

Abstract
Many human-disease associated amino acid residues (DARs) appear as the wild-type in other species. This phenomenon is commonly explained by the presence of compensatory residues in these other species that alleviate the deleterious effects of the DARs. The general validity of this hypothesis, however, is unclear, because few compensatory residues have been identified. Here we test the compensation hypothesis by assembling and analyzing 1,077 DARs located in 177 proteins of known crystal structures. Because destabilizing protein structures is a primary reason why DARs are deleterious, we focus on protein stability in this analysis. We discover that, in species where a DAR represents the wild-type, the destabilizing effect of the DAR is generally lessened by the observed amino acid substitutions in the spatial proximity of the DAR. This and other findings provide genome-scale evidence for the compensation hypothesis and have important implications for understanding epistasis in protein evolution and for using animal models of human diseases.
Keywords: disease mutation, epistasis, evolution, intramolecular interaction, protein stability
Introduction
It was first reported in 2002 that a number of human disease-associated amino acid residues (DARs) appear as the wild-type in the laboratory mouse and various other species (Kondrashov et al. 2002; Waterston et al. 2002). For example, mutation from Gly to Ser at amino acid position 471 of human androgen receptor causes the complete androgen insensitivity syndrome, characterized by feminization of genetic males, but Ser is the wild-type residue (WTR) in both mouse and rat (Gao and Zhang 2003). Uncovering the cause of this interesting phenomenon can help understand both the molecular basis of human disease and the mechanisms of protein evolution. We previously reported that these special DARs are not enriched in associations with late-onset or mild diseases and that their wild-type status in nonhuman species is not attributable to founder effects as one might hypothesize in the case of the laboratory mouse (Gao and Zhang 2003). Instead, it was proposed from the very beginning (Kondrashov et al. 2002) and is now widely believed (Gao and Zhang 2003; Kulathinal et al. 2004; Ferrer-Costa et al. 2007; Baresic et al. 2010) that human DARs can become WTRs in other species because of the presence in these species of compensatory residues that alleviate the deleterious effects of the DARs. Nevertheless, because potential compensatory residues have been identified in only a few cases (Kondrashov et al. 2002), the general validity of the compensation hypothesis remains unclear. For two reasons, protein structural analysis may provide significant insights. First, a primary mechanism by which DARs cause diseases is reducing protein structural stability (Yue et al. 2005). Second, compensatory residues of a DAR likely reside in the same protein as the DAR and interact with the DAR (Poon et al. 2005; Davis et al. 2009; Baresic et al. 2010), and thus may be detected through structural analysis. Here, we assemble a large set of structurally mapped DARs that appear as the wild-type in at least one nonhuman species and test whether the potential compensatory residues in the spatial neighborhood of the DARs mitigate the destabilizing effects of the DARs in the nonhuman species.
Results
Protein Stability Reduction Caused by DARs
We began with 51,920 DARs from the Human Gene Mutation Database (Stenson et al. 2003) and Universal Protein Resource (UniProt) (UniProt Consortium 2011). Among them, 9,212 DARs were mapped to 579 unique human protein structures from the Protein Data Bank (PDB) (Berman 2008). Of these structurally mapped DARs, 1,077 appear as the wild-type in the one-to-one orthologous proteins of at least one nonhuman species (Altenhoff et al. 2011) and thus are called wt-DARs. Although wt-DARs are often referred to as compensated pathogenic deviations (Kondrashov et al. 2002) in the literature, we avoid the use of this term because it equates a phenomenon (DAR observed as the wild-type in other species) with one of its potential causes (compensation). The remaining 8,135 DARs are referred to as regular DARs, or rg-DARs. We used Rosetta (Kellogg et al. 2011) to predict the change in human protein stability upon mutation from the WTR to the corresponding DAR (ΔΔG = ΔGDAR − ΔGWTR). The more positive ΔΔG is, the bigger the stability reduction is. Thus, ΔΔG is referred to as the stability reduction upon mutation. The median ΔΔG for mutations to wt-DARs is 1.44 Rosetta energy unit (REU), which is equivalent to ∼0.79 kcal/mol according to a linear conversion model (supplementary fig. S1, Supplementary Material online). This amount is significantly smaller than the median ΔΔG (4.09 REU or ∼2.25 kcal/mol) for mutations to rg-DARs (P < 10−41, Mann–Whitney U test; fig. 1), consistent with an earlier observation that mutations to wt-DARs have on average weaker impacts on structural stabilities than mutations to rg-DARs (Ferrer-Costa et al. 2007).
