

Abstract
Archaeplastida (=Kingdom Plantae) are primary plastid-bearing organisms that evolved via the endosymbiotic association of a heterotrophic eukaryote host cell and a cyanobacterial endosymbiont approximately 1,400 Ma. Here, we present analyses of cyanobacterial and plastid genomes that show strongly conflicting phylogenies based on 75 plastid (or nuclear plastid-targeted) protein-coding genes and their direct translations to proteins. The conflict between genes and proteins is largely robust to the use of sophisticated data- and tree-heterogeneous composition models. However, by using nucleotide ambiguity codes to eliminate synonymous substitutions due to codon-degeneracy, we identify a composition bias, and dependent codon-usage bias, resulting from synonymous substitutions at all third codon positions and first codon positions of leucine and arginine, as the main cause for the conflicting phylogenetic signals. We argue that the protein-coding gene data analyses are likely misleading due to artifacts induced by convergent composition biases at first codon positions of leucine and arginine and at all third codon positions. Our analyses corroborate previous studies based on gene sequence analysis that suggest Cyanobacteria evolved by the early paraphyletic splitting of Gloeobacter and a specific Synechococcus strain (JA33Ab), with all other remaining cyanobacterial groups, including both unicellular and filamentous species, forming the sister-group to the Archaeplastida lineage. In addition, our analyses using better-fitting models suggest (but without statistically strong support) an early divergence of Glaucophyta within Archaeplastida, with the Rhodophyta (red algae), and Viridiplantae (green algae and land plants) forming a separate lineage.
Keywords: origin of plastids, phylogeny, Cyanobacteria, Archaeplastida
Introduction
The Archaeplastida (=Kingdom Plantae) are plastid-bearing eukaryotes that are direct descendants of the primary endosymbiotic capture of a cyanobacterium that occurred approximately 1,400 Ma. Primary plastids are typically photosynthetic organelles that today are found in three lineages of the Archaeplastida, namely, Glaucophyta (glaucophytes), Rhodophyta (red algae), and Chloroplastida (=Viridiplantae: green algae and plants) (Keeling 2004). Although the origin of the Archaeplastida lineage has been studied extensively, due to the antiquity of the symbiotic event the exact phylogenetic relationship of primary plastids to extant cyanobacterial diversity has proven difficult to determine, and currently several competing hypotheses exist (Deusch et al. 2008; Falcón et al. 2010; Criscuolo and Gribaldo 2011; Dagan et al. 2013). In this study, we seek to clarify the best-supported hypothesis for the origin of the eukaryotic plastid lineage based on sequence analysis of plastid genes and their cyanobacterial homologues.
Cyanobacteria form a monophyletic group nested within the crown of the eubacterial domain (Blank 2004). They assume a variety of shapes and forms, including unicellular, multicellular, and colony-forming species, which have been classified into five morphological groups based upon increasing complexity (designated sections I–V in Rippka et al. [1979]). Molecular phylogenies have largely recircumscribed these groups and identified some novel but coherent phylogenetic lineages (Honda et al. 1999; Turner et al. 1999; Criscuolo and Gribaldo 2011) referred to here as follows: GBACT, UNIT+ (as in Criscuolo and Gribaldo [2011] plus Cyanothece PCC7425 and Acaryochloris marina), SPM-3, SO-6, OSC-2, and NOST-1. The groups GBACT, UNIT+, SPM-3, and SO-6 all contain unicellular members which reproduce by budding or binary fission and form morphological section I. Although OSC-2 and NOST-1 both contain filamentous taxa, only the latter have specialized cells (nitrogen-fixing heterocysts), and are consequently placed in sections III and IV, respectively. Sections II and V contain additional unicellular and filamentous species, respectively, but are not represented in our study due to lack of available genomic data.
Phylogenetic studies based on the analysis of ribosomal DNA tend to find plastids branching early within the cyanobacterial clade, often with close ties to the unicellular species of section I, but with low statistical support for relationships among major lineages (Bhattacharya and Medlin 1995; Nelissen et al. 1995; Turner et al. 1999; Reyes-Prieto et al. 2010). Similarly, a study based on the analysis of protein-coding nucleotide gene data and rRNA found the plastid lineage branching with nitrogen-fixing unicells of group SPM-3 in section I (Falcón et al. 2010). Analyses of amino acid data, on the other hand, tend to recover trees with the unicellular GBACT group diverging first (and paraphyletically) followed by a split between plastids and the remaining Cyanobacteria (Rodríguez-Ezpeleta et al. 2005). In the most comprehensive study to date, these same relationships were obtained with strong statistical support (Criscuolo and Gribaldo 2011). In contrast, evidence based on gene sequence similarity and presence or absence of cyanobacterial genes in the nuclear genomes of primary plastid-bearing taxa suggest that plastids are deeply nested within extant cyanobacterial diversity and most closely related to heterocyst-forming, filamentous taxa of sections IV (Deusch et al. 2008) or sections IV and V (Dagan et al. 2013).
Relationships among the three major lineages of the Archaeplastida have been difficult to resolve robustly. Indeed, all three possible combinations of relationships have found some support (albeit weak) from molecular analyses (Bhattacharya and Medlin 1995; Nelissen et al. 1995; Criscuolo and Gribaldo 2011). The most common hypothesis supported by the plastid and cyanobacterial protein-coding gene data is that the Glaucophyta branch first and form the sister lineage to the Rhodophyta and Chlorophyta (Turner et al. 1999; Moreira et al. 2000; Martin et al. 2002). This hypothesis is most-parsimoniously consistent with the ancestral presence of a peptidoglycan cell wall in the glaucocystophytes (Aitken and Stanier 1979), having been inherited from the cyanobacterial ancestor, and its subsequent loss in a common ancestor of the red and green lineages. Nevertheless, an extensive genomic survey by Rodríguez-Ezpeleta et al. (2005) provided contradictory evidence for the relationships among the primary lineages. In the study, analyses of plastid and cyanobacterial proteins gave the typical early-branching Glaucophyta topology, while a phylogeny based on nuclear-encoded protein sequences resulted in a tree where Rhodophyta was the earliest-branching lineage. In contrast, in the study of Criscuolo and Gribaldo (2011), analyses of plastid and cyanobacterial proteins, and their codon-degenerated nucleotide equivalents, resulted in the Rhodophyta diverging first, while cyanobacterial genes and their plant nuclear homologues resulted in the Glaucophyta as the earliest-diverging lineage when considering codon-degenerated nucleotide data and the Rhodophyta as the earliest-diverging lineage when using amino acids.
Although a robust resolution of the phylogenetic position of the plastid lineage within the Cyanobacteria has previously been obtained in only a single study (Criscuolo and Gribaldo 2011), the result is generally congruent with the majority of prior studies based on phylogenetic analyses of amino acid and rRNA data, suggesting it is most likely the correct result for these data. However, the same cannot be said for the relationships among the primary plant lineages and their phylogenetic resolution remains difficult to obtain with confidence and without conflict among data partitions. Such phylogenetic ambiguity and, in extreme cases, strong conflict among data partitions, is in part due to the technical limitations of current phylogenetic theory and practice in relation to the reconstruction of deep evolutionary divergences (Jeffroy et al. 2006; Philippe et al. 2011). In general, the more ancient the phylogeny, the more difficult it is to infer. This is due to the effect of mutational saturation because the greater the divergence time between two taxa, the more likely it is that multiple nucleotide substitutions will have occurred, especially at sites not under selective pressure. Assuming infinite time, a stable mutational process, and no selection, the nucleotide found at a given site can be modeled as a random variable reflecting the nucleotide composition frequency distribution (Yang 2006). However, the compositions may well differ over the tree, and in that case the phylogenetic signal of mutationally saturated data is effectively replaced by a potentially misleading signal driven by nucleotide composition. Consequently, even in the absence of selection, divergent taxa may share character states by convergence (homoplasy), especially when their genomes have similar overall nucleotide compositions (Stayton 2008). In particular, the selectively neutral changes in protein-coding gene sequences as a result of codon-degeneracy make the accurate resolution of deep relationships with these data error prone because phylogeny reconstruction programs may mistake convergent character states for synapomorphies. Conversely, the existence of selection on the structure and functions of proteins is likely to mean amino acid sequences are less prone to the problems associated with mutational saturation, but susceptible to convergent selective forces. These observations, plus the greater number of character states (20 amino acids in proteins versus 4 bases in nucleotide sequences) has lead to the common advice that amino acid sequences should be used when analyzing ancient relationships (Simmons et al. 2004).
Despite the theoretical advantages of using amino acid data, the computational burden imposed by the greater dimensionality of amino acid substitution models means that the analysis of amino acid data typically requires the use of empirically derived substitution rates which may, or may not, fit the data well. Consequently, the number of taxa included in such analyses may need to be limited, especially when using complex mixture models and nonstationary models. These considerations are particularly important when modeling substitutional change at deeper phylogenetic levels, as often there are theoretical and practical trade-offs to be made concerning the amount, and type, of data to analyze and the complexity of the models to be applied. Considering the wide variety of phylogenetic methods and models available, it is perhaps not surprising that phylogenetic conflict is often observed between phylogenetic analyses. Phylogenetic conflict arising between analyses of the same data can usually be resolved by considering the fit of the model to the data (Huelsenbeck and Crandall 1997)—better-fitting model are to be preferred a priori. However, phylogenetic conflict between analyses of different representations of the same data having evolved under a single evolutionary history—nucleotide protein-coding gene sequences and their amino acid translations—is of a fundamentally different type and requires a theoretical justification for preferring a solution.
