (Sigma), and picrotoxin (Sigma) were dissolved in water.
Statistics.
All data are mean ± SEM. Comparisons were made using the Student's t test, using neurons pooled from multiple mice.
Results
We used an optogenetic approach to investigate the corelease of glutamate and acetylcholine by striatal cholinergic interneurons onto FSIs. ChR2 fused to the fluorescent reporter mCherry was targeted to cholinergic interneurons by injecting the Cre-dependent virus AAV-DIO-ChR2-mCherry into the dorsal striatum of choline ChAT-Cre mice. Immunohistochemistry demonstrated selective expression of ChR2-mCherry in ChAT-positive striatal neurons (Fig. 1A).
Figure 1.
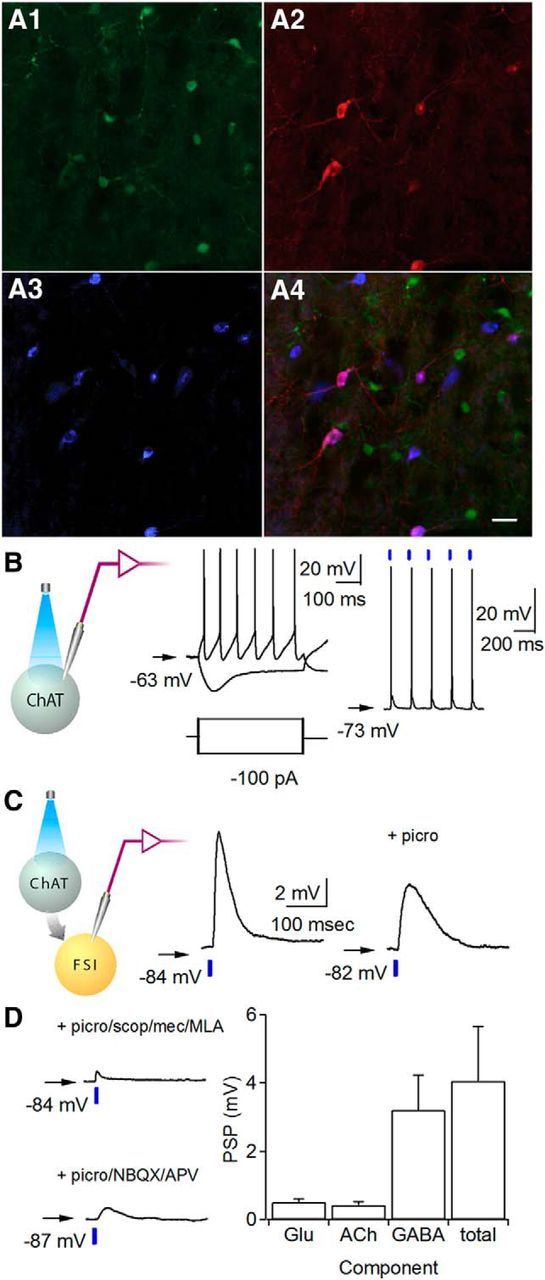
Optogenetic activation of striatal cholinergic interneurons elicits complex PSPs in FSIs. A1–A4, Striatal slices in ChAT-Cre::Lhx6-GFP mouse injected with AAV-DIO-ChR2-mCherry. Immunohistochemistry for Lhx6-GFP (A1), mCherry (A2), ChAT (A3), and all three (A4). Scale bar, 50 μm. B, Left, Recording configuration. Whole-cell current-clamp recordings from mCherry-positive cholinergic interneurons. Membrane potential responses to injection of ± 100 pA (left) or a train of 5 ms pulses of blue light (470 nm) at 5 Hz (right, blue bar). C, Left, Schematic of recording configuration. Representative light-evoked synaptic potentials in whole-cell current-clamp recordings from striatal FSIs before (center) and after (right) addition of picrotoxin (picro; 50 μm). D, Upper left, Presumed glutamatergic responses recorded in the presence of picrotoxin, scopolamine (scop; 10 μm), mecamylamine (mec; 5 μm), and methyllycaconitine (MLA; 10 nm). Lower left, Presumed cholinergic responses recorded in the presence of picrotoxin, NBQX, and APV. Right, Average PSP amplitude attributable to glutamate (Glu), acetylcholine (ACh), or GABA, and the total PSP. Arrow indicates resting membrane potential.