Fig. 1.
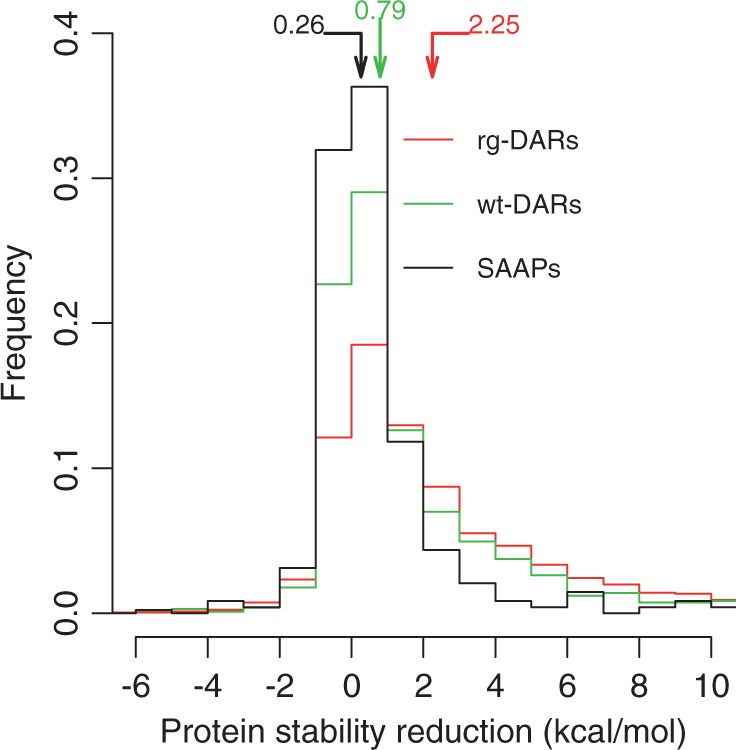
Frequency distributions of human protein stability reduction (ΔΔG) caused by mutations to human single amino acid polymorphisms (SAAPs) with minor allele frequencies (MAFs) > 0.01 (black), disease-associated residues that appear as the wild-type in at least one nonhuman species (wt-DARs) (green), and other disease-associated residues (rg-DARs) (red). The samples include 482 SAAPs, 1,077 wt-DARs, and 8,124 of the 8,135 rg-DARs (11 rg-DARs are not included because Rosetta failed to complete the computations in 72 h). Protein stability reduction is expressed in kcal/mol estimated from REU by linear regression (supplementary fig. S1, Supplementary Material online). Arrows indicate median values of the distributions. The three distributions are all significantly different from one another (P < 10−14, Mann–Whitney U test).
That wt-DARs impose milder destabilizing effects than rg-DARs has two reasons. First, wt-DARs are more similar to WTRs than are rg-DARs in physicochemical properties (Ferrer-Costa et al. 2007). Second, the structural positions of wt-DARs and rg-DARs may be different such that the same type of mutation has different destabilizing effects when leading to wt-DARs versus rg-DARs. To explore this possibility, we analyzed, among all 380 possible types of amino acid changes, the 128 types that are observed in mutations to both wt-DARs and rg-DARs in our data set (supplementary table S1, Supplementary Material online). Among these 128 types, 13 showed a significantly smaller median ΔΔG for mutations to wt-DARs than mutations to rg-DARs (P < 0.05, Mann–Whitney U test; supplementary table S1, Supplementary Material online), whereas none showed the opposite pattern. Thus, for some mutation types, wt-DARs are located at positions with milder stability impacts than rg-DARs. Furthermore, there is a negative correlation between sample size and log(P value) in the above Mann–Whitney U test (supplementary fig. S2, Supplementary Material online), suggesting that more mutation types would show the same significant trend as the 13 mutation types should the samples be larger. Thus, there is indeed evidence that on average wt-DARs are located at positions that have milder stability impacts than are rg-DARs.