In this study, we attempt to clarify the evolutionary origin of plastids from within the cyanobacterial lineage, and to resolve relationships among the three Archaeplastida lineages. We contrast analyses of combined cyanobacterial and plastid protein-coding genes that are performed on nucleotide sequences and their corresponding amino acid translations. We demonstrate strong conflicting phylogenetic evidence between the two data types and apply nonhomogeneous composition models as well as novel data-recoding techniques to identify the causes of the phylogenetic incongruence. Further, we argue that the analyses based on amino acids are to be preferred as we identify a lineage-specific composition bias in the nucleotide data that is due to differing mutation biases acting on neutral synonymous substitutions.
Results and Discussion
Experimental Design
Data sets consisting of homologous plastid protein-coding genes were constructed, analyzed, and compared to identify taxonomic incongruences. Seventy-five protein-coding gene matrices were concatenated and translated to their amino acid sequences—the concatenated 75 gene nucleotide data set (“cg75”) and their amino acid translations (“cp75”) had a one-to-one, codon-to-amino acid, correspondence (further details of the labeling of data sets are given in the legend of table 1). Phylogenetic analyses of the concatenated data sets were contrasted and strongly conflicting results identified. Further analyses using codon-degeneracy recoding techniques that partially or completely remove the phylogenetic signal associated with synonymous substitutions were performed to resolve conflicts and identify the origin of the plastid lineage.
Table 1.
Summary of Phylogenetic Support Values.
| Analysis | Plastids Sister to |
SO-6 | UNIT+ | Glaucophyta Sister to |
|||
|---|---|---|---|---|---|---|---|
| “core” | OSC-2 | Prochlorococcus | Monophyletic | Monophyletic | Rhodophyta | Viridiplantae | |
| cg75_mlboot | −0.72 | 0.72 | −0.72 | 0.72 | −0.70 | 0.59 | −0.59 |
| cp75_mlboot | 0.99 | −0.99 | −1.00 | 1.00 | 1.00 | 0.70 | −0.70 |
| cp75_stat | 1.00 | −1.00 | −1.00 | 1.00 | 1.00 | 1.00 | −1.00 |
| cp75_CAT | 1.00 | −1.00 | −1.00 | 1.00 | 1.00 | −0.94 | −0.94 |
| cp75_NDCH | 1.00 | −1.00 | −1.00 | 1.00 | 1.00 | −0.71 | −0.71 |
| cg75_stat | −1.00 | 1.00 | −1.00 | 1.00 | −1.00 | 1.00 | −1.00 |
| cg75_NDCH | −1.00 | 1.00 | −1.00 | 1.00 | 1.00 | 1.00 | −1.00 |
| cg75_degen3 | −1.00 | 0.89 | −1.00 | 1.00 | 1.00 | 0.98 | −0.98 |
| cg75_degen | 0.98 | −0.98 | −1.00 | 1.00 | 1.00 | 0.80 | −0.80 |
| cg75_degenLR3 | 0.95 | −0.95 | −1.00 | 1.00 | 1.00 | 0.83 | −0.83 |
| cg75_degen1LR | −0.91 | 0.91 | −0.91 | 0.91 | −0.74 | 0.45 | −0.45 |
| cg75_degen12S | −0.92 | −0.92 | 0.78 | −0.92 | −0.91 | −0.82 | 0.82 |
Note.—BPs (“mlboot”) or posterior probabilities are shown for relationships (columns) for selected analyses (rows). A positive value is the support for the relationship or a negative value is the support for its most supported conflicting node, where appropriate. When the relationship is a sister-group relationship, the support value reported is the lowest among those of the two monophyletic sister groups and of the clade formed by those two groups. The “core” refers to the “core-cyanobacteria” group defined as all Cyanobacteria present in our taxonomic sampling except the early-diverging GBACT taxa. The “degen*” analyses are performed under standard ML, but a proportion of the signal associated with codon synonymy is suppressed by recoding some of the codon positions where codon degeneracy exists (supplementary table S1, Supplementary Material online): “1LR” designates the signal associated with first codon position synonymy among leucine and arginine codons; “12S” designates the signal associated with first and second codon position synonymy among serine codons; “3” designates the signal associated with third codon position synonymy among all codons families. “CAT” designates the site-heterogeneous composition model implemented in Phylobayes. “stat” indicates a stationary composition model and “NDCH” designates the nonstationary (tree-heterogeneous) composition model implemented in P4.
Analyses of Nucleotides and Amino Acids Produce Conflicting Phylogenetic Trees for Plastid and Cyanobacterial Relationships
Analyses of the nonrecoded nucleotide data (“cg75”) yielded a topology where the plastids are nested within a paraphyletic grade of cyanobacterial taxa, which would imply that plastids evolved from an already relatively differentiated branch of extant cyanobacterial diversity (fig. 1; supplementary fig. S1: “cg75_stat” composition homogeneous Markov chain Monte Carlo [MCMC] 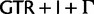 and supplementary fig. S2: “cg75_NDCH” NDCH [node-discrete composition heterogeneity model: Foster (2004); Cox et al. (2008)] MCMC, Supplementary Material online). Although the maximum likelihood (ML) bootstrap and the composition homogeneous MCMC analyses resulted in identical trees, the nodes of the paraphyletic grade are poorly supported in the ML bootstrap analyses.
and supplementary fig. S2: “cg75_NDCH” NDCH [node-discrete composition heterogeneity model: Foster (2004); Cox et al. (2008)] MCMC, Supplementary Material online). Although the maximum likelihood (ML) bootstrap and the composition homogeneous MCMC analyses resulted in identical trees, the nodes of the paraphyletic grade are poorly supported in the ML bootstrap analyses.
Fig. 1.

ML bootstrap analysis of the protein-coding gene data set “cg75” and 50% majority-rule consensus tree of 200 ML (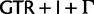 ) bootstrap trees. Values above the branches are BPs. Colors indicate taxonomic groups (supplementary table S1, Supplementary Material online): Bacteria (purple), Cyanobacteria (blue), Glaucophyta (orange), Rhodophyta (red), and Viridiplantae (green). Note that Prochlorococcus is attracted to the Archaeplastida clade causing lower support values between the two points of attachment.
) bootstrap trees. Values above the branches are BPs. Colors indicate taxonomic groups (supplementary table S1, Supplementary Material online): Bacteria (purple), Cyanobacteria (blue), Glaucophyta (orange), Rhodophyta (red), and Viridiplantae (green). Note that Prochlorococcus is attracted to the Archaeplastida clade causing lower support values between the two points of attachment.
In contrast to the analyses of the nucleotide data, those of the amino acid data (“cp75”) result in trees where the plastids belong to an earlier divergence of the Cyanobacteria (fig. 2: ML bootstrap “cp75_mlboot” and supplementary fig. S3: “cp75_stat,” supplementary fig. S4: “cp75_CAT,” and supplementary fig. S5: “cp75_NDCH,” Supplementary Material online). These trees distinguish two groups of Cyanobacteria that we name here for convenience: “GBACT” (those taxa that diverged earlier than the plastids and its sister-clade) and “core-cyanobacteria” (UNIT+, OSC-2, SO-6, NOST-1, and SPM-3 that form a sister-group to the plastids). The sister-group relationship between plastids and core-cyanobacteria is well supported by ML bootstrap analyses (99% bootstrap proportion [BP], fig. 2) and when more complex evolution models are used (CAT model: supplementary fig. S4: “cp75_CAT,” and NDCH model, and supplementary fig. S5, “cp75_NDCH,” Supplementary Material online).
Fig. 2.

ML bootstrap analysis of the protein data set “cp75” and 50% majority-rule consensus tree of 200 ML (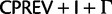 ) bootstrap trees. Values above branches are BPs. Colors indicate taxonomic group (refer legend of fig. 1).
) bootstrap trees. Values above branches are BPs. Colors indicate taxonomic group (refer legend of fig. 1).
In summary, our analyses consistently show a conflict between protein-coding genes and their direct translations into proteins with respect to the origin of plastids even when sophisticated and better-fitting models are used.
Character Recoding and Deletion Strategies
In addition to using better-fitting models to overcome reconstruction artifacts, potentially misleading phylogenetic signal can be eliminated by deleting or recoding parts of the data prior to analysis. Here, we use a codon-degeneracy recoding technique that partially or completely removes phylogenetic signal associated with synonymous substitutions (Regier et al. 2010; Criscuolo and Gribaldo 2010, 2011; Zwick et al. 2012; Rota-Stabelli et al. 2013; Cox et al. 2014). Such a method is appropriate because even in the absence of a selection coefficient discriminating among synonymous codons (“major codon preference” [Akashi et al. 1998]), a genome-wide directional mutation bias can drive a codon usage bias among synonymous codons that can vary across a phylogenetic tree and lead to reconstruction artifacts if not modeled correctly. In the “Bacterial, Archaeal, and Plant Plastid” genetic code, most synonymous codons only differ by a single substitution at the third codon position. The exceptions are the codons of leucine (Leu), arginine (Arg), and serine (Ser). In the case of Leu or Arg, the codons may also differ by a substitution in the first codon position (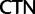 and
and  codon families for Leu and
codon families for Leu and 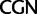 and
and 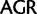 for Arg), whereas Ser codons may also differ by substitutions at the first and second codon positions (
for Arg), whereas Ser codons may also differ by substitutions at the first and second codon positions (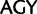 and
and 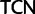 ) (for synonymous codon ambiguity codes see Criscuolo and Gribaldo 2010). Between synonymous Ser codons belonging to the
) (for synonymous codon ambiguity codes see Criscuolo and Gribaldo 2010). Between synonymous Ser codons belonging to the 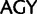 and
and 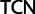 codon families, a minimal transformation series requires two point mutations and the amino acid has to change to either a threonine (Thr,
codon families, a minimal transformation series requires two point mutations and the amino acid has to change to either a threonine (Thr, 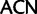 codon family) or a cysteine (Cys,
codon family) or a cysteine (Cys, 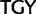 ) intermediate.
) intermediate.
A common method used to reduce the artifacts associated with synonymous substitutions is simply to remove all third codon positions from the data matrix, because these are the positions where the majority of synonymous codons differ. However, these data deletions also removes nonsynonymous changes that occur at third codon positions, that is, Phe (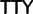 )
)  Leu (
Leu ( ), Met (
), Met (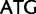 )
)  Ile (
Ile (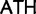 ), Tyr (
), Tyr (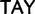 )
)  stop (
stop (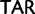 ), His (
), His (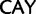 )
)  Gln (
Gln (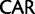 ), Asn (
), Asn (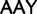 )
)  Lys (
Lys (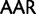 ), Asp (
), Asp (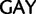 )
)  Glu (
Glu (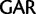 ), Cys (
), Cys (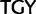 )
)  stop (
stop (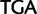 ), Cys (
), Cys (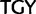 )
)  Trp (
Trp (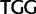 ), Trp (
), Trp (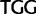 )
)  stop (
stop (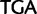 ), and Ser (
), and Ser (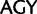 )
)  Arg (
Arg (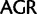 ). Similarly, by deleting sites associated with synonymous substitutions among Leu, Arg, and Ser (Inagaki and Roger 2006) considerable amounts of phylogenetic signal from nonsynonymous substitutions at the same sites is removed. To alleviate this problem, here we use a codon recoding method that eliminates the phylogenetic signal associated with synonymous substitutions by recoding nucleotides in codon triplets with IUPAC ambiguity codes so that all synonymous codon variants coding for a particular amino acid are represented by a single degenerate triplet (e.g., Thr:
). Similarly, by deleting sites associated with synonymous substitutions among Leu, Arg, and Ser (Inagaki and Roger 2006) considerable amounts of phylogenetic signal from nonsynonymous substitutions at the same sites is removed. To alleviate this problem, here we use a codon recoding method that eliminates the phylogenetic signal associated with synonymous substitutions by recoding nucleotides in codon triplets with IUPAC ambiguity codes so that all synonymous codon variants coding for a particular amino acid are represented by a single degenerate triplet (e.g., Thr: 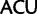 ,
, 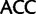 ,
, 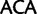 ,
, 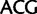

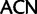 ; Leu:
; Leu:  ,
,  ,
, 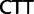 ,
, 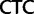 ,
,  ,
,  ).
).
We used this method to generate a matrix where the signal associated with synonymy at third codon positions is removed, by applying the above recoding at third codon positions. This matrix should constitute a slight improvement with respect to the more common practice consisting in removing third codon positions because it preserves part of the signal associated with nonsynonymous substitutions. That matrix was subjected to ML bootstrap analysis (“cg75_degen3” in table 1). But as previously stated, Leu, Arg, and Ser codons families have synonymous substitutions also at first positions, and even at second positions in the case of Ser. Our recoding technique allows the removal of the signal associated with these synonymous substitutions without at the same time loosing all signals present in the first and second position. Therefore, we generated a matrix where the codons were fully recoded, and performed ML bootstrap on it (“cg75_degen” in supplementary table S1, Supplementary Material online). The comparison of the results of “cg75_degen” and “cg75_degen3” should allow to gauge the effect of synonymous substitutions at Leu, Arg, and Ser first and second codon positions alone.
Our recoding technique is different from that used by Regier et al. (2010) and Zwick et al. (2012) who used the software Degen1 which distinguishes between the two codon variant families of Ser (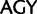 and
and 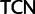 ), whereas in our recoding scheme these same codons were degenerated to
), whereas in our recoding scheme these same codons were degenerated to 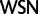 . As the mutational paths connecting the
. As the mutational paths connecting the 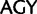 and
and 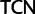 Ser codon families imply two nucleotide changes, it has been argued by Regier et al. (2010) that the Ser synonymous substitutions may carry a signal less likely to be misleading than the other synonymous substitutions. To assess this prediction, two further recoded matrices were generated. The first in which all synonymous substitutions were eliminated using ambiguity codes except for those at first and second positions of Ser codons and a second, complementary, matrix where the only recoded characters were those involved in Ser codon synonymy at first and second positions. ML bootstrap analyses were performed on these two recoded matrices (“cg75_degenLR3” and “cg75_degen12S” in supplementary table S1, Supplementary Material online). Finally, to assess the phylogenetic signal among substitutions at first position of Leu and Arg codons alone, a matrix was generated where only substitutions among codon variants of these amino acids at first positions were recoded with degenerate ambiguity codes, and analyzed it under ML bootstrap (“cg75_degen1LR” in supplementary table S1, Supplementary Material online). The recoding operations were performed using python scripts based on methods implemented in P4.
Ser codon families imply two nucleotide changes, it has been argued by Regier et al. (2010) that the Ser synonymous substitutions may carry a signal less likely to be misleading than the other synonymous substitutions. To assess this prediction, two further recoded matrices were generated. The first in which all synonymous substitutions were eliminated using ambiguity codes except for those at first and second positions of Ser codons and a second, complementary, matrix where the only recoded characters were those involved in Ser codon synonymy at first and second positions. ML bootstrap analyses were performed on these two recoded matrices (“cg75_degenLR3” and “cg75_degen12S” in supplementary table S1, Supplementary Material online). Finally, to assess the phylogenetic signal among substitutions at first position of Leu and Arg codons alone, a matrix was generated where only substitutions among codon variants of these amino acids at first positions were recoded with degenerate ambiguity codes, and analyzed it under ML bootstrap (“cg75_degen1LR” in supplementary table S1, Supplementary Material online). The recoding operations were performed using python scripts based on methods implemented in P4.
Compositional Effects at Third Codon Positions Can Explain Some, but Not All, of the Observed Incongruence
Third codon positions are the sites at which composition biases driven by mutation biases are most likely to be apparent. This is because the preference for a particular nucleotide at a third codon position can typically be accommodated within a family of synonymous codons (i.e., coding for a single amino acid), and therefore the nucleotide composition at third codon positions is less constrained by selection at the amino acid level than at first and second codon positions, and more susceptible to mutation pressure.
Codon-degeneracy recoding of third codon positions (“cg75_degen3” supplementary fig. S6, Supplementary Material online), or the use of the composition heterogeneous NDCH model (“cg75_NDCH” supplementary fig. S2, Supplementary Material online) both result in the expected recovery of a monophyletic UNIT+ cyanobacterial group. These results contrast with composition homogeneous analyses using un-recoded third codon positions (i.e., fig. 1, “cg75_stat,” and supplementary fig. S1, Supplementary Material online) where the UNIT+ group is paraphyletic with the low 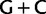 composition A. marina forming a clade with other low
composition A. marina forming a clade with other low 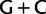 taxa such as SPM-3, NOST-1, OSC-2, and plastids (fig. 3).
taxa such as SPM-3, NOST-1, OSC-2, and plastids (fig. 3).
Fig. 3.

Simplified ML bootstrap tree for the recoded protein-coding gene data set “cg75_degen12S” and 50% majority-rule consensus tree of 200 ML (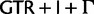 ) bootstrap trees. Clades are labeled by their group label were possible. The codon usage bias and
) bootstrap trees. Clades are labeled by their group label were possible. The codon usage bias and 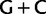 proportions at the three codon positions of the original “cg75” data set (i.e., without recoding) are presented to the right of the taxa (average values are given for summarized groups). This tree was chosen to display codon usage biases and G + C proportions because it seems to exemplify reconstruction errors induced by compositional effects. The topology of this tree somewhat correlates with composition and codon usages biases. Codon usage bias among Leu, Ser, and Arg is measured as the
proportions at the three codon positions of the original “cg75” data set (i.e., without recoding) are presented to the right of the taxa (average values are given for summarized groups). This tree was chosen to display codon usage biases and G + C proportions because it seems to exemplify reconstruction errors induced by compositional effects. The topology of this tree somewhat correlates with composition and codon usages biases. Codon usage bias among Leu, Ser, and Arg is measured as the 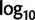 of the unbiased ratio between the usage of the two families of codons where the number of occurrences of codons of a family is divided by the number of possible codons in that family (2 or 4). Codon family labels:
of the unbiased ratio between the usage of the two families of codons where the number of occurrences of codons of a family is divided by the number of possible codons in that family (2 or 4). Codon family labels:  ,
,  ;
; 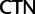 ,
,  ;
; 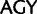 ,
, 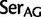 ;
; 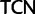 ,
, 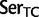 ;
; 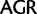 ,
,  ; and
; and 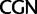 ,
,  . The codon bias representation is inspired by figure 1 of Inagaki and Roger (2006). Values above branches are BPs. Colors indicate taxonomic group (refer legend of fig. 1). *Prochlorococcus is an abbreviation of Prochlorococcus marinus (SO-6).
. The codon bias representation is inspired by figure 1 of Inagaki and Roger (2006). Values above branches are BPs. Colors indicate taxonomic group (refer legend of fig. 1). *Prochlorococcus is an abbreviation of Prochlorococcus marinus (SO-6).
Moreover, it is notable that two composition vectors are optimally required by the NDCH model to fit the nucleotide data and that these composition vectors correspond to high and low 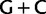 biases (i.e., 65% and 23%). Groupings on the basis of shared biases in
biases (i.e., 65% and 23%). Groupings on the basis of shared biases in 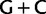 richness are commonly observed and are the signature of inaccurate reconstruction methods (see for instance fig. 1A of Jeffroy et al. [2006]).
richness are commonly observed and are the signature of inaccurate reconstruction methods (see for instance fig. 1A of Jeffroy et al. [2006]).
Our results suggest the existence of phylogenetic artifacts when analyzing the protein-coding genes data set due to mutation-driven lineage-specific composition biases residing in third codon positions of protein-coding genes. This observation justifies the removal of third codon positions, but recoding the data using ambiguity codes instead avoids discarding all signals present at third codon positions, which might in some cases improve the accuracy of the reconstruction over a simple removal. Even so, recoding third codon positions does not resolve the phylogenetic conflict with the amino acid data concerning the position of plastids within Cyanobacteria.
Compositional Effects at First Codon Positions also Affect Which Topology Is Obtained
Further codon-degeneracy recoding analyses described later strongly suggest that the discordance between analyses based on nucleotides and analyses based on amino acids is due in part to synonymous substitutions at first codon positions among synonymous variants in the Leu (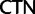 /
/ ) and Arg (
) and Arg (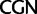 /
/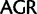 ) codon families. Removing the signal associated with these synonymous substitutions by ambiguity recoding, together with the signal associated with codon synonymy at third codon positions, results in the recovery of a sister-group relationship between plastids and core-cyanobacteria as with the amino acid data (supplementary fig. S7, Supplementary Material online, “cg75_degenLR3” in table 1). In the topology obtained by the analysis of nonrecoded nucleotide data (fig. 1, “cg75_mlboot”), plastids are sister to OSC-2, which are characterized by a lower
) codon families. Removing the signal associated with these synonymous substitutions by ambiguity recoding, together with the signal associated with codon synonymy at third codon positions, results in the recovery of a sister-group relationship between plastids and core-cyanobacteria as with the amino acid data (supplementary fig. S7, Supplementary Material online, “cg75_degenLR3” in table 1). In the topology obtained by the analysis of nonrecoded nucleotide data (fig. 1, “cg75_mlboot”), plastids are sister to OSC-2, which are characterized by a lower 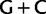 proportion at first codon positions than other Cyanobacteria (fig. 3). Because plastids have the lowest
proportion at first codon positions than other Cyanobacteria (fig. 3). Because plastids have the lowest 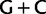 content at first codon positions in the data, it is possible that their grouping with OSC-2 is an artifact due to convergent nucleotide compositions. Removing only the first codon position signal associated with synonymous substitutions among codon variants in both the Arg and Leu codon families, while keeping all third codon position signal, results in a topology similar to the one obtained when the nonrecoded data is analyzed (supplementary fig. S8, Supplementary Material online, “cg75_degen1LR” in table 1). This signal is therefore only partly responsible for the conflict between nucleotide and amino acid analyses, and may be a reflection of the lower
content at first codon positions in the data, it is possible that their grouping with OSC-2 is an artifact due to convergent nucleotide compositions. Removing only the first codon position signal associated with synonymous substitutions among codon variants in both the Arg and Leu codon families, while keeping all third codon position signal, results in a topology similar to the one obtained when the nonrecoded data is analyzed (supplementary fig. S8, Supplementary Material online, “cg75_degen1LR” in table 1). This signal is therefore only partly responsible for the conflict between nucleotide and amino acid analyses, and may be a reflection of the lower 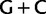 composition bias at first codon positions than at third codon positions (fig. 3).
composition bias at first codon positions than at third codon positions (fig. 3).
Phylogenetic Effects of Substitutions between Serine Codon Families
Unlike the two Arg (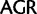 /
/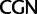 ) and the two Leu (
) and the two Leu (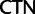 /
/ ) codon families, which differ by a single nucleotide substitution at the first codon position (
) codon families, which differ by a single nucleotide substitution at the first codon position (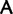

 and
and 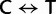 , respectively), the two Ser codon families (
, respectively), the two Ser codon families (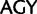 /
/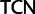 ) differ by two nucleotide substitutions, both transversions, one each at the first and second codon positions (
) differ by two nucleotide substitutions, both transversions, one each at the first and second codon positions (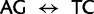 ). The most direct mutational paths—with single discrete nucleotide substitutions—between the two Ser codon families imply intermediate Thr or Cys codons. The biochemical properties of the amino acids, as well as rates estimated from empirical data, suggest that it is easy to substitute a Thr for a Ser, or a Ser for a Cys (Rota-Stabelli et al. 2013). However, simultaneous nucleotide substitutions at the first two codon positions may also occur (Kosiol et al. 2007) and have been previously implied from observations of empirical data (Averof et al. 2000). The latter observation would suggest that the rate of Ser codon family interconversion (
). The most direct mutational paths—with single discrete nucleotide substitutions—between the two Ser codon families imply intermediate Thr or Cys codons. The biochemical properties of the amino acids, as well as rates estimated from empirical data, suggest that it is easy to substitute a Thr for a Ser, or a Ser for a Cys (Rota-Stabelli et al. 2013). However, simultaneous nucleotide substitutions at the first two codon positions may also occur (Kosiol et al. 2007) and have been previously implied from observations of empirical data (Averof et al. 2000). The latter observation would suggest that the rate of Ser codon family interconversion (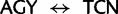 ) is reduced relative to other synonymous interfamily codon conversions requiring only a single synonymous nucleotide substitution. However, these two simultaneous synonymous substitutions may also be subject to mutational biases and therefore could be phylogenetically misleading (Rota-Stabelli et al. 2013).
) is reduced relative to other synonymous interfamily codon conversions requiring only a single synonymous nucleotide substitution. However, these two simultaneous synonymous substitutions may also be subject to mutational biases and therefore could be phylogenetically misleading (Rota-Stabelli et al. 2013).
To investigate the rate of substitutions between synonymous codon families of Ser, Arg, and Leu, we recoded the data so that each codon family was individually distinguished and applied a 23 character state amino acid model in which we used the following notations: 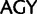 :
: 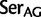 ;
; 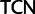 :
: 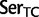 ;
; 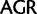 :
:  ;
; 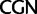 :
:  ;
;  :
:  ; and
; and 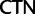 :
:  . A 21-state amino acid model was previously used by Zwick et al. (2012), which distinguished only between Ser codon family variant (
. A 21-state amino acid model was previously used by Zwick et al. (2012), which distinguished only between Ser codon family variant (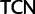 and
and 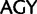 ), whereas Rota-Stabelli et al. (2013) used a similar 23-state substitution matrix as our model but with a mixture model of composition vectors (Phylobayes CAT-model variant). Exchange rates (rate parameters sensu Swofford et al. [1996], the products of the mean instantaneous substitution rate [μ], and the relative rate parameters) were estimated from a P4 MCMC chain using a
), whereas Rota-Stabelli et al. (2013) used a similar 23-state substitution matrix as our model but with a mixture model of composition vectors (Phylobayes CAT-model variant). Exchange rates (rate parameters sensu Swofford et al. [1996], the products of the mean instantaneous substitution rate [μ], and the relative rate parameters) were estimated from a P4 MCMC chain using a 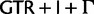 model with fixed topology, α parameter of the Γ distribution, and proportion of invariable sites, the values of which were taken from the optimal ML results obtained by RAxML under a
model with fixed topology, α parameter of the Γ distribution, and proportion of invariable sites, the values of which were taken from the optimal ML results obtained by RAxML under a 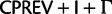 model on the standard 20 state amino acid data. As expected, the three highest estimated substitution rates, of a total of 253 rates, were due to implied synonymous substitutions between the Arg codon families (
model on the standard 20 state amino acid data. As expected, the three highest estimated substitution rates, of a total of 253 rates, were due to implied synonymous substitutions between the Arg codon families (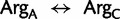 : 2,0061.9), the Leu codon families (
: 2,0061.9), the Leu codon families (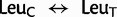 : 1,5275.5), and the Ser codon families (
: 1,5275.5), and the Ser codon families (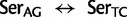 : 4,393.5; supplementary table S2, Supplementary Material online). In comparison, the mean substitution rate between amino acids (i.e., due to nonsynonymous substitutions) was 241 (76 median). The estimated substitution rates between Ser codons variants and Thr or Cys intermediates were the following:
: 4,393.5; supplementary table S2, Supplementary Material online). In comparison, the mean substitution rate between amino acids (i.e., due to nonsynonymous substitutions) was 241 (76 median). The estimated substitution rates between Ser codons variants and Thr or Cys intermediates were the following: 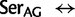 Thr: 2,262.4 (0.51
Thr: 2,262.4 (0.51  rate [
rate [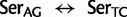 ], ranked 6th highest);
], ranked 6th highest); 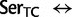 Thr: 955.8 (0.22
Thr: 955.8 (0.22 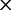 rate[
rate[ ], ranked 18th highest);
], ranked 18th highest); 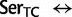 Cys: 923.6 (0.21
Cys: 923.6 (0.21 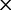 rate[
rate[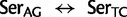 ], ranked 19th highest); and
], ranked 19th highest); and 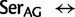 Cys: 729.8 (0.17
Cys: 729.8 (0.17 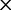 [
[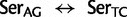 ], ranked 25th highest). According to these estimations, synonymous substitutions between Ser codon family variants (
], ranked 25th highest). According to these estimations, synonymous substitutions between Ser codon family variants (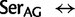
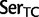 ) occur at a much higher rate than nonsynonymous substitutions, indeed, nearly twice the rate of the highest rate between Ser and either of Thr or Cys. It is not known, however, to what extent the
) occur at a much higher rate than nonsynonymous substitutions, indeed, nearly twice the rate of the highest rate between Ser and either of Thr or Cys. It is not known, however, to what extent the 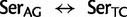 rate also captures mutational paths involving undetected Thr or Cys intermediates that may be short-lived due to their being selectively deleterious (Averof et al. 2000). Nevertheless, it is clear that synonymous substitutions among Ser codon families are frequent and therefore at least potentially subject to mutational biases that could lead to phylogenetic artifacts if not accounted for in the substitution model. Moreover, while the exchange rate between Ser codon family variants is much lower than between the codon variants of Arg and Leu, the rate of Ser codon family variant exchange is still higher than the highest nonsynonymous exchange rate between Lys
rate also captures mutational paths involving undetected Thr or Cys intermediates that may be short-lived due to their being selectively deleterious (Averof et al. 2000). Nevertheless, it is clear that synonymous substitutions among Ser codon families are frequent and therefore at least potentially subject to mutational biases that could lead to phylogenetic artifacts if not accounted for in the substitution model. Moreover, while the exchange rate between Ser codon family variants is much lower than between the codon variants of Arg and Leu, the rate of Ser codon family variant exchange is still higher than the highest nonsynonymous exchange rate between Lys  Arg (exchange rate: 3969.3) and considerably larger than most nonsynonymous rates (supplementary table S2, Supplementary Material online). Indeed, nonsynonymous exchange rates vary widely, suggesting that the amino acids involved in the most frequent exchanges might also be the most noisy, and warrant either synonymizing or eliminating from the data set; Lys–Arg would be a candidate for this as its exchange rate is especially high.
Arg (exchange rate: 3969.3) and considerably larger than most nonsynonymous rates (supplementary table S2, Supplementary Material online). Indeed, nonsynonymous exchange rates vary widely, suggesting that the amino acids involved in the most frequent exchanges might also be the most noisy, and warrant either synonymizing or eliminating from the data set; Lys–Arg would be a candidate for this as its exchange rate is especially high.
In our analyses, including only the synonymous substitutions associated with Ser codon family interconversion did not alter the topology (compare supplementary fig. S9, “cg75_degen” in table 1 and supplementary fig. S7, “cg75_degenLR3” in table 1, Supplementary Material online). However, removing only the synonymous substitutions associated with Ser codon family interconversion resulted in a likely artifactual topology where Prochlorococcus is placed as sister-group to the plastids, and Rhodophyta are attracted to a more basal position within plastids (supplementary fig. S10, Supplementary Material online, “cg75_degen12S” in table 1). These observations suggest that synonymous Ser codon family interconversion and the implied underlying composition biases did not contribute to the misleading phylogenetic signal. This conclusion is further supported by observing composition base ratios at individual codon positions (supplementary figs. S11–S13, Supplementary Material online). If Ser codon family interconversion were responsible for an artifactual attraction between Prochlorococcus and the plastids, we would expect to see among these taxa either a 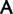 bias at the first position correlated with at
bias at the first position correlated with at  bias at the second position, or a
bias at the second position, or a 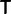 bias at the first position correlated with a
bias at the first position correlated with a  bias at the second position. However, such biases are not evident in the plots. What is evident is a
bias at the second position. However, such biases are not evident in the plots. What is evident is a 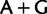 bias present in Prochlorococcus (and Trichodesmium of the OSC-2 group) and plastids at third codon positions (supplementary fig. S13, Supplementary Material online), and a preference for
bias present in Prochlorococcus (and Trichodesmium of the OSC-2 group) and plastids at third codon positions (supplementary fig. S13, Supplementary Material online), and a preference for  over
over  at first positions among the same taxa (supplementary fig. S11, Supplementary Material online). These observations and the correlation between
at first positions among the same taxa (supplementary fig. S11, Supplementary Material online). These observations and the correlation between 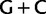 richness, codon usage biases, and the topology shown in figure 3, strongly suggest that the attraction between Prochlorococcus and Rhodophyta is an artifact due to convergent composition biases at first and third codon positions. Moreover, because the artifactual attraction between Prochlorococcus and Rhodophyta occurs when the signal associated with substitutions between Ser codon families is negated, these same substitutions contain accurate historical signal that is sufficient to overcome some of the negative effects of an underlying composition bias not associated with Ser codons. In contrast, Rota-Stabelli et al. (2013), in a study of arthropod relationships, observed that the inclusion of the synonymous Ser signal had a negative effect on the accuracy of the phylogenetic results as their removal resulted in increased model dependency whereby a better-fitting model recovered a tree similar to that obtained when using amino acid data—a tree they considered more likely to be correct (c.f. Zwick et al. 2012). Hence, our finding that the synonymous Ser signal appears historically accurate for our data does not suggest a principle more generally applicable to other data.
richness, codon usage biases, and the topology shown in figure 3, strongly suggest that the attraction between Prochlorococcus and Rhodophyta is an artifact due to convergent composition biases at first and third codon positions. Moreover, because the artifactual attraction between Prochlorococcus and Rhodophyta occurs when the signal associated with substitutions between Ser codon families is negated, these same substitutions contain accurate historical signal that is sufficient to overcome some of the negative effects of an underlying composition bias not associated with Ser codons. In contrast, Rota-Stabelli et al. (2013), in a study of arthropod relationships, observed that the inclusion of the synonymous Ser signal had a negative effect on the accuracy of the phylogenetic results as their removal resulted in increased model dependency whereby a better-fitting model recovered a tree similar to that obtained when using amino acid data—a tree they considered more likely to be correct (c.f. Zwick et al. 2012). Hence, our finding that the synonymous Ser signal appears historically accurate for our data does not suggest a principle more generally applicable to other data.
Mitigating the Effects of Composition Biases due to Synonymous Substitutions
We identify in our data a composition bias introduced by synonymous substitutions at first (Leu and Arg) and third codon positions of protein-coding genes as the source of phylogenetic conflict with analyses based on the protein translations of the same genes. These observations indicate that the evolutionary mechanisms underlying synonymous and nonsynonymous substitutions are different in these data and should in principle be modeled differently. However, a model that distinguishes between substitution types does not currently exist. Although codon models allow separate transition/transversion ratios (κ), the composition, be it nucleotide (Muse and Gaut 1994) or codon (Goldman and Yang 1994), is still homogeneous among substitution types, as is the case with conventional nucleotide models based on the general time-reversible model (Rodriguez et al. 1990), including the NDCH model.
In our study, the composition bias at third codon positions is correlated with tree structure. The biases vary gradually across the nucleotide-based tree from the root, through a grade of cyanobacterial taxa, to the plastid clade. Standard models of substitution assume a stationary composition across the tree, and the violation of this assumption can be expected to induce phylogenetic artifacts. Moreover, in this study the use of a nonstationary composition model (NDCH) was unable to mitigate the principal effects of the composition bias: perhaps because the studied taxa have a gradually varying composition rather than belonging to discrete composition categories as assumed by the NDCH model. However, by recoding codons with nucleotide ambiguity codes so as to eliminate the signal associated with synonymous substitutions between codon variants, we were able to obtain an essentially congruent tree to that based on the translated amino acids by using standard composition-stationary models. Synonymous substitutions undoubtedly contain phylogenetic signal at a relatively shallow phylogenetic depth, but because of their freedom from selective constraint, they occur rapidly and are therefore prone to mutation biases. Consequently, depending on the presence, distribution, and strength of such biases, synonymous substitutions may cause reconstruction difficulties. In our study, they are clearly the source of a phylogenetic artifact when using standard phylogenetic models. It is particularly noteworthy that the source of the conflict was not just the third codon positions alone—which are often removed from analyses due to their presumed substitutional saturation—but also the synonymous substitutions in the first positions of Leu and Arg codons that are by themselves sufficient to induce the phylogenetic artifact. Furthermore, although in our study substitutions between Ser codon families (which may be effectively synonymous if occurring in tandem) had a beneficial effect on the topology, this may not always be the case, especially if the codon bias reinforces an already underlying mutational bias (Rota-Stabelli et al. 2013).
Relationships among the Cyanobacteria and Plastids
The data recoding analyses performed here show that the conflicting results observed between analyses based on nucleotide data and those based on the corresponding amino acid translations are caused by the combined effects of composition biases affecting first and third codon positions in the nucleotide data. However, compared with the nucleotide analyses, the analyses of amino acid data were more robust to the methods used. Consequently, we suggest the sister-group relationship between plastids and a monophyletic core-cyanobacteria consisting of the NOST-1, UNIT, SO-6, OSC-2, and SPM-3 groups, observed both in the amino acid analyses and codon-degeneracy recoded analyses, is the better-supported hypothesis based on these data. This result supports those obtained by other studies based on amino acid data (Rodríguez-Ezpeleta et al. 2005; Criscuolo and Gribaldo 2011) and contradicts those obtained using nucleotides data which placed plastids most-closely related to nitrogen-fixing unicells of group SPM-3 in section I (Falcón et al. 2010). The same early-branching Archaeplastida tree based on amino acids was obtained by Dagan et al. (2013) (their supplementary fig. S6), however, the result was attributed to long-branch attraction due to the presence of significantly greater numbers of unique substitutions on branches in the plastid clade. In the analyses presented here which use better-fitting composition heterogeneous models, we were not able to find evidence to support the hypothesis that the early-branching of the Archaeplastida within the cyanobacteria is an artifact. Consequently, our analyses do not support a close relationship between the Archaeplastida and filamentous, heterocyst-forming, cyanobacteria of section IV (Deusch et al. 2008) or sections IV and V (Dagan et al. 2013) based on evidence of gene content similarity, or the presence of starch-like storage polysaccharides typically of plastids in unicellular diazotrophic cyanobacteria of section V (Deschamps et al. 2008; Ball et al. 2011).
Most of our phylogenetic analyses support the monophyly of the cyanobacterial groups NOST-1, UNIT+, SO-6, OSC-2, and SPM-3 (Honda et al. 1999; Turner et al. 1999; Criscuolo and Gribaldo 2011), whereas GBACT, is consistently resolved as paraphyletic, diverging before the core-cyanobacteria and plastids. Cases where UNIT+ or SO-6 are nonmonophyletic can be attributed to composition-induced reconstruction artifacts. Although relationships between groups of core-cyanobacteria vary among analyses, when effects of nucleotide composition heterogeneity are eliminated (either by analyzing amino acid data or by means of codon degeneracy recoding) the tree is compatible with a first divergence of UNIT+, and sister group relationships between OSC-2 and SO-6 and between NOST-1 and SPM-3.
In our analyses, an earliest-branching position of Rhodophyta within Archaeplastida (with Glaucophyta and Viridiplantae forming sister-groups) is typically associated with an attraction to Prochlorococcus due to shared composition bias and codon usage patterns (fig. 3). Prochlorococcus is a member of the S0-6 group, and, as argued earlier, likely forms a monophyletic core-cyanobacterial group (with NOST-1, UNIT+, SO-6, and SPM-3) sister to the plastids. Most other analyses support a sister-group relationship between Glaucophyta and Rhodophyta (figs. 1 and 2). Nevertheless, the Glaucophyta are identified as the earliest-branching lineage in the analysis of amino acid data using both the CAT and the NDCH models (supplementary figs. S4 and S5, Supplementary Material online), and as these models have a better fit (as determined by the estimated marginal likelihoods) than standard homogeneous models and are designed to overcome artifacts caused by amino acid composition heterogeneity across sites or across taxa, these trees are the preferred solution. In addition, to the evidence from amino acid data, the early branching of Glaucophyta is also congruent with some analyses of nuclear gene data (Turner et al. 1999; Moreira et al. 2000; Martin et al. 2002; Chan et al. 2011) and several morphological and metabolic characters, such as the plesiomorphic retention of a peptidoglycan-containing cell wall that is absent in Rhodophyta and Viridiplantae (Steiner and Löffelhardt 2002; McFadden and Dooren 2004; Weber et al. 2006), and the plesiomorphic absence of both plastidial phosphate-translocator (Price et al. 2012) and nuclear-encoded pigment-binding proteins (Wolfe et al. 1994; Price et al. 2012), which are present in the Rhodophyta and Viridiplantae.
Materials and Methods
Taxon Selection
Taxa were selected to obtain as comprehensive a coverage as possible of the Archaeplastida (Viridiplantae, Rhodophyta, and Glaucophyta) and Cyanobacteria phylogenetic tree, at the same time minimizing the total number of selected taxa due the computation burden of the analyses being performed. To this end, we conducted preliminary analyses of the Cyanobacteria on all 115 cyanobacterial ribosomal small subunit (SSU) sequences available in the NCBI GenBank in October 2010. We then inferred Neighbor-Joining trees using log determinant (NJLD) distances and ML trees using the programs P4 (Foster 2004) and RAxML (Stamatakis 2006), respectively. All further NJLD and ML trees were obtained using these programs. Using these trees, we selected 16 taxa to represent the Cyanobacteria groups: 2 GBACT (Gloeobacter violaceus and Synechococcus JA33Ab), 3 UNIT+ (A. marina, Thermosynechococcus elongatus, and Cyanothece PCC7425), 2 NOST-1 (Nostoc punctiforme and Nostoc PCC7120), 1 OSC-2 (Trichodesmium erythraeum), 3 SO-6 (Synechococcus elongatus, Synechococcus RCC307, and Prochlorococcus marinus), and 5 SPM-3 (Synechococcus PCC7002, Synechocystis PCC6803, Microcystis aeruginosa, Cyanothece PCC7822, and Cyanothece PCC8801). For convenience, we refer to cyanobacterial groups GBACT, NOST-1, UNIT+, SO-6, OSC-2, and SPM-3 adapting the notation used in Criscuolo and Gribaldo (2011), itself based on the notations originally designated by Turner et al. (1999) and Honda et al. (1999). The latter five groups we refer to as the “core-cyanobacteria” as they often appear as a monophyletic group (Criscuolo and Gribaldo 2011). We further selected 18 plant taxa to represent the Archaeplastida lineage: 1 Glaucophyta (Cyanophora paradoxa), 4 Rhodophyta (Cyanidium caldarium, Cyanidioschyzon merolae, Porphyra purpurea, and Gracilaria tenuistipitata), and 13 Viridiplantae (Ostreococcus tauri, Pyramimonas parkeae, Pycnococcus provasolii, Monomastix oke1, Micromonas RCC299, Chlamydomonas reinhardtii, Scenedesmus obliquus, Chlorella vulgaris, Bryopsis hypnoides, Nephroselmis olivacea, belonging to Chlorophyta, and Chlorokybus atmophyticus, Zygnema circumcarinatum, Aneura mirabilis, belonging to the Streptophyta). Eight bacterial outgroup taxa were chosen, including phototrophs with a rudimentary photosynthesis-related machinery (Heliobacterium modesticaldum, Bacillus subtilis, Bdellovibrio bacteriovorus, Novosphingobium aromaticivorans, Rhodopseudomonas palustris, Chlorobium limicola, Herpetosiphon aurantiacus, and Roseiflexus castenholzii). Taxonomy and NCBI genome accession numbers are presented in supplementary table S3 (Supplementary Material online).
Data were retrieved and stored in a local PostGreSQL (8.4.7) relational database with the BioSQL (1.0.1) schema. For each of the chosen taxa, gene and the corresponding protein BLAST databases were constructed using makeblastdb (NCBI BLAST 2.2.24+).
Data Selection and Data Set Assembly
We obtained a preliminary set of 78 loci by performing a search for gene name terms identified in the genome annotations of the 18 selected plastid-bearing taxa, and selecting those loci that occurred in 8 or more taxa. Amino acid data sets for all taxa of the selected loci were aligned using MUSCLE 3.8 (Edgar 2004) and sequences that were inconsistent with the rest of the alignment (i.e., misaligned due to mistaken annotation) were removed. At this stage, two loci (ycf1 and ycf20) were excluded due to lack of data. To search for proteins that were present but lacked annotation (or were mis-annotated), we built amino acid protein models of the selected loci using HMMER 3.0 (Finn et al. 2011), and performed a HMMER search of the amino acid BLAST databases of taxa with unidentified loci. The cut-off e value of the HMMER search was chosen individually for each locus after consideration of the results. Specifically, all best scoring sequences were considered provided the ratio between the score of a sequence and the score of the next best-ranked sequence remained greater than 0.75. Some flexibility was allowed to this cut-off criterion so that at least one sequence for each taxon was considered. In total, 414 additional sequences were found using this method (supplementary table S4, Supplementary Material online). The identified amino acid sequences were then re-aligned using Muscle 3.8 and inspected by eye a second time to exclude inconsistent and mis-aligned sequences. Sequences that were an exact duplicate of a sequence already present were also excluded. Additionally, we also attempted to identify taxon gene sequences missing for individual loci by constructing a consensus sequence of nucleotide sequences that corresponded to the updated amino acid data sets using Biopython 1.55 (Cock et al. 2009), and performing a nucleotide BLAST analysis of taxon gene BLAST databases. Using this method, we identified a further 33 sequences (supplementary table S4, Supplementary Material online). To identify missing gene sequences that had been relocated from the plastid to the nuclear compartment through the mechanism of endosymbiotic gene transfer, we searched the on-line NCBI “nt” database repository with BLAST using the “Entrez” interface of Biopython with the previously constructed nucleotide consensus sequences used as query sequences for each locus. Using this method, we identified a further 14 sequences (supplementary table S4, Supplementary Material online). In total, 76 loci with sequences having at least 20 of the 34 ingroup taxa were retained for further analysis. This set of 76 selected loci included 23 ribosomal proteins, 30 photosystem genes, plus a further 23, mainly “housekeeping,” genes (supplementary table S4, Supplementary Material online).
Phylogenetic Analyses of Individual Loci
Nucleotide gene sequences of the 76 loci were aligned using TranslatorX 1.1 (Abascal et al. 2010). An initial estimation of the set of ambiguously aligned sites was obtained using Gblocks 0.91b (Castresana 2000) and then re-assessed by eye. All ambiguously aligned sites, and the other sites within the codons where the ambiguously aligned sites occur, were excluded from further analysis. Gene sequence data sets were translated into their corresponding amino acid protein data sets using the translate function of Biopython and the “Bacterial, Archaeal, and Plant Plastid” genetic code (NCBI translation table 11).
For each of the 76 nucleotide gene alignments, we inferred NJLD trees and conducted ML bootstrap analyses (1,000 replicates), the latter with model types identified using MrModeltest 2.3 (Nylander 2004) and the Akaike information criterion (AIC) criterion. All selected models included a parameter describing a discrete gamma-distribution of among-site rates (four categories) and a parameter for a proportion of invariant sites, when found to be optimal. All model parameters were optimized during analysis unless otherwise stated, the exception being the exchange rates of empirical protein models. Where optimal models were not implemented in particular software, we used the implemented model with the highest AIC score. Each inferred NJLD and ML tree was inspected for implied instances of gene duplication. Using this information, and by comparing the tree topologies to preliminary NJLD and ML trees based on SSU rRNA, we selected a likely orthologous sequences for each taxon and discarded the remaining paralogues. Furthermore, we assessed the trees for potential cases of lateral gene transfer by noting cases of well supported but taxonomically incongruous relationships that were not pertinent to the specific relationships under study and discarded these taxa from both the nucleotide and amino acid data sets. We considered taxa aberrant when they contradicted the monophyly of the following clades at  95% bootstrap support: outgroup Bacteria, ingroup taxa, Archaeplastida, Glaucophyta, Rhodophyta, and Viridiplantae.
95% bootstrap support: outgroup Bacteria, ingroup taxa, Archaeplastida, Glaucophyta, Rhodophyta, and Viridiplantae.
For each gene data set, we performed a composition homogeneous MCMC analyses under the best-fitting evolutionary model with at least one million generations using P4 (Foster 2004). The MCMC chains were continued if the log likelihood values appeared not to have plateaued or the ESS (effective sampling size) sampling values were less than 300. Using the χ2 test statistic and a simulated posterior predictive distribution for the null distribution sampled from the MCMC, we tested whether the composition obtained with the model was sufficient to explain the variation in composition across the tree using a one-tailed area probability test (the P value was deemed significant if <0.05). When the model composition failed the test, we added supplementary composition vectors step-wise using the NDCH model implemented in P4 until the composition test was passed (i.e., until the model was able to generate simulated data with a composition heterogeneity statistically compatible with that of the real data). Marginal log likelihoods were calculated using equation 16 of Newton and Raftery (1994) as implemented in P4. The same procedure was used in all MCMC NDCH analyses. Each inferred MCMC NDCH tree was again assessed for aberrant taxonomic relationships as described earlier. Following this inspection, the cyst locus was discarded due to lack of data, reducing the number of loci to 75. The size of each finalized gene data set, the numbers of included taxa, and the numbers of included sites are presented in supplementary table S5, Supplementary Material online. NCBI GenBank accession numbers of all included sequences are noted in supplementary table S6, Supplementary Material online.
Optimal model types were re-assessed for the updated gene data sets using MrModeltest and ML bootstrap and MCMC NDCH model analyses repeated. Optimal protein models were assessed for each amino acid data set using ProtTest 2.4 (Abascal et al. 2005) and ML bootstrap and MCMC NDCH model analyses were performed as described earlier. The loci and the corresponding selected models are provided in supplementary table S7, Supplementary Material online.
Concatenated Data Analyses
We constructed two combined data matrices, one by concatenating all 75 genes (“cg75”) and the other their 75 translations into proteins (“cp75”). Optimal model types for these data sets were estimated using MrModeltest (for “cg75”) and ProtTest (for “cp75”). ML bootstrap analyses (200 pseudo-replicates each with 2 searches) were performed using RAxML under the optimal data-homogeneous and stationary composition models available for each of the two concatenated data sets (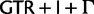 for nucleotides and
for nucleotides and 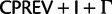 for amino acids, “cg75_mlboot” and “cp75_mlboot” in supplementary table S1, Supplementary Material online). Bayesian analyses were performed with P4 using the optimal data-homogeneous and stationary composition models available for each of the two concatenated data sets (
for amino acids, “cg75_mlboot” and “cp75_mlboot” in supplementary table S1, Supplementary Material online). Bayesian analyses were performed with P4 using the optimal data-homogeneous and stationary composition models available for each of the two concatenated data sets (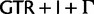 for nucleotides and
for nucleotides and 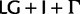 for amino acids) following the procedure described for the individual loci, but with a tolerance for lower ESS values (nucleotide-based analysis “cg75_stat” and amino acid-based analysis “cp75_stat” in supplementary table S1, Supplementary Material online). Site-homogeneous with nonstationary composition NDCH analyses were performed with P4 by adding composition vectors to the above settings, following the procedure described for the individual loci (nucleotide-based analysis “cg75_NDCH” and amino acid-based analysis “cp75_NDCH” in supplementary table S1, Supplementary Material online). An across-site composition heterogeneous Bayesian analysis was performed using the
for amino acids) following the procedure described for the individual loci, but with a tolerance for lower ESS values (nucleotide-based analysis “cg75_stat” and amino acid-based analysis “cp75_stat” in supplementary table S1, Supplementary Material online). Site-homogeneous with nonstationary composition NDCH analyses were performed with P4 by adding composition vectors to the above settings, following the procedure described for the individual loci (nucleotide-based analysis “cg75_NDCH” and amino acid-based analysis “cp75_NDCH” in supplementary table S1, Supplementary Material online). An across-site composition heterogeneous Bayesian analysis was performed using the  model implemented in Phylobayes 3.3 (Lartillot and Philippe 2004) (options “-cat -lg -dgam 4”) on the amino acid data set (“cp75_CAT” in supplementary table S1, Supplementary Material online). It was conducted using 2 MCMC chains and the automatic stopping criterion based on the computation of convergence statistics between 2 chains (maxdiff > 0.3 and effective size > 50, checking every 100 cycles, options “-s -nchain 2 100 0.3 50”).
model implemented in Phylobayes 3.3 (Lartillot and Philippe 2004) (options “-cat -lg -dgam 4”) on the amino acid data set (“cp75_CAT” in supplementary table S1, Supplementary Material online). It was conducted using 2 MCMC chains and the automatic stopping criterion based on the computation of convergence statistics between 2 chains (maxdiff > 0.3 and effective size > 50, checking every 100 cycles, options “-s -nchain 2 100 0.3 50”).
Supplementary Material
Supplementary figures S1–S13 and tables S1–S7 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org).
Acknowledgments
The authors thank four anonymous reviewers for their helpful criticism. This work was supported by a Fundação para a Ciência e a Tecnologia (FCT, Portugal) grant PTDC/BIA-BCM/099565/2008 to C.J.C.; the European Regional Development Fund (ERDF) through the COMPETE – Operational Programme Competitiveness and national funds through FCT (PEst-C/MAR/LA0015/2011) to Centro de Ciências do Mar, Faro, Portugal (CCMar); and the European Research Council Advanced Investigator Programme to T.M.E.
References
- Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21(9):2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38(2 Suppl):W7–W13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken A, Stanier RY. Characterization of peptidoglycan from the cyanelles of Cyanophora paradoxa. J Gen Microbiol. 1979;112(2):219–223. [Google Scholar]
- Akashi H, Kliman RM, Eyre-Walker A. Mutation pressure, natural selection, and the evolution of base composition in Drosophila. Genetica. 1998 102/103:49–60. [PubMed] [Google Scholar]
- Averof M, Rokas A, Wolfe KH, Sharp PM. Evidence for a high-frequency of simultaneous double-nucleotide substitutions. Science. 2000;287(5456):1283–1386. doi: 10.1126/science.287.5456.1283. [DOI] [PubMed] [Google Scholar]
- Ball S, Colleoni C, Cenci U, Raj JN, Tirtiaux C. The evolution of glycogen and starch metabolism in eukaryotes gives molecular clues to understand the establishment of plastid endosymbiosis. J Exp Bot. 2011;62:1775–1801. doi: 10.1093/jxb/erq411. [DOI] [PubMed] [Google Scholar]
- Bhattacharya D, Medlin L. The phylogeny of plastids: a review based on comparisons of small-subunit ribosomal RNA coding regions. J Phycol. 1995;31(4):489–498. [Google Scholar]
- Blank CE. Evolutionary timing of the origins of mesophilic sulphate reduction and oxygenic photosynthesis: a phylogenomic dating approach. Geobiology. 2004;2(1):1–20. [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Chan CX, Yang EC, Banerjee T, Yoon HS, Martone PT, Estevez JM, Bhattacharya D. Red and green algal monophyly and extensive gene sharing found in a rich repertoire of red algal genes. Curr Biol. 2011;21(4):328–333. doi: 10.1016/j.cub.2011.01.037. [DOI] [PubMed] [Google Scholar]
- Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJL. Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25(11):1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CJ, Foster PG, Hirt RP, Harris SR, Embley MT. The archaebacterial origin of eukaryotes. Proc Natl Acad Sci U S A. 2008;105(51):20356–20361. doi: 10.1073/pnas.0810647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CJ, Li B, Foster PG, Embley TM, Civáň P. Conflicting phylogenies for early land plants are caused by composition biases among synonymous substitutions. Syst Biol. 2014;63:272–279. doi: 10.1093/sysbio/syt109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo A, Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10:210. doi: 10.1186/1471-2148-10-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo A, Gribaldo S. Large-scale phylogenomic analyses indicate a deep origin of primary plastids within Cyanobacteria. Mol Biol Evol. 2011;28(11):3019–3032. doi: 10.1093/molbev/msr108. [DOI] [PubMed] [Google Scholar]
- Dagan T, Roettger M, Stucken K, Landan G, Koch R, Major P, Gould SB, Goremykin VV, Rippka R, Tandeau de Marsac N, et al. Genomes of Stigonematalean cyanobacteria (subsection V) and the evolution of oxygenic photosynthesis from prokaryotes to plastids. Genome Biol Evol. 2013;5(1):31–44. doi: 10.1093/gbe/evs117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps P, Colleoni C, Nakamura Y, Suzuki E, Putaux JL, Buléon A, Haebel S, Ritte G, Steup M, Falcón LI, et al. Metabolic symbiosis and the birth of the plant kingdom. Mol Biol Evol. 2008;25:536–548. doi: 10.1093/molbev/msm280. [DOI] [PubMed] [Google Scholar]
- Deusch O, Landan G, Roettger M, Gruenheit N, Kowallik KV, Allen JF, Martin W, Dagan T. Genes of cyanobacterial origin in plant nuclear genomes point to a heterocyst-forming plastid ancestor. Mol Biol Evol. 2008;25(4):748–761. doi: 10.1093/molbev/msn022. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcón LI, Magallón S, Castillo A. Dating the cyanobacterial ancestor of the chloroplast. ISME J. 2010;4(6):777–783. doi: 10.1038/ismej.2010.2. [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(2 Suppl):W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PG. Modeling compositional heterogeneity. Syst Biol. 2004;53(3):485–495. doi: 10.1080/10635150490445779. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11(5):725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Honda D, Yokota A, Sugiyama J. Detection of seven major evolutionary lineages in Cyanobacteria based on the 16S rRNA gene sequence analysis with new sequences of five marine Synechococcus strains. J Mol Evol. 1999;48(6):723–739. doi: 10.1007/pl00006517. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Crandall KA. Phylogeny estimation and hypothesis testing using maximum likelihood. Annu Rev Ecol Syst. 1997;28:437–466. [Google Scholar]
- Inagaki Y, Roger AJ. Phylogenetic estimation under codon models can be biased by codon usage heterogeneity. Mol Phylogenet Evol. 2006;40(2):428–434. doi: 10.1016/j.ympev.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Jeffroy O, Brinkmann H, Delsuc F, Philippe H. Phylogenomics: the beginning of incongruence? Trends Genet. 22. 2006;(4):225–231. doi: 10.1016/j.tig.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Keeling PJ. Diversity and evolutionary history of plastids and their hosts. Am J Bot. 2004;91(10):1481–1493. doi: 10.3732/ajb.91.10.1481. [DOI] [PubMed] [Google Scholar]
- Kosiol C, Holmes I, Goldman N. An empirical codon model for protein sequence evolution. Mol Biol Evol. 2007;24(7):1464–1479. doi: 10.1093/molbev/msm064. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21(6):1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- Martin W, Rujan T, Richly E, Hansen A, Cornelsen S, Lins T, Leister D, Stoebe B, Hasegawa M, Penny D. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc Natl Acad Sci U S A. 2002;99(19):12246–12251. doi: 10.1073/pnas.182432999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden GI, van Dooren GG. Evolution: red algal genome affirms a common origin of all plastids. Curr Biol. 2004;14(13):R514–R516. doi: 10.1016/j.cub.2004.06.041. [DOI] [PubMed] [Google Scholar]
- Moreira D, Le Guyader H, Philippe H. The origin of red algae and the evolution of chloroplasts. Nature. 2000;405(6782):69–72. doi: 10.1038/35011054. [DOI] [PubMed] [Google Scholar]
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11(5):715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- Nelissen B, Van de Peer Y, Wilmotte A, De Wachter R. An early origin of plastids within the cyanobacterial divergence is suggested by evolutionary trees based on complete 16S rRNA sequences. Mol Biol Evol. 1995;12(6):1166–1173. doi: 10.1093/oxfordjournals.molbev.a040289. [DOI] [PubMed] [Google Scholar]
- Newton MA, Raftery AE. Approximate Bayesian inference with the weighted likelihood bootstrap. J Roy Stat Soc B Met. 1994;56(1):3–48. [Google Scholar]
- Nylander JAA. Mr Modeltest v2 [Program distributed by the author] Uppsala (Sweeden): Evolutionary Biology Centre, Uppsala University; 2004. [Google Scholar]
- Philippe H, Brinkmann H, Lavrov DV, Littlewood DTJ, Manuel M. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 2011;9(3):e1000602. doi: 10.1371/journal.pbio.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DC, Chan CX, Yoon HS, Yang EC, Qiu H, Weber APM, Schwacke R, Gross J, Blouin NA, Lane C, et al. Cyanophora paradoxa genome elucidates origin of photosynthesis in algae and plants. Science. 2012;335:843–847. doi: 10.1126/science.1213561. [DOI] [PubMed] [Google Scholar]
- Regier JC, Shultz JW, Zwick A, Hussey A, Ball B, Wetzer R, Martin JW, Cunningham CW. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature. 2010;463:1079–1083. doi: 10.1038/nature08742. [DOI] [PubMed] [Google Scholar]
- Reyes-Prieto A, Yoon HS, Moustafa A, Yang EC, Andersen RA, Boo SM, Nakayama T, Ishida K, Bhattacharya D. Differential gene retention in plastids of common recent origin. Mol Biol Evol. 2010;27(7):1530–1537. doi: 10.1093/molbev/msq032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippka R, Deruelles J, Waterbury JB, Herdman M, Stanier RY. Generic assignments, strain histories and properties of pure cultures of Cyanobacteria. J Gen Microbiol. 1979;111(1):1–61. [Google Scholar]
- Rodriguez F, Oliver JL, Marin A, Medina JR. The general stochastic model of nucleotide substitution. J Theor Biol. 1990;142(4):485–501. doi: 10.1016/s0022-5193(05)80104-3. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Ezpeleta N, Brinkmann H, Burey SC, Roure B, Burger G, Löffelhardt W, Bohnert HJ, Philippe H, Lang BF. Monophyly of primary photosynthetic eukaryotes: green plants, red algae, and glaucophytes. Curr Biol. 2005;15(14):1325–1330. doi: 10.1016/j.cub.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Rota-Stabelli O, Lartillot N, Philippe H, Pisani D. Serine codon-usage bias in deep phylogenomics: pancrustacean relationships as a case study. Syst Biol. 2013;62(1):121–133. doi: 10.1093/sysbio/sys077. [DOI] [PubMed] [Google Scholar]
- Simmons MP, Carr TG, O’Neill K. Relative character-state space, amount of potential phylogenetic information, and heterogeneity of nucleotide and amino acid characters. Mol Phylogenet Evol. 2004;32(3):913–926. doi: 10.1016/j.ympev.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stayton CT. Is convergence surprising? An examination of the frequency of convergence in simulated datasets. J Theor Biol. 2008;252(1):1–14. doi: 10.1016/j.jtbi.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Steiner JM, Löffelhardt W. Protein import into cyanelles. Trends Plant Sci. 2002;7(2):72–77. doi: 10.1016/s1360-1385(01)02179-3. [DOI] [PubMed] [Google Scholar]
- Swofford DL, Olsen GJ, Waddell PJ, Hillis DM. 1996. Phylogenetic inference. In: Hillis DM, Moritz C, Mable BK, editors. Molecular systematics, 2nd ed. Sunderland (MA): Sinauer Associates. p. 407–514. [Google Scholar]
- Turner S, Pryer KM, Miao VPW, Palmer JD. Investigating deep phylogenetic relationships among Cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 1999;46(4):327–338. doi: 10.1111/j.1550-7408.1999.tb04612.x. [DOI] [PubMed] [Google Scholar]
- Weber AP, Linka M, Bhattacharya D. Single, ancient origin of a plastid metabolite translocator family in plantae from an endomembrane-derived ancestor. Eukaryot Cell. 2006;5(3):609–612. doi: 10.1128/EC.5.3.609-612.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe GR, Cunningham FX, Durnfordt D, Green BR, Gantt E. Evidence for a common origin of chloroplasts with light-harvesting complexes of different pigmentation. Nature. 1994;367:566–568. [Google Scholar]
- Yang Z. Computational molecular evolution. Oxford Series in Ecology and Evolution. Oxford: Oxford University Press; 2006. [Google Scholar]
- Zwick A, Regier JC, Zwickl DJ. Resolving discrepancy between nucleotides and amino acids in deep-level arthropod phylogenomics: differentiating serine codons in 21-amino-acid models. PLoS One. 2012;7(11):e47450. doi: 10.1371/journal.pone.0047450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.