

Abstract
The first microRNAs (miRNAs) were identified as essential, conserved regulators of gene expression, targeting the same genes across nearly all bilaterians. However, there are also prominent examples of conserved miRNAs whose functions appear to have shifted dramatically, sometimes over very brief periods of evolutionary time. To determine whether the functions of conserved miRNAs are stable or dynamic over evolutionary time scales, we have here defined the neutral turnover rates of short sequence motifs in predicted primate 3′-UTRs. We find that commonly used approaches to quantify motif turnover rates, which use a presence/absence scoring in extant lineages to infer ancestral states, are inherently biased to infer the accumulation of new motifs, leading to the false inference of continually increasing regulatory complexity over time. Using a maximum likelihood approach to reconstruct individual ancestral nucleotides, we observe that binding sites of conserved miRNAs in fact have roughly equal numbers of gain and loss events relative to ancestral states and turnover extremely slowly relative to nearly identical permutations of the same motif. Contrary to case studies showing examples of functional turnover, our systematic study of miRNA binding sites suggests that in primates, the regulatory roles of conserved miRNAs are strongly conserved. Our revised methodology may be used to quantify the mechanism by which regulatory networks evolve.
Keywords: molecular evolution, motif turnover, miRNA evolution, regulatory network, molecular phylogeny, parsimony
Introduction
Prominent studies have shown experimentally that some conserved microRNAs (miRNAs) can regulate the same targets over deep evolutionary time (Pasquinelli et al. 2000; Moss and Tang 2003; Kucherenko et al. 2012; La Torre et al. 2013), yet most conserved miRNAs have no experimentally identified conserved target genes and are dispensable in Caenorhabditis elegans under laboratory conditions, suggesting that the function of these conserved miRNAs may be changing or hidden (Chen and Rajewsky 2007; Miska et al. 2007). Of the 21–23 nucleotides of a mature miRNA that might potentially base pair to regulate target mRNAs, only the first seven to eight nucleotides are essential (Lewis et al. 2003; Brennecke et al. 2005). This region has come to be known as the “seed” of miRNA target interactions and has been used as a basis for computationally predicting potential interactions between miRNAs and their target mRNAs. In support of a model of the conserved roles of miRNAs, Farh et al. (2005) have used these computational predictions to show that miRNA binding sites are conserved relative to random sequence within 3′-UTRs. This signature of relative conservation has served as a basis to improve miRNA targeting prediction, implemented through the popular miRNA target predictor TargetScan (Lewis et al. 2003, 2005). In addition to this work, evolutionary analyses have shown that SNPs in miRNA binding sites are rare relative to other SNP classes and that miRNAs themselves appear to be deeply conserved as a class (Chen and Rajewsky 2006a, 2006b). Together, this literature suggests that miRNAs are conserved regulators of key developmental processes.
Simultaneously, many of these same studies and others also show evidence that the processes miRNAs participate in are changing. Since the first miRNAs were discovered, very few appear to be similarly essential to developmental processes (Miska et al. 2007) or to participate in targeting interactions that are well conserved, even for miRNAs that are themselves extremely well-conserved (Chen and Rajewsky 2007; Gao 2010). In previous studies, which used a comparative approach to examine 3′-UTRs derived from human, dog, mouse, rat, and chicken to find the conservation levels of miRNA targets (as defined by miRNA seed complementarity), 90% of potential miRNA binding sites are estimated to be nonconserved (Lewis et al. 2005; Farh et al. 2005; Xie et al. 2005, as summarized in Hiard et al. 2010, fig. 2A). Within these nonconserved target sites, many appear to be functional in vitro (Farh et al. 2005), and evolutionary studies have estimated that 30–50% of nonconserved miRNA binding sites may be functional (Chen and Rajewsky 2006b).
Fig. 2.