The observation that wt-DARs are less destabilizing than rg-DARs suggests that the mechanism mitigating the deleterious effects of DARs in nonhuman species has a limited power. As a comparison, we also computed the average ΔΔG for mutations to known common single amino acid polymorphisms (SAAPs) in humans (i.e., with allele frequencies >0.01) (Sherry et al. 2001), which should be mostly neutral. As expected, this ΔΔG (median = 0.47 REU or ∼0.26 kcal/mol) is significantly lower than that for wt-DARs (P < 10−14; fig. 1).
Testing the Compensation Hypothesis
Intramolecular compensatory residues may appear anywhere in a protein to mitigate protein stability reduction caused by a wt-DAR, because protein stability is contributed by all residues. However, spatially neighboring residues of the wt-DAR can have strong stabilizing effects via noncovalent bonds. Furthermore, it is currently infeasible to examine the potential compensatory effects of a large number of residues simultaneously, whereas examining these residues one by one requires the information of the order with which these residues emerged in evolution, which is difficult to obtain. Thus, in this study, we focused on only the spatial neighborhood of a wt-DAR when examining potential compensatory residues. For reasons detailed in Materials and Methods, we considered all residues that are within 4 Å from a focal residue to be its neighboring residues, where the distance between two residues is measured by the shortest spatial distance of their nonhydrogen atoms. We found that, in 94.6% of the cases when a DAR is the wild-type in a species, the neighboring residues are not identical between that species and human; these cases were subject to further analysis.
Let us use the example of plasminogen to illustrate our analysis (fig. 2). Plasminogen is the precursor of plasmin, which dissolves the fibrin of blood clots. Normal humans have Arg at amino acid position 532 of plasminogen, but mutation to His at this position causes plasminogen deficiency (Online Mendelian Inheritance in Man or OMIM: 217090), characterized by decreased serum plasminogen activity. Interestingly, His is the wild-type in the giant panda. Four neighboring residues of this DAR differ between wild-type human and giant panda and are candidate compensatory residues. We computed the stability reduction caused by the mutation from Arg to His in the human structure (ΔΔG1; fig. 2A). We also computed the corresponding stability reduction caused by the same mutation in the “pandanized” human structure where all neighboring residues are of the panda version (ΔΔG2; fig. 2B). Consistent with the compensation hypothesis, ΔΔG2 (−4.43 REU or ∼−2.43 kcal/mol) is substantially smaller than ΔΔG1 (1.19 REU or ∼0.65 kcal/mol), suggesting that one or more of the four neighboring residues in panda that differ from human are compensatory. The negative ΔΔG2 suggests that the replacement of Arg with His increases the panda plasminogen stability and thus may have been beneficial. As a negative control, we considered horse, in which Arg is the wild-type. We computed the stability reduction caused by the mutation from Arg to His in the “horsenized” human structure where all neighboring residues are of the horse version (ΔΔG3; fig. 2C). As expected, ΔΔG3 (2.99 REU or ∼1.65 kcal/mol) is not smaller than ΔΔG1, indicating that the smaller ΔΔG2, compared with ΔΔG1, is not due to random substitutions. We caution, however, that ΔΔG prediction is notoriously difficult and that Rosetta and other top ranked prediction programs have only moderate accuracies (Khan and Vihinen 2010; Thiltgen and Goldstein 2012). Consequently, ΔΔG comparison for any individual case may not be reliable; only comparisons based on large samples are trustable.
Fig. 2.
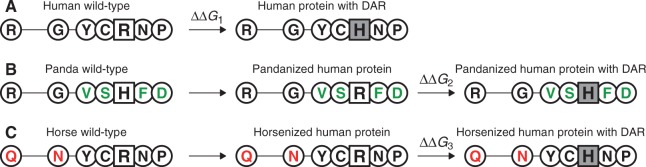
Testing the compensation hypothesis for the disease-associated residue (DAR) at position 532 of human plasminogen (UniProt accession number: P00747). The DAR site and its orthologous site in nonhuman species are squared, and the DAR is shaded. Spatial neighbors of the DAR site, shown as circles, are identified using the human plasminogen model (2KNF in PDB as the template). (A) Wild-type sequence in human (P00747) and the stability reduction (ΔΔG1) of the human plasminogen caused by mutation from the wild-type (R) to the DAR (H). (B) Panda wild-type plasminogen (G1MBX3), “pandanized” human plasminogen, and the stability reduction (ΔΔG2) of the pandanized human plasminogen caused by mutation from the human wild-type (R) to the DAR (H). The neighboring residues in panda that differ from those in human are shown in green. (C) Horse wild-type plasminogen (F6USP9), “horsenized” human plasminogen, and the stability reduction (ΔΔG3) of the horsenized human plasminogen caused by mutation from the human wild-type (R) to the DAR (H). The neighboring residues in horse that differ from those in human are shown in red. Sequence alignment is provided in supplementary figure S6, Supplementary Material online.