To physiologically confirm ChR2 expression in cholinergic interneurons, we performed whole-cell current-clamp recordings of mCherry-positive neurons in acute striatal slices. These neurons showed intrinsic properties typical of cholinergic interneurons, including spontaneous action potential firing and a voltage sag with hyperpolarizing current (Fig. 1B). To test ChR2 function, brief (5 ms) blue light (470 nm) pulses were delivered to the slice and mCherry-positive neurons fired single action potentials in response to each light pulse (Fig. 1B).
To target FSIs for whole-cell recordings, we used Lhx6-GFP mice, which express the green fluorescent reporter in a subset of GABAergic interneurons, including striatal FSIs (Gittis et al., 2010). Optogenetic activation of cholinergic interneurons with blue light pulses triggered depolarizing postsynaptic potentials (PSPs) in FSIs (Fig. 1C,D; 4.0 ± 1.6 mV, n = 5 neurons; n = 3 mice). The PSPs likely have a cholinergic component, but may also include monosynaptic glutamatergic and disynaptic GABAergic components, similar to those found in MSNs (Witten et al., 2010; English et al., 2011; Higley et al., 2011). Subsequent recordings were performed in the presence of the GABAA antagonist picrotoxin to block disynaptic GABAergic responses, which produced a PSP of 1.2 ± 0.2 mV (Fig. 1C,D; n = 15 neurons; n = 6 mice). In additional experiments, we isolated cholinergic and glutamatergic components using a mixture of either cholinergic antagonists (scopolamine, mecamylamine, and methyllycaconitine) or glutamatergic antagonists (NBQX and APV), respectively. The presumed cholinergic component measured 0.4 ± 0.1 mV (Fig. 1D; n = 12 neurons; n = 4 mice), and was blocked by the addition of cholinergic antagonists, while the presumed glutamatergic component measured 0.5 ± 0.1 mV (Fig. 1D; n = 6 neurons; n = 3 mice), and was blocked by the addition of glutamate receptor antagonists. These experiments indicate that optogenetic stimulation of cholinergic interneurons depolarizes FSIs, a portion of which is glutamate mediated.
To further characterize light-activated glutamatergic responses, we performed voltage-clamp recordings of FSIs in the presence of cholinergic and GABAergic antagonists. At hyperpolarized holding potentials, light pulses elicited EPSCs measuring 12 ± 1 pA (Fig. 2A; n = 20 neurons; n = 13 mice), which were abolished by glutamatergic antagonists NBQX and APV (Fig. 2A, n = 5 neurons), confirming that activation of cholinergic interneurons triggers glutamate release onto FSIs. At depolarized holding potentials, a substantial EPSC was also detected (Fig. 2A). To determine whether this outward current is carried by AMPA or NMDA receptors, we measured light-evoked EPSCs at different holding potentials before and after applying APV. Under control conditions, the current–voltage (I–V) curve showed substantial outward currents at depolarized potentials, while in the presence of APV, the EPSC showed an inwardly rectifying current–voltage relationship (Fig. 2A). Thus, optogenetic stimulation of cholinergic interneurons results in both AMPA and NMDA receptor-mediated EPSCs in FSIs.
Figure 2.

Optogenetic stimulation of cholinergic interneurons triggers VGLUT3-dependent glutamatergic currents. Recordings performed in picrotoxin, mecamylamine, scopolamine, and methyllycaconitine to isolate glutamatergic currents. A, Whole-cell voltage-clamp recordings of FSIs from ChAT-Cre::Lhx6-GFP mice injected with AAV-DIO-ChR2-mCherry. Left, Recording configuration. Representative light-evoked EPSC recorded at −70 mV, before (black) and after (gray) application of the AMPA antagonist NBQX (5 μm). Center, Representative light-evoked EPSCs at holding potentials of −80 mV (black) and +60 mV (gray). Right, Current–voltage relationship of EPSCs before (open symbols; n = 6 neurons) and after (filled symbols) application of APV (50 μm). B, Whole-cell voltage-clamp recordings from striatal FSIs in Lhx6-GFP mice following cortical injection of AAV-hSyn-ChR2-YFP. Left, Recording configuration. Center, Representative light-evoked EPSCs at holding potentials of −80 mV (black) and +60 mV (gray). Right, Current–voltage relationship of EPSCs (n = 6 neurons). C, Light-evoked currents in ChAT-Cre::Lhx6-GFP::VGLUT3 KO mice following striatal injection of AAV-DIO-ChR2-mCherry. Left, Recording configuration. Center, Representative light-evoked EPSC at −70 mV. Right, Glutamatergic EPSC amplitudes in WT (n = 19 neurons, small black circles) versus VGLUT3 KO mice (n = 8 neurons, red circle). Large filled symbols show average ± SEM in each group. Ctl, control. *p ≤ 0.05.
The substantial NMDA-mediated EPSC was surprising, as intrastriatal electrical stimulation, presumed to recruit excitatory cortical and thalamic afferents, evokes inwardly rectifying EPSCs in FSIs, with little to no NMDA-mediated component (Gittis et al., 2010). The inwardly rectifying AMPA-mediated EPSCs are mediated by GluA2-lacking AMPA receptors on FSIs (Gittis et al., 2010, 2011). To verify the NMDA-mediated EPSC was not an artifact of optogenetic stimulation, we stimulated cortically derived glutamatergic inputs onto FSIs using ChR2. The sensorimotor cortices of Lhx6-GFP mice were injected with AAV-hSyn-ChR2-YFP to express ChR2 in cortical neurons, some of which sent axons into the dorsal striatum. Illuminating such synaptic terminals evoked EPSCs, which were inward at hyperpolarized potentials and nearly absent at depolarized potentials (Fig. 2B). Cortically derived light-activated EPSCs in striatal FSIs showed an inwardly rectifying I–V curve (Fig. 2B), comparable to EPSCs generated with intrastriatal electrical stimulation. These findings suggest that glutamate released by cholinergic interneurons activates both AMPA and NMDA receptors on FSIs, while glutamate released by cortical afferents activates only AMPA receptors. The AMPA-mediated EPSC in both cases is inwardly rectifying, consistent with the presence of GluA2-lacking AMPA receptors.
While the detection of glutamate-mediated currents after optogenetic activation of cholinergic interneurons is consistent with the idea that cholinergic interneurons release glutamate, cholinergic interneurons may trigger another neuron population to release glutamate. Recent studies demonstrated that striatal dopamine terminals release glutamate (Hnasko et al., 2010), though this appears to be confined to the nucleus accumbens (Stuber et al., 2010). To confirm glutamate was derived from cholinergic interneurons, rather than dopamine neurons, we took advantage of the fact that these two neuron populations express different VGLUTs. Dopamine neurons express VGLUT2 (Kawano et al., 2006; Stuber et al., 2010), while striatal cholinergic interneurons express VGLUT3 (Fremeau et al., 2002; Gras et al., 2002). If cholinergic interneurons indeed release glutamate, light-evoked EPSCs should depend upon VGLUT3. Optogenetic activation of cholinergic interneurons in ChAT-Cre::Lhx6-GFP::VGLUT3 KO mice did not elicit measurable glutamatergic EPSCs in FSIs (Fig. 2C; 0.4 ± 0.2 pA, n = 8 neurons; n = 3 mice; p = 0.0001 compared with WT). These results suggest cholinergic interneurons release glutamate monosynaptically onto FSIs.
While pharmacological studies have shown cholinergic modulation of FSIs (Koós and Tepper, 2002), fast cholinergic currents in FSIs have not been measured directly. To measure nicotinic receptor-mediated neurotransmission, we recorded light-activated currents in FSIs in the presence of a drug mixture that blocked both GABAergic and glutamatergic transmission (picrotoxin, NBQX, and APV). We observed a slow inward light-activated current averaging 14 ± 5 pA with a τdecay of 87 ± 9 ms (Fig. 3A,B; n = 9 neurons; n = 6 mice). The cholinergic EPSC was not blocked by methyllycaconitine (n = 3 neurons, data not shown), but was blocked by mecamylamine (Fig. 3A; n = 6 neurons, n = 4 mice; p = 0.04, paired t test), indicating that non-α7 nicotinic acetylcholine receptors mediate the cholinergic EPSC in FSIs.
Figure 3.

Optogenetic stimulation of cholinergic interneurons triggers VGLUT3-dependent monosynaptic nicotinic currents. Whole-cell voltage-clamp recordings from striatal FSIs recorded in the presence of picrotoxin, NBQX, and APV to isolate cholinergic currents. A, ChAT-cre::Lhx6-GFP mice injected with AAV-DIO-ChR2-mCherry in dorsal striatum. Left, Recording configuration. Center, Representative light-activated currents recorded from an FSI at −70 mV before (black) and after (orange) application of mecamylamine. Right, EPSC amplitudes before and after addition of mecamylamine (mec; n = 6 neurons); filled symbols represent average ± SEM. B, ChAT-cre::Lhx6-GFP::VGLUT3 KO mice injected with AAV-DIO-ChR2-mCherry. Left, Recording configuration. Center, Representative light-activated current at a holding potential of −70 mV. Right, Cholinergic EPSC amplitudes in FSIs from WT (black symbols; n = 9 neurons) and VGLUT3 KO mice (red symbols, n = 6 neurons). Filled symbols represent average ± SEM. *p ≤ 0.05.
To test the hypothesis that VGLUT3 increases the amount of acetylcholine packaged and released by vesicles in cholinergic interneurons, we measured light-activated nicotinic receptor-mediated currents in FSIs in VGLUT3 KO mice. Interestingly, cholinergic currents in VGLUT3 KO mice measured only 1.3 ± 0.4 pA (n = 6 neurons; n = 3 mice), compared with14 ± 5 pA in WT mice (n = 9 neurons; n = 4 mice, p = 0.05 for comparison; Fig. 3B). These results indicate that deletion of VGLUT3 in cholinergic interneurons strongly attenuates nicotinic signaling in FSIs.
Optogenetic activation of cholinergic interneurons triggers inhibitory GABAergic postsynaptic currents (IPSCs) in MSNs by a disynaptic mechanism (Witten et al., 2010; English et al., 2011; Nelson et al., 2014). Since the disynaptic IPSC depends on cholinergic neurotransmission, we hypothesized that it might also be decreased in VGLUT3 KO mice. We compared light-evoked disynaptic IPSCs in slices from WT and VGLUT3 KO mice. In WT slices, single light pulses triggered large IPSCs (Fig. 4A; 2134 ± 564 pA; n = 7 neurons; n = 5 mice), whereas in VGLUT3 KO slices, IPSCs had significantly smaller amplitudes (Fig. 4B; 324 ± 80 pA; n = 16 neurons; n = 6 mice, p < 0.0001). As predicted from these smaller IPSCs, light pulses had little effect on MSN firing in the VGLUT3 KO slices, in contrast to the robust inhibition of MSN firing in WT slices (Fig. 4A,B). These results demonstrate monosynaptic cholinergic currents as well as acetylcholine-dependent disynaptic inhibitory currents are substantially reduced in the absence of VGLUT3. Together, these findings implicate both cholinergic interneurons and more specifically, VGLUT3, in striatal synaptic transmission involving glutamate, acetylcholine, and GABA.
Figure 4.

Light-evoked disynaptic inhibitory currents in MSNs depend on VGLUT3. ChAT-Cre animals injected with AAV-DIO-ChR2-mCherry in dorsal striatum. Whole-cell recordings of striatal MSNs measured in the presence of NBQX and APV to isolate GABAergic currents. Voltage-clamp recordings with cesium chloride-based internal solution, resulting in inward inhibitory currents at −70 mV. A, Left, Recording configuration. Center, Representative light-evoked IPSC from a WT mouse. Right, Light-evoked voltage response during spiking (induced with direct current injection). B, Left, Representative light-evoked IPSC in a VGLUT3 KO. Middle, Light-evoked voltage response during spiking (induced with direct current injection). Right, Light-evoked IPSC amplitudes in MSNs from WT (black symbols, n = 7 neurons) and VGLUT3 KO (red symbols, n = 16 neurons) animals. Filled symbols represent average ± SEM. In current-clamp traces, arrow indicates resting membrane potential. *p ≤ 0.05.
Discussion
We demonstrate that optogenetic activation of dorsal striatal cholinergic interneurons triggers corelease of glutamate and acetylcholine onto FSIs, causing small amplitude excitatory synaptic events. Surprisingly, we find that glutamate released by cholinergic interneurons activates both AMPA and NMDA receptors, compared with glutamate released by corticostriatal afferents, which activates exclusively AMPA receptors. For the first time, we have also directly measured cholinergic currents between these two sparse interneuron populations, and find they are mediated by non-α7-containing nicotinic receptors. Finally, we now show that both nicotinic currents in FSIs and disynaptic GABAergic currents in MSNs are markedly attenuated in the absence of VGLUT3.
Glutamate corelease by striatal cholinergic neurons may have several roles. First, although glutamate release onto FSIs generates small responses unlikely to generate action potentials, it uniquely activates NMDA receptors. Prior structural evidence suggests that the cholinergic inputs onto FSIs are distinct from other major glutamatergic inputs (Chang and Kita, 1992; Fujiyama et al., 2004). Interestingly, optogenetic stimulation of cholinergic interneurons also elicits a larger NMDA receptor-mediated EPSC in MSNs than does stimulation of cortical glutamatergic inputs (Higley et al., 2011). Thus, direct activation of NMDA receptors by cholinergic interneurons may support distinct forms of synaptic plasticity in both FSIs and MSNs, though the direct glutamatergic response is small in both cell types.
Our findings support a second, and likely more important, role for VGLUT3: synergistic loading of glutamate and acetylcholine into synaptic vesicles. Glutamate transport is thought to enhance the pH gradient across the vesicle membrane, which in turn increases the packaging of acetylcholine by the proton-dependent VAChT located on the same vesicles (Gras et al., 2008). Thus, copackaging is predicted to enhance cholinergic neurotransmission. Consistent with this prediction, we show marked reductions in the amplitude of cholinergic currents in VGLUT3 KO mice. Importantly, this reduction in cholinergic transmission also extends to the disynaptic inhibition of MSNs. Large, light-evoked disynaptic IPSCs in MSNs were reported in two prior studies (English et al., 2011; Nelson et al., 2014), and a barrage of IPSCs was observed in another study (Witten et al., 2010). Cholinergic interneurons may themselves receive disynaptic inhibition (Sullivan et al., 2008). IPSCs in MSNs appear to be driven by activation of nicotinic receptors on neurogliaform cells (English et al., 2011) and striatal dopamine terminals, but not FSIs (Nelson et al., 2014). With this finding in mind, the reduction in disynaptic inhibition of MSNs in the absence of VGLUT3 is likely due to reductions in nicotinic, rather than glutamatergic transmission. The physiologic significance of VGLUT3 and glutamate release in cholinergic interneurons may thus be due to invigoration of striatal cholinergic signaling, and regulation of the larger disynaptic responses. BAC transgenic lines using the ChAT locus have shown increased VAChT expression and behavioral abnormalities (Kolisnyk et al., 2013; Nagy and Aubert, 2013). Though a potential change in acetylcholine release in our mice may alter the magnitude of light-evoked cholinergic responses, the release is not significant enough to compensate for reductions in cholinergic signaling in the absence of VGLUT3. Thus, we do not believe it fundamentally changes our qualitative description of glutamate–acetylcholine corelease.
VGLUT3 KO mice are hyperactive at night and this behavior can be normalized by systemic anticholinesterase treatment (Gras et al., 2008). It was hypothesized that loss of VGLUT3 decreased cholinergic signaling and caused hyperactivity. Our data indeed show that VGLUT3 KO mice have attenuated striatal cholinergic transmission. However, recent studies of conditional VAChT KO mice indicate that specific deletion of VAChT in cholinergic interneurons does not affect locomotor activity (Guzman et al., 2011). In light of these observations, glutamate signaling by cholinergic interneurons may also be required for normal locomotor activity. Since glutamate currents evoked in FSIs are not of sufficient magnitude to cause firing of these neurons, the glutamate may have a more modulatory role through activation of NMDA receptors on FSIs. Future experiments will address more specifically the behavioral impact of VGLUT3 expression by cholinergic interneurons.
Footnotes
The work in this article was funded by the A.P. Giannini Research fellowship (A.B.N.), National Institute of Neurological Disorders and Stroke (NINDS) R01 NS064984 (A.C.K.), and NINDS RO1 NS082650 (R.P.S.). We thank Nora Hammack and Frances Farrimond for technical assistance.
The authors report no conflicting financial interests.
References
- Chang HT, Kita H. Interneurons in the rat striatum: relationships between parvalbumin neurons and cholinergic neurons. Brain Res. 1992;574:307–311. doi: 10.1016/0006-8993(92)90830-3. [DOI] [PubMed] [Google Scholar]
- El Mestikawy S, Wallén-Mackenzie A, Fortin GM, Descarries L, Trudeau LE. From glutamate co-release to vesicular synergy: vesicular glutamate transporters. Nat Rev Neurosci. 2011;12:204–216. doi: 10.1038/nrn2969. [DOI] [PubMed] [Google Scholar]
- English DF, Ibanez-Sandoval O, Stark E, Tecuapetla F, Buzsáki G, Deisseroth K, Tepper JM, Koos T. GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat Neurosci. 2011;15:123–130. doi: 10.1038/nn.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/S0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH. The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci U S A. 2002;99:14488–14493. doi: 10.1073/pnas.222546799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyama F, Kuramoto E, Okamoto K, Hioki H, Furuta T, Zhou L, Nomura S, Kaneko T. Presynaptic localization of an AMPA-type glutamate receptor in corticostriatal and thalamostriatal axon terminals. Eur J Neurosci. 2004;20:3322–3330. doi: 10.1111/j.1460-9568.2004.03807.x. [DOI] [PubMed] [Google Scholar]
- Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci. 2010;30:2223–2234. doi: 10.1523/JNEUROSCI.4870-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Leventhal DK, Fensterheim BA, Pettibone JR, Berke JD, Kreitzer AC. Selective inhibition of striatal fast-spiking interneurons causes dyskinesias. J Neurosci. 2011;31:15727–15731. doi: 10.1523/JNEUROSCI.3875-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, El Mestikawy S. A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci. 2002;22:5442–5451. doi: 10.1523/JNEUROSCI.22-13-05442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras C, Amilhon B, Lepicard EM, Poirel O, Vinatier J, Herbin M, Dumas S, Tzavara ET, Wade MR, Nomikos GG, Hanoun N, Saurini F, Kemel ML, Gasnier B, Giros B, El Mestikawy S. The vesicular glutamate transporter VGLUT3 synergizes striatal acetylcholine tone. Nat Neurosci. 2008;11:292–300. doi: 10.1038/nn2052. [DOI] [PubMed] [Google Scholar]
- Guzman MS, De Jaeger X, Raulic S, Souza IA, Li AX, Schmid S, Menon RS, Gainetdinov RR, Caron MG, Bartha R, Prado VF, Prado MA. Elimination of the vesicular acetylcholine transporter in the striatum reveals regulation of behaviour by cholinergic-glutamatergic co-transmission. PLoS Biol. 2011;9:e1001194. doi: 10.1371/journal.pbio.1001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, Gasnier B, Giros B, El Mestikawy S. The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J Neurosci. 2001;21:RC181. doi: 10.1523/JNEUROSCI.21-22-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Gittis AH, Oldenburg IA, Balthasar N, Seal RP, Edwards RH, Lowell BB, Kreitzer AC, Sabatini BL. Cholinergic interneurons mediate fast VGluT3-dependent glutamatergic transmission in the striatum. PLoS One. 2011;6:e19155. doi: 10.1371/journal.pone.0019155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Edwards RH. Neurotransmitter corelease: mechanism and physiological role. Annu Rev Physiol. 2012;74:225–243. doi: 10.1146/annurev-physiol-020911-153315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD, Rayport S, Edwards RH. Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron. 2010;65:643–656. doi: 10.1016/j.neuron.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano M, Kawasaki A, Sakata-Haga H, Fukui Y, Kawano H, Nogami H, Hisano S. Particular subpopulations of midbrain and hypothalamic dopamine neurons express vesicular glutamate transporter 2 in the rat brain. J Comp Neurol. 2006;498:581–592. doi: 10.1002/cne.21054. [DOI] [PubMed] [Google Scholar]
- Kolisnyk B, Guzman MS, Raulic S, Fan J, Magalhães AC, Feng G, Gros R, Prado VF, Prado MA. ChAT-ChR2-EYFP mice have enhanced motor endurance but show deficits in attention and several additional cognitive domains. J Neurosci. 2013;33:10427–10438. doi: 10.1523/JNEUROSCI.0395-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Dual cholinergic control of fast-spiking interneurons in the neostriatum. J Neurosci. 2002;22:529–535. doi: 10.1523/JNEUROSCI.22-02-00529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim K, Zhou ZJ. Role of ACh-GABA cotransmission in detecting image motion and motion direction. Neuron. 2010;68:1159–1172. doi: 10.1016/j.neuron.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy PM, Aubert I. B6eGFPChAT mice overexpressing the vesicular acetylcholine transporter exhibit spontaneous hypoactivity and enhanced exploration in novel environments. Brain Behav. 2013;3:367–383. doi: 10.1002/brb3.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Hammack N, Yang CF, Shah NM, Seal RP, Kreitzer AC. Striatal Cholinergic interneurons drive GABA release from dopamine terminals. Neuron. 2014;82:63–70. doi: 10.1016/j.neuron.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Qin C, Hu F, Tan J, Qiu L, Zhao S, Feng G, Luo M. Habenula “cholinergic” neurons co-release glutamate and acetylcholine and activate postsynaptic neurons via distinct transmission modes. Neuron. 2011;69:445–452. doi: 10.1016/j.neuron.2010.12.038. [DOI] [PubMed] [Google Scholar]
- Seal RP, Akil O, Yi E, Weber CM, Grant L, Yoo J, Clause A, Kandler K, Noebels JL, Glowatzki E, Lustig LR, Edwards RH. Sensorineural deafness and seizures in mice lacking vesicular glutamate transporter 3. Neuron. 2008;57:263–275. doi: 10.1016/j.neuron.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD, Hnasko TS, Britt JP, Edwards RH, Bonci A. Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J Neurosci. 2010;30:8229–8233. doi: 10.1523/JNEUROSCI.1754-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MA, Chen H, Morikawa H. Recurrent inhibitory network among striatal cholinergic interneurons. J Neurosci. 2008;28:8682–8690. doi: 10.1523/JNEUROSCI.2411-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]