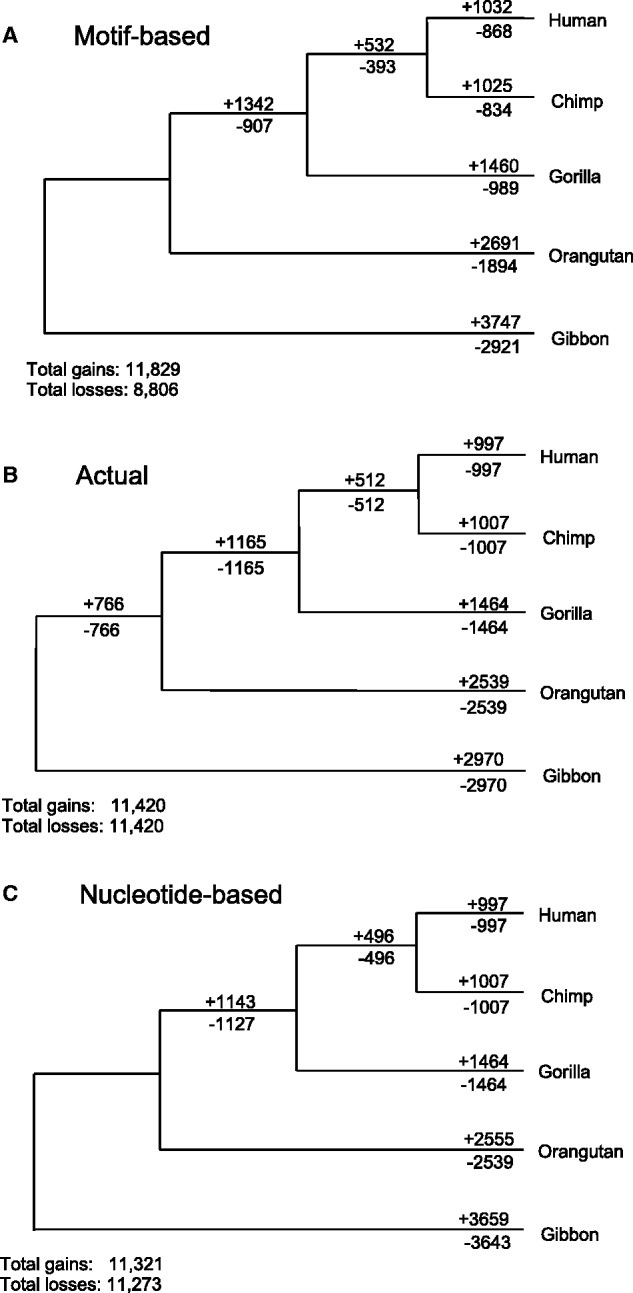
Turnover rates of 8mers in a long, neutrally evolving simulated primate sequence. In (A), the motif-based approach finds many more gain events than losses. In (B), we show the actual numbers of turnover events that occurred globally in this neutrally evolving sequence, in which gains and losses are balanced as every 8mer lost from an ancestor is also a gain of a new 8mer in the descendant. In (C), the maximum likelihood approach to ancestral reconstruction finds similar gain events to loss events, but slightly underestimates overall gains and losses relative to actual events. Because DNAML creates a trifurcation at the root, some turnover events from the common ancestor of Human, Chimp, Gorilla, and Orangutan are misattributed to Gibbon.
Some of the most compelling evidence that miRNAs may play nonconserved roles comes from a study of the members of a recently speciated clade of cichlids with dramatically divergent behavior, morphology, and diet but nearly identical genomes (so much so that the majority of SNPs within species are also shared between them). Genotyping of these species revealed both a significantly elevated occurrence and divergence of SNPs in predicted miRNA binding sites relative to the surrounding 3′-UTR and the rest of the genome. These results suggest that the modifications underlying speciation in these fish may be driven in part by changes in miRNA regulation (Loh et al. 2010).
Similar restructuring of miRNA regulatory networks has been seen in insects, through an evolutionary and experimental study of the miR10/100 family of miRNAs, which found that the strand from which the mature form of this conserved miRNA hairpin is derived has shifted at least three times during insect evolution, completely changing the functional targets of this conserved gene (Griffiths-Jones et al. 2011).
One explanation for the conservation of miRNAs despite shifts in the target genes invokes a model in which miRNAs may improve the robustness of gene regulatory networks by participating in feedback loops that buffer the genome against perturbations (Wu et al. 2009). According to this model, removing miRNAs does not necessarily produce any particular phenotype but makes phenotypes less stable inenvironments that fluctuate, a process known as canalization. miR-7 has been demonstrated to participate in this type of role in the neuronal specification of D. melanogaster, as flies lacking miR-7 develop abnormally only at fluctuating temperatures (Li et al. 2009). Interestingly, the robustness of these networks has been shown to contribute to the overall evolvability of systems by permitting phenotypes to vary and thus to be exposed to natural selection - allowing miRNAs in these roles to indirectly modulate the pace of evolution (Wu et al. 2009). Indeed, through multigenerational selection experiments, it is shown that miR-9a, another miRNA that ensures the precise neuronal specification in Drosophila (Li et al. 2006), dampens the impact of genomic diversity on variability of cell behavior (Cassidy et al. 2013). This model does not predict the pace at which miRNAs change their targets but only suggests that traits that vary will be less heavily regulated by miRNAs than canalized traits. Under this model, the loss of individual binding sites reduces the overall canalization of pathways, making them less robust and more heritably variable, whereas the gain of binding sites confers the opposite effect. In this way, strong positive or negative selection may operate on binding sites without any immediately observable phenotypes.
As an alternative explanation for changes in regulatory networks, and the increase in regulatory complexity inferred from reconstructed ancestral states, it has been proposed that regulatory complexity evolved not out of increased selective pressure but due to its absence (Lynch 2007). Under this model, increases in regulatory complexity are compensations for weakly deleterious alleles that cannot be purged effectively by purifying selection. This model leads to the hypothesis that changes in miRNA targeting will be expected to be quite frequent, and to lend themselves to the gradual, constant accumulation of new miRNA binding sites, with little selective pressure for the loss of existing interactions.
Quantifying the rate at which miRNA binding sites evolve has important implications for discovering the purpose of miRNA regulatory networks, yet even individual studies such as those described above show contradictory evidence as to whether fast or slow turnover rates predominate in the targeting of mRNAs by miRNAs. Although we know of only one other study that has undertaken a systematic survey of miRNA binding site turnover rates (Xu et al. 2013), several studies have examined the turnover rate of miRNAs themselves by scoring extant species as having or not having particular miRNAs and extrapolating ancestral states accordingly (Nozawa et al. 2010, 2012; Meunier et al. 2013; Xiao et al. 2013). These motif-based ancestral reconstruction studies show a systematic increase in the number of miRNAs over evolutionary time.
We have here applied existing approaches and devised alternative methods to more accurately survey the rates at which primate 3′-UTRs gain and lose the binding sites necessary for miRNA targeting. In this way, we can differentiate between models proposing that miRNAs are changing their functions rapidly via positive selection, are strongly conserved via purifying selection, or are evolving neutrally via drift.
Results and Discussion
Approaches and Terminology
Initially, we applied an ancestral reconstruction model which we have termed the “motif-based” approach. For these purposes, we defined the presence of a motif in a given lineage as an exact match to a given sequence (an arbitrarily chosen eight base pair stretch of nucleotides with no initial reference to function) at a given position in a 3′-UTR alignment. Using this definition, we assigned each species as either having or not having the motif of interest and used these values to infer ancestral states most parsimonious with the observed distribution of the motif in extant lineages. Ancestors inferred not to have a motif whose descendants possessed a motif at the site in question were considered as “gain” events in the descendants, whereas ancestors inferred to have a motif whose descendants did not possess the motif were considered to have “lost” this motif. This process was repeated across all possible eight nucleotide motifs and all positions of all 3′-UTRs. Note that in this model, only the presence or absence of a motif is inferred in the ancestor and not the ancestral sequence itself. Therefore, it is possible in this case for two sister species that differ by a single nucleotide to have an inferred ancestor that is inferred not to have either descendant state and for a motif to be inferred as arising without destroying any existing motif in the ancestor.
We also implemented a second approach, termed a “nucleotide-based” ancestral reconstruction, which uses a per-nucleotide ancestral motif reconstruction approach to infer ancestral motif states. Note that unlike the motif-based ancestral reconstruction, with this method ancestral sequences can be queried directly, and every loss of a particular motif by point mutation in an ancestral sequence can be alternatively defined as the gain of the new descendant motif.
The Undercounting of Losses by the Motif-Based Parsimony Approach
The motif-based approach which we initially used to determine the turnover rates of miRNA binding sites was designed in a similar way to previous studies conducted on miRNAs themselves (Nozawa et al. 2010, 2012; Meunier et al. 2013; Xiao et al. 2013). Although there are variations in methodology, all of these approaches use the presence or absence of a gene in extant species to infer presence or absence of the gene in ancestral lineages. Our motif-based approach yielded results suggesting that miRNA binding sites, like miRNA genes, are gained much more frequently than they are lost, leading to a net gain of miRNA binding sites over time. However, when this work was repeated with all motifs of length 8, including those not thought to have any function, the bias remained (table 1, fig. 1A). These results were quite unexpected; in the absence of insertions and deletions (which we excluded from analysis), every motif lost from an ancestral sequence should be replaced with a different motif in a descendant created by a point substitution, such that although individual motif types might be selected for or against, gains and losses should be identical overall.
Table 1.
Parsimony Approaches Scoring Motifs as Present/Absent Undercount Losses.
| Data Set | Species Surveyed | Gains | Losses |
|---|---|---|---|
| Meunier et al. (2013)a | Human, macaque, mouse, oppossum, platypus, chicken | 719 | 140 |
| Nozawa et al. (2010)a | (Drosophila) melanogaster, simulans, sechellia, yakuba, erecta, ananassae, pseudoobscura, persimilis, willistoni, mojavensis, virilis, grimshawi | 101 | 48 |
| Nozawa et al. (2012)a | Arabidopsis, papaya, poplar, Medicago, soybean, grape, rice, Sorghum, maize, moss, green algae | 743 | 77 |
| Xiao et al. (2013)a | Oryza Sativa, Phoenix Dactylifera, Populus Trichocarpa, Malus domestica, Glycine max, Solanum lycopersicum, Citrus sinensis, Arabidopsis thaliana | 167 | 4 |
| Current study, experimental primate parsimonyb | Human, chimp, gorilla, orangutan, gibbon | 8,337,944 | 6,087,687 |
| Current study, simulated neutral primate parsimonyc | Human, chimp, gorilla, orangutan, gibbon | 11,829 | 8,806 |
aEarlier studies examining the turnover rates of miRNA genes find many more gain events than losses.
bWhen using these methods, more gains than losses are inferred for the turnover rates of miRNA binding sites in the 3′-UTRs of primates.
cSimulated neutral data sets (which have been simulated to have identical gain and loss rates in reality) also infer more gains than losses.
Fig. 1.

Two methods for inferring turnover rates in syntenically defined primate 3′-UTRs. In (A), the ancestral states of 3′-UTRs (containing or not containing a binding site) are inferred using presence/absence calls in extant species at the level of motifs, and gains and losses are in turn inferred from the inferred ancestors. Losses appear to occur much less frequently than gains. In (B), a revised maximum likelihood approach using individual reconstructed ancestral nucleotides was applied to primate 3′-UTRs. The skew toward gains is completely ameliorated.
To further examine these results, we simulated neutral data whose ancestry could be directly queried, with speciation times based on those estimated for primates. Using our initial approach, we found that in this neutral data set, the apparent rate of miRNA binding site gain still dramatically outpaced the rate of binding site loss (fig. 2A). When we used the known ancestors to determine actual turnover rates, it was immediately apparent that our initial method dramatically undercounts loss events (fig. 2B). A more detailed analysis revealed that the inferred discrepancy between binding site loss events and gain events is due to a saturation of mutations in the underlying sequence and inherent differences in the effects of this saturation on gain and loss events. In instances in which multiple independent mutations produce the same outcome in two groups of species sharing a common ancestor, the assumption of infinite sites is violated, and approaches assuming that every mutation occurs at a separate site will misinfer a single event in the lineage leading to the ancestor (fig. 3). This applies equally to both gain and loss substitutions. However, when gain and loss events are scored at the level of motifs, the probability of multiple convergent gain events in the descendants of a common ancestor lacking a motif is much less than the probability of multiple convergent loss events in an ancestor possessing a motif. This is because convergent loss events in two descendant lineages can occur through any two mutations anywhere within the existing motif, whereas convergent gain events from the same inherited ancestral sequence require the same mutation to occur twice at the same site (fig. 3). Because this probability of independent convergent losses is inherently much greater than the probability of independent convergent gains, any method that initially scores a motif only as present or absent will misinfer multiple loss events as a single loss much more often than it misinfers multiple gain events as a single gain, in a manner that becomes increasingly evident as the sequence divergence between species increases and the likelihood of convergent substitutions within a motif increases accordingly. This correlation of levels of bias with evolutionary divergence can be observed both experimentally (see table 1) and via simulation (data not shown). Moreover, because our motif-based approach infers only the presence or absence of a motif in the ancestral sequence, in cases of multiple mutations within a single region, an ancestral sequence can be inferred as not being any one motif, resulting in an inference of multiple de novo motifs having arisen at this location without any corresponding losses in the undefined ancestor.
Fig. 3.
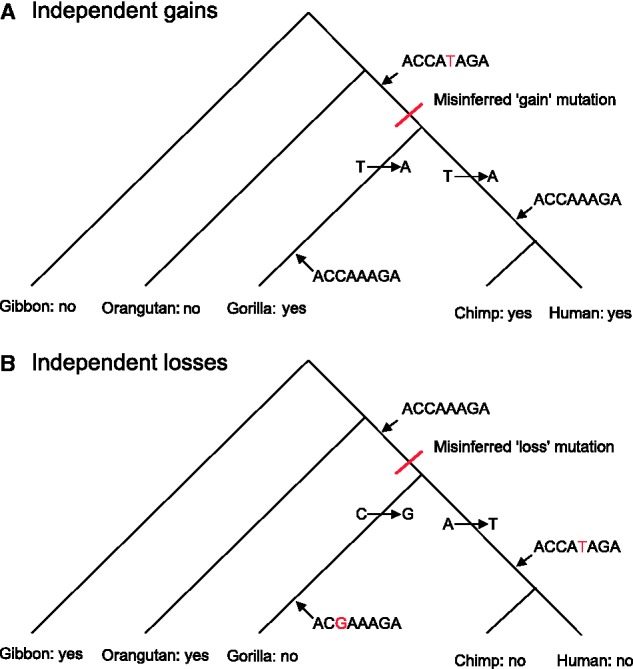
Independent gains are less likely than independent losses. Although parsimony approaches may misinfer independent gain substitutions in multiple descendants as single events (A) and independent loss substitutions as single events (B), independent gains in an ancestral sequence one step away from being a binding site can only occur by one type of substitution (represented by two independent T–>A events at position 5), whereas independent loss substitutions can occur by any event anywhere within an existing site (here represented by C–>G at position 3 and A–>T at position 5). Independent, misinferred loss substitutions are therefore much more likely to occur than independent, misinferred gains.
Correctly Counting Gains and Losses
To correct for the bias induced by the motif-based approach, we implement a nucleotide-based model (using the maximum likelihood based program dnaml) to reconstruct ancestral sequence states and directly measure gain and loss events relative to these ancestral sequences (Felsenstein 2013). By implementing an explicit model of nucleotide mutation likelihoods, the maximum likelihood model partially corrects for the undercounting of loss and gain events due to multiple substitutions at the same site. By reconstructing individual nucleotides rather than only scoring for absence of a particular binding site, this approach is able to explicitly define ancestral motifs, forcing every gain of a new motif to come about through the loss of an ancestral sequence. This approach is also able to count different nucleotide mutations causing the loss of the same motif as independent events, thus avoiding the misinference of a single ancestral loss in these cases and normalizing the undercounting of loss events to be the same as that of gain events. With this modified approach, we revisited our simulated data set to reconstruct ancestral nucleotides and observed turnover rates that closely reproduced those found when using the known ancestors (fig. 2C). Using this approach, gains and losses were correctly inferred to occur in equal proportion (with minor discrepancies due to the misinference of ambiguous nucleotides in some of the ancestral sequences), and when this revised approach was applied to our experimental data set, gains and losses were also inferred in equal proportions (fig. 1B).
Inferring the Evolutionary Rate of miRNA Binding Sites
When short sequences corresponding to miRNA binding sites were empirically ranked relative to other sequences one mutational step away—to mirror the underlying evolutionary pressures operating on nucleotide composition as closely as possible (see Materials and Methods)—we observed that miRNA binding sites corresponding to a large number of well-conserved miRNAs had both lower loss rates and lower gain rates than any other members of their cohorts of nearly identical sequences. When searching for this pattern across all short sequences, we found that both 7mers and 8mers corresponding to miRNAs have a significantly greater proportion of slow ranking gain and loss rates than that of the overall pool of short sequences. This result was most pronounced in the case of 8mers, in which, out of the set of 93 well-conserved miRNAs, seven had both gain rates and loss rates slower than all sequences one mutational step away, whereas only 60 out of all 65,536 8mers met this criteria in the full pool (empirical binomial P value 2.46 × 10−12) as shown in table 2. When presented graphically, it is immediately apparent that 8mers corresponding to conserved miRNAs undergo gain and loss events much less frequently than the overall pool of 8mers (fig. 4). We also examined various other metrics of binding site turnover rates (gain and loss rates slower than the median, gain rates slower and loss rates faster than the median, gain rates faster and loss rates slower than the median, gain and loss rates both faster than the median, and gain and loss rates faster than all sequences one mutational step away) and found that binding sites of conserved miRNAs were much more likely than other sequences to be slow evolving and less likely to be fast evolving. Consistent with experimental evidence that 8mers are the biologically relevant determinants of miRNA-mediated gene regulation (Brennecke et al. 2005) and that the ninth nucleotide does not contribute to seed binding, 9mer results were markedly less significant than those of 7mers and 8mers, with 8mers showing the most significant results.
Table 2.
Enrichment Levels of miRNA Binding Sites Having a Given Turnover Rank.
| 7mera | 8mera | 9mera | |
|---|---|---|---|
| Total conserved miRNAs | 94 | 93 | 92 |
| Total short sequences | 16,384 | 65,536 | 262,144 |
| Gain rank slowest and loss rank slowest | 4/94 vs. 24/16,384 (P = 1.26E-005*) | 7/93 vs. 60/65,536 P = 4.768E-012*) | 0/92 vs. 388/262,144 (P = 1) |
| Gain rank < median rank and loss rank < median rank | 60/94 vs. 4,854/16,384 (P = 7.47E-012*) | 54/93 vs. 15,736/65,536 (P = 2.459E-012*) | 45/92 vs. 63,850/262,144 (P = 2.98E-007*) |
| Gain rank < median rank and loss rank > median rank | 3/94 vs. 3,291/16,384 P = 0.9999997889) | 5/93 vs. 13,315/65,536 (P = 0.9999900675) | 21/92 vs. 64,558/262,144 (P = 0.6937253547) |
| Gain rank > median rank and loss rank < median rank | 15/94 vs. 3,427/16,384 (P = 0.9083059651) | 14/93 vs. 13,396/65,536 (P = 0.9261940449) | 18/92 vs. 64,332/262,144 (P = 0.893040712) |
| Gain rank > median rank and loss rank > median rank | 12/94 vs. 4,812/16,384 (P = 0.9999594283) | 5/93 vs. 16,118/65,536 (P = 0.9999998491) | 8/92 vs. 69,404/262,144 (P = 0.9999954775) |
| Gain rank fastest and loss rank fastest | 0/94 vs. 16/16,384 (P = 1) | 0/93 vs. 35/65,536 P = 1) | 0/92 vs. 85/262,144 (P = 1) |
aFor every seed length, conserved miRNAs were defined as those whose seed exists as a miRNA in human, mouse, and zebrafish. This resulted in 94, 93, and 92 “real” miRNAs for 7mer, 8mer, and 9mer seeds, respectively. For each seed length, every seed's turnover rates were ranked relative to sequences one mutational step away, with a rank of 1 corresponding to the slowest turnover rates and the highest rank corresponding to the fastest turnover rates.
*Significant P values. P values were calculated using binomial distributions on the empirical data. Low probabilities near 0 indicate that having the same or more events occur by chance in a random draw from the data set is extremely unlikely, whereas high probabilities near 1 indicate that observing this number of events or more in a random draw from the data set is virtually guaranteed.
Fig. 4.
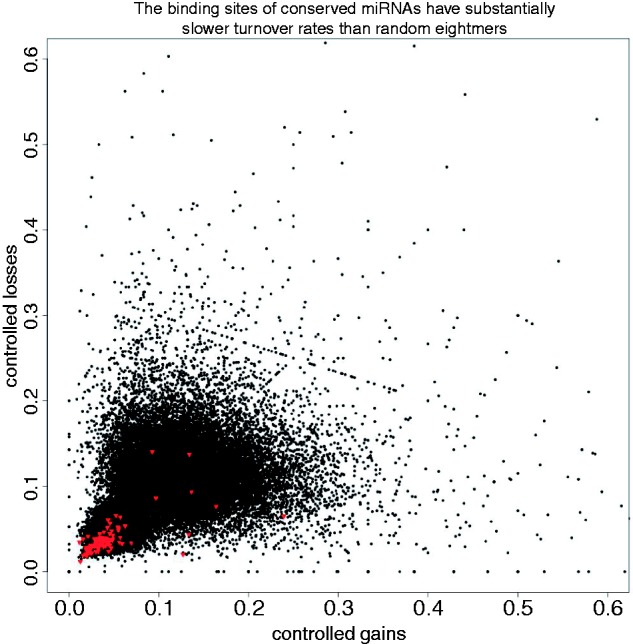
The binding sites of conserved miRNAs have substantially slower turnover rates than random 8mers. After normalizing gain and loss rates by total number of sites, it is apparent that the binding sites of conserved miRNAs (in red) have slower turnover rates than those of other 8mer sequences in 3′-UTRs.
Biological Implications
Having found a strong signal of slower overall turnover rates for the binding sites of strongly conserved miRNAs relative to our data set as a whole, it is notable that although crystal structures only show evidence for interactions between nucleotides 2–8 of mature miRNAs and target mRNAs (Faehnle et al. 2013), binding sites defined as the reverse complement of nucleotides 1–8 had a stronger signal than any other seed examined. These results cannot be explained by any neutral byproduct of the eight nucleotide at position 1, as adding a nucleotide at position 9 abolishes significance completely.
Previous work has found increased signatures of conservation for mRNAs containing an “A” opposite the first nucleotide of the mature miRNAs (Brennecke et al. 2005; Lewis et al. 2005), but as there is a strong bias toward “U” at this position in the mature miRNA, it is unclear whether this signature of conservation is due to base pairing or some other factor. To understand the nature of this interaction, we examined the subset of conserved miRNA 8mer seeds whose first nucleotide is not a “U.” We found that out of 32 such miRNAs, 21 of the binding sites for these miRNAs had reduced turnover rates when modified to contain an “A” opposite the first nucleotide relative to the unmodified binding sites. Although individual reductions in turnover rate were only modest and nonsignificant, the binomial probability of 21 reduced turnover rates in 32 trials occurring by chance is 6.16 × 10−7 (supplementary table S1, supplementary file S1, Supplementary Material online). These results lend support to the notion that an “A” opposite the first nucleotide has a general stabilizing effect on the interaction between miRNAs and mRNA targets through mechanisms other than base pairing.
Our data set contains well-conserved miRNAs whose binding sites are turning over faster than most sequences one mutational step away, as well as some whose binding sites exhibit unbalanced gain and loss rates. Strong skews in the turnover rates of particular well-conserved miRNAs may represent potential candidates for miRNAs with newly evolved functions. Some of the 60 8mers in the overall data set with slower gain and loss ranks than all other sequences one mutational step away may likewise correspond to the motifs of undiscovered 3′-UTR regulators with strongly constrained function.
Contrary to theoretical arguments advocating the neutral expansion of complex regulatory networks, and case studies of newly evolved regulatory circuits that may underlie adaptive changes in miRNA targeting, our empirical results on a large number of well-conserved miRNAs show that in general, the regulatory networks of well-established miRNAs are neither expanding nor contracting within primates, and there is no significant enrichment for miRNA binding sites with rapid turnover. Instead, we find evidence of strong purifying selection against both gains of new sites and loss of existing ones. Taken as a class, the binding sites of well-conserved miRNAs change extremely slowly, suggesting that the evolutionary niche played by miRNAs in primates is not largely driven by positive selection, but by the maintenance of conserved essential biological functions, many of which may be as yet undiscovered. These conserved functions may serve to canalize or modify gene expression levels. Although our study cannot directly address the evolutionary dynamics that play out in other taxa, the accumulation of more experimentally annotated 3′-UTRs across closely related species groups should make this possible in the near future.
Conclusions
Our results suggest that the eighth nucleotide at position 1 of miRNA seeds may impart specificity to miRNA targeting, that the binding sites of well-conserved miRNAs are governed by strong purifying selection in primates, and that the functions of well-conserved miRNAs are therefore also likely to be strongly conserved.
Methodologically, our study makes several improvements over previous approaches to the quantification of motif turnover rate dynamics. By examining every nucleotide for maximum likelihood reconstruction rather than applying a binary gain/loss condition, we effectively correct for inherently biased gain/loss ratios of motif-based analyses that have been previously interpreted in the literature as indicative of a general accumulation of regulatory complexity. By correcting turnover rates for the number of occurrences of each short sequence, and ranking each sequence relative to its nearest mutational neighbors, our method corrects not only for differences in mononucleotide and dinucleotide composition, but higher order effects out to the full length of the sequence under investigation.
Although we here report overall turnover rates of individual well-conserved miRNAs, our analysis may productively be reexamined to derive the turnover rates of particular species or categories of target genes or expanded to include the turnover rates of poorly conserved miRNAs. With slight modification, our methods may be extended to transcription factor binding site turnover rates and those of other motifs. We caution against the application of these methodologies to sequences predicted by synteny to widely diverged species, as alignable material decreases rapidly with phylogenetic distance even within primates.
Materials and Methods
Data Sets
Our experimental data set was curated from primate genomes using MAF alignments to the gorilla genome (to take advantage of the most current primate genomes) curated on the UCSC genome browser (Kent et al. 2002). We compared these alignments with annotated human genes and used the alignment program LASTZ (Harris 2007) to filter out human proteins with low coverage or duplicated sequence (see supplementary methods in supplementary file S2, Supplementary Material online). The 3′-UTRs of the resulting set of genes were aligned with ambiguous nucleotides inserted to separate discontinuous regions in the MAF alignment, and regions with gaps or ambiguous nucleotides in some species were masked to ambiguous nucleotides in all species.
A second primate data set was simulated under a neutral model of evolution to evaluate whether underlying biases exist in different methods of counting miRNA binding site turnover events. This data set was created with the program SFS_CODE (Hernandez 2008, see supplementary methods in supplementary file S2 and supplementary table S2 in supplementary file S1, Supplementary Material online) and consisted of 30,000 nucleotides of simulated sequence, with speciation events added using primate divergence times estimated by TimeTree (Hedges et al. 2006) to approximate the experimental primate data.
Defining the miRNA “Seeds” and Control Motifs
We analyzed the turnover rates of all seven base pair, eight base pair, and nine base pair sequences within 3′-UTRs (which we have termed 7mers, 8mers, and 9mers, respectively, or k-mers as a general term for these sequences of length k). In the case of 7mers, we defined a subset of putatively functional miRNA “seed” binding sites as those 7mers corresponding to the reverse complement of nucleotides 2–8 in the mature miRNA. The reverse complement of nucleotides 1–8 was defined as putatively functional for 8mers, and the reverse complement of nucleotides 1–9 was used for 9mers. In all cases, well-conserved mature miRNA seeds were defined as those annotated in miRbase 18 as a miRNA seed in human, mouse, and zebrafish (Kozomara and Griffiths-Jones 2014).
Motif-Based Parsimony Approach.
We implemented a parsimony approach modeled on previous motif turnover studies (Nozawa et al. 2010, 2012; Meunier et al. 2013; Xiao et al. 2013) in which each location in an aligned 3′-UTR having at least one instance of a given short “seed” sequence—excluding those regions of 3′-UTR with gaps in any of the species—was examined to determine which species contained the full-length seed sequence at that location (coded as a “1”) and which did not (“0”). Subsequently, we fit a most parsimonious interpretation of the ancestral gain and loss substitutions needed to fit the observed presence/absence values to the known species. Locations in the 3′-UTR in which multiple types of substitutions led to the same overall level of parsimony were not analyzed. When examining all 3′-UTRs in aggregate for a given 7mer, 8mer, or 9mer, each branch in the phylogeny had a cumulative number of inferred gain and loss events. By adding together the gains and losses across all branches, we were able to infer a total number of gain and loss events for a given k-mer across the phylogeny. In this way, the overall turnover rates of miRNA binding sites could be compared with the turnover rates of other 7mers, 8mers, and 9mers.
Nucleotide-Based Maximum likelihood Approach.
As a separate approach, we inferred the ancestral states of each nucleotide using the maximum likelihood implementation of dnaml from the phylip phylogeny package (Felsenstein 2013). As before, seed sequences corresponding to miRNA binding sites were analyzed, as well as other sequences of identical length, for 7mers, 8mers, and 9mers. Gain and loss events are assigned directly by comparing the aligned inferred ancestral sequences to the descendants.
Because frequently occurring motifs in 3′-UTRs have a higher probability of observing turnover events than rare ones (see supplementary fig., supplementary file S3, Supplementary Material online), we calculated a normalized turnover rate by dividing the number of gain or loss events of each sequence by the number of total sites observed for that sequence. To control for the effects of nucleotide composition on turnover rate, we compared the normalized turnover rates of every short sequence to those of a cohort of short sequences within one base substitution of the sequence of interest, and assigned each short sequence a gain rank, loss rank, and total number of occurrences rank relative to this cohort. All scripts used to generate the data can be found online at https://github.com/alfredsimkin/AMTA (last accessed April 19, 2014) (Ancestral Motif Turnover Analysis).
Supplementary Material
Supplementary files S1–S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Matt Rasmussen, Andrew Grimson, Jaaved Mohammed, Chip Aquadro, Xiaoping Zhu, and members of both the University of Massachusetts Medical School (UMMS) and École Polytechnique Fédérale de Lausanne (EPFL) communities for the suggestion of the maximum likelihood model of individual nucleotide reconstruction and other useful feedback. This work was supported by grants from the Swiss National Science Foundation, a European Research Council (ERC) Starting Grant to J.D.J., and the National Institutes of Health grant R01NS066586 to F.-B.G.
References
- Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005;3:e85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy JJ, Jha AR, Posadas DM, Giri R, Venken KJ, Ji J, Jiang H, Bellen HJ, White KP, Carthew RW. miR-9a minimizes the phenotypic impact of genomic diversity by buffering a transcription factor. Cell. 2013;155:1556–1567. doi: 10.1016/j.cell.2013.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Rajewsky N. Deep conservation of microRNA-target relationships and 3’UTR motifs in vertebrates, flies, and nematodes. Cold Spring Harb Symp Quant Biol. 2006a;71:149–56. doi: 10.1101/sqb.2006.71.039. [DOI] [PubMed] [Google Scholar]
- Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet. 2006b;38:1452–1456. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8:93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- Faehnle CR, Elkayam E, Haase AD, Hannon GJ, Joshua-Tor L. The making of a slicer: activation of human argonaute-1. Cell Rep. 2013;3:1901–1909. doi: 10.1016/j.celrep.2013.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP [updated 2013 May 30; cited 2014 April 19] 2013 Available from: http://evolution.genetics.washington.edu/phylip.html. [Google Scholar]
- Gao FB. Context-dependent functions of specific microRNAs in neuronal development. Neural Dev. 2010;5:25. doi: 10.1186/1749-8104-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Hui JHL, Marco A, Ronshaugen M. MicroRNA evolution by arm switching. EMBO Rep. 2011;12:172–7. doi: 10.1038/embor.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS. Improved pairwise alignment of genomic DNA [PhD thesis] University Park Pennsylvania: The Pennsylvania State University; 2007. [Google Scholar]
- Hedges S, Blair JD, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22:2971–2972. doi: 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- Hernandez RD. A flexible forward simulator for populations subject to selection and demography. Bioinformatics. 2008;24:2786–2787. doi: 10.1093/bioinformatics/btn522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiard S, Charlier C, Coppieters W, Georges M, Baurain D. Patrocles: a database of polymorphic miRNA-mediated gene regulation in vertebrates. Nucleic Acids Res. 2010;38:D640–D651. doi: 10.1093/nar/gkp926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler AD. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D69. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucherenko MM, Barth J, Fiala A, Shcherbata HR. Steroid-induced microRNA let-7 acts as a spatio-temporal code for neuronal cell fate in the developing Drosophila brain. EMBO J. 2012;31:4511–4523. doi: 10.1038/emboj.2012.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Torre A, Georgi S, Reh TA. Conserved microRNA pathway regulates developmental timing of retinal neurogenesis. Proc Natl Acad Sci U S A. 2013;110:E2362–E2370. doi: 10.1073/pnas.1301837110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Lewis BP, I-hung S, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW. A microRNA imparts robustness against environmental fluctuation during development. Cell. 2009;137:273–282. doi: 10.1016/j.cell.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang F, Lee JA, Gao FB. MicroRNA-9a ensures the precise specification of sensory organ precursors in Drosophila. Genes Dev. 2006;20:2793–2805. doi: 10.1101/gad.1466306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh YE, Yi SV, Streelman JT. Evolution of microRNAs and the diversification of species. Genome Biol Evol. 2010;3:55–65. doi: 10.1093/gbe/evq085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. The evolution of genetic networks by non-adaptive processes. Nat Rev Genet. 2007;8:803–813. doi: 10.1038/nrg2192. [DOI] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, Bartel DP, Ambros VR, Horvitz HR. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS Genet. 2007;3:e215. doi: 10.1371/journal.pgen.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss EG, Tang L. Conservation of the heterochronic regulator Lin-28, its developmental expression and microRNA complementary sites. Dev Biol. 2003;258:432–442. doi: 10.1016/s0012-1606(03)00126-x. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Müller P, et al. Conservation of the sequence and temporal expression of Let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- Meunier J, Lemoine F, Soumillon M, Liechti A, Weier M, Guschanski K, Hu H, Khaitovich P, Kaessmann H. Birth and expression evolution of mammalian microRNA genes. Genome Res. 2013;23:34–45. doi: 10.1101/gr.140269.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa M, Miura S, Nei M. Origins and evolution of microRNA genes in Drosophila species. Genome Biol Evol. 2010;2:180–189. doi: 10.1093/gbe/evq009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa M, Miura S, Nei M. Origins and evolution of microRNA genes in plant species. Genome Biol Evol. 2012;4:230–239. doi: 10.1093/gbe/evs002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CI, Yang S, Tang T. Evolution under canalization and the dual roles of microRNAs: a hypothesis. Genome Res. 2009;19:734–743. doi: 10.1101/gr.084640.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Xia W, Yang Y, Mason AS, Lei X, Ma Z. Characterization and evolution of conserved microRNA through duplication events in date palm (Phoenix dactylifera) PLoS One. 2013;8:e71435. doi: 10.1371/journal.pone.0071435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3’ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhang R, Shen Y, Liu G, Lu X, Wu C-I. The evolution of evolvability in microRNA target sites in vertebrates. Genome Res. 2013;23:1810–1816. doi: 10.1101/gr.148916.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.