We conducted the same analyses for a large set of wt-DARs. For each wt-DAR, we averaged ΔΔG2 from multiple species if the DAR is found to be the wild-type in multiple species. We then compared the average ΔΔG2 with the corresponding ΔΔG1. Overall, ΔΔG2 (median = 1.23 REU or ∼0.68 kcal/mol) is significantly smaller than ΔΔG1 (median = 1.59 REU or ∼0.87 kcal/mol) (P < 10−7, Wilcoxon signed-rank test; fig. 3). For each wt-DAR, ΔΔG1 − ΔΔG2 measures the stabilizing effect of the neighboring residues from the species where the DAR is the wild-type. A positive value of (ΔΔG1 − ΔΔG2) indicates that those neighboring residues are compensatory. In spite of the statistically significant difference between ΔΔG1 and ΔΔG2, the median of (ΔΔG1 − ΔΔG2) is rather small (0.17 REU or 0.09 kcal/mol). We found that in fact 52.7% of the wt-DARs have ΔΔG1 < 1 kcal/mol, which are not conventionally considered to be destabilizing (Tokuriki and Tawfik 2009). For those wt-DARs considered to be destabilizing (ΔΔG1 > 1 kcal/mol), the median of (ΔΔG1 − ΔΔG2) is 1.03 REU or ∼0.56 kcal/mol (P < 10−10, fig. 3). Because some proteins harbor many more wt-DARs than do other proteins, we also respectively averaged ΔΔG1 and ΔΔG2 values from different wt-DARs in the same protein before comparison, but the results were similar (P < 0.003; P < 0.007 for destabilizing wt-DARs; supplementary fig. S3, Supplementary Material online).
Fig. 3.
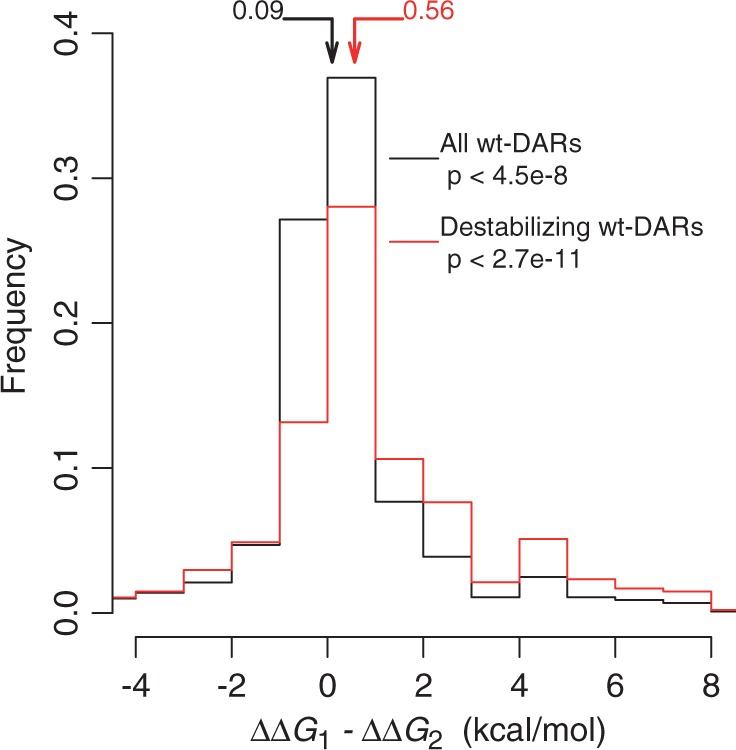
Frequency distribution of the difference in protein stability reduction upon mutation from a human WTR to a DAR in the absence (ΔΔG1) and presence (ΔΔG2) of neighboring residues from a species where the DAR is the wild-type. The larger the difference, the greater the compensation effect. Destabilizing wt-DARs have ΔΔG1 > 1 kcal/mol. Arrows indicate median values of the corresponding distributions. For both distributions, ΔΔG1 − ΔΔG2 is significantly biased toward positive values, as indicated by the P values from the Wilcoxon signed-rank test.
To compare ΔΔG3 and ΔΔG2, we focused on destabilizing wt-DARs. For each wt-DAR, we need a pair of species whose WTRs are the same as the human DAR and the human WTR, respectively. We chose those species pairs that have the same numbers of neighboring residue differences from the human protein. This requirement reduced our sample size substantially but allowed a fair comparison between ΔΔG3 and ΔΔG2. We found that ΔΔG2 remains significantly smaller than ΔΔG1 (P = 0.02; fig. 4), whereas ΔΔG3 is not significantly different from ΔΔG1 (P > 0.5; fig. 4). Furthermore, ΔΔG2 is significantly smaller than ΔΔG3 (P < 0.01; fig. 4). Thus, as predicted by the compensation hypothesis, the compensatory effects are bestowed by the neighboring residues in species where the human DARs are the wild-type, but not by the neighboring residues in species where the human WTRs are the wild-type.
Fig. 4.
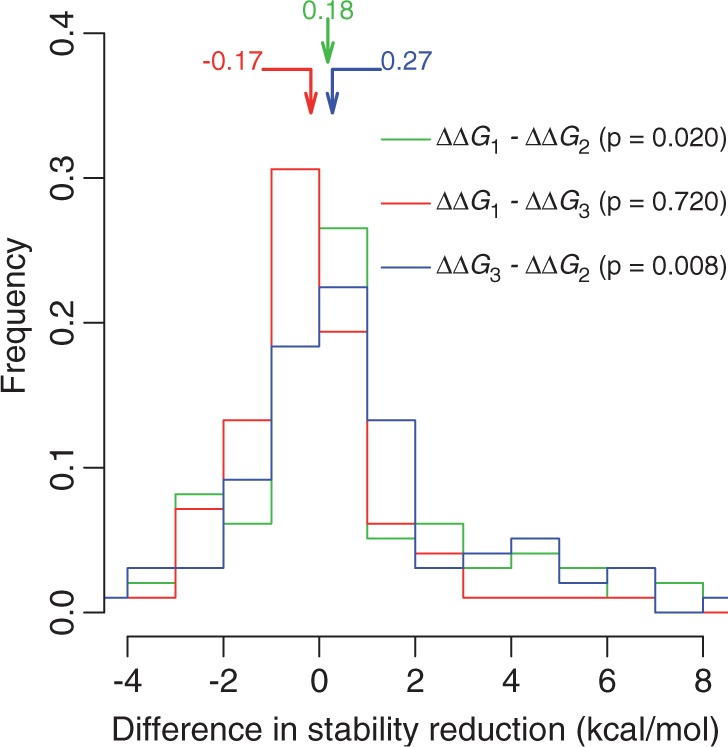
Frequency distribution of the difference in protein stability reduction upon mutation from a human WTR to a DAR among various genetic backgrounds. ΔΔG1, in the human background (see fig. 2A); ΔΔG2, in the presence of neighboring residues from a species where the DAR is the wild-type (see fig. 2B); ΔΔG3, in the presence of neighboring residues from a nonhuman species where the human WTR is the wild-type (see fig. 2C). The P values are from one-tail Wilcoxon signed-rank test. A total of 314 pairs of WTRs and destabilizing DARs are examined.
Compensatory Effects Extend to Amino Acids Similar to DARs
If the above detected compensatory effects of neighboring residues are due to physical interactions between the neighboring residues and the DARs, the compensatory effects may also exert on amino acids that are physicochemically similar to the DARs. Because the greater the physicochemical similarity between two amino acids, the higher the substitution rate between them in evolution (Miyata et al. 1979; Zhang 2000), we used the PAM250 substitution matrix (Dayhoff et al. 1978) to gauge physicochemical similarities between amino acids. For each DAR, we identified the non-WTR amino acid(s) that the DAR will most likely be replaced with in evolution according to PAM250 and referred to it as DAR-like (DARL). There may be more than one DARL if several amino acids are equally likely to replace the DAR. Similarly, for each WTR, we identified the non-DAR amino acid(s) that the WTR will most likely be replaced with in evolution (WTRL). If the WTRL set and DARL set identified for a WTR and its DAR overlap, we do not consider the case further. We then examined the stability reduction caused by mutation from WTR to DARL in the human protein (ΔΔG1) and the corresponding stability reduction in the presence of the neighboring residues from a species in which the DAR is the wild-type (ΔΔG2). As predicted, the compensatory effects of the neighboring residues also exert on DARLs (P < 10−6; fig. 5A). By contrast, no such effect for WTRLs is detectable (P > 0.2; fig. 5B).
Fig. 5.
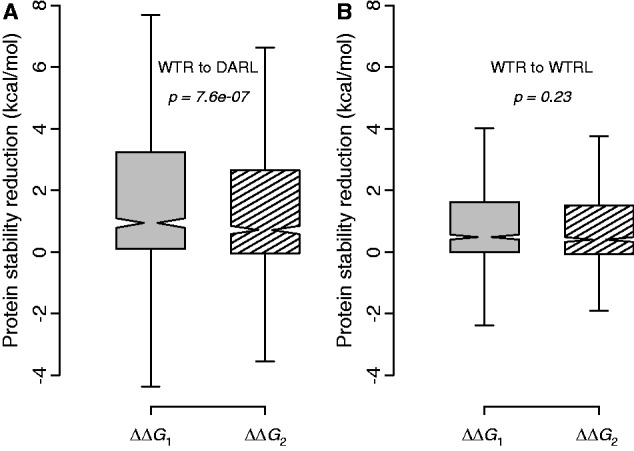
Protein stability reduction upon mutation. (A) Distribution of protein stability reduction upon mutation from a human WTR to a residue that is physicochemically similar to a DAR in the absence (ΔΔG1, gray bar) and presence (ΔΔG2, striped bar) of neighboring residues from a species where the DAR is the wild-type. (B) Distribution of protein stability reduction upon mutation from a human WTR to a residue that is physicochemically similar to the WTR (WTRL) in the absence (ΔΔG1, gray bar) and presence (ΔΔG2, striped bar) of neighboring residues from a species where the DAR is the wild-type. The P values are from Wilcoxon signed-rank test. A total of 590 pairs of WTRs and DARs are examined in each panel.
Discussion
Taken together, our results provide genome-scale evidence that, in species where DARs appear as the wild-type, residues at the spatial proximities of the DARs mitigate their deleterious effects in destabilizing the protein structures. Because reducing protein stability is a primary mechanism by which DARs cause diseases, our findings support the hypothesis that compensatory residues render the otherwise unacceptable DARs acceptable in evolution.
A few biologically or medically important protein families have been intensively crystallized, whereas most other protein families have few members with solved structures. To examine whether our results have been influenced by this imbalanced data, we focused on a subset of protein structures with pairwise sequence identity <60%. We found that our results in figure 3 can be repeated by this subset of data (supplementary fig. S4, Supplementary Material online), suggesting that the compensation hypothesis is supported robustly by many protein families rather than a few. It is worth pointing out that Rosetta predictions of ΔΔG are not always accurate (Kellogg et al. 2011), which limits the statistical power of our analysis, but also means that our conclusions are likely to be conservative.
Despite the detection of statistically significant compensatory effects, the median difference between ΔΔG1 and ΔΔG2 is quite small even for destabilizing wt-DARs (0.56 kcal/mol), indicating that the overall compensatory effect detected is small. Although the actual compensation may be larger if some compensatory residues are outside the 4 Å neighborhood examined, even the small compensatory effect detected could have appreciable impacts. Because wild-type proteins are only marginally stable (folding energy = −3 to −10 kcal/mol) (Tokuriki and Tawfik 2009) and mutations to destabilizing wt-DARs have a median ΔΔG of 3.54 kcal/mol, proteins with wt-DARs could become marginally unstable (ΔG > 0 kcal/mol). When ΔG ∼ 0, a small change in ΔG could result in a substantial change in the fraction of folded protein molecules. For example, a wild-type protein with ΔG = −3 kcal/mol has >99% of molecules folded under 37 °C (see Materials and Methods). Upon mutation to an average destabilizing wt-DAR (ΔΔG = 3.54 kcal/mol), folded protein molecules drop to 30% (ΔG = 0.54 kcal/mol). With the help of the detected median compensatory effect (ΔΔG = −0.56 kcal/mol), the fraction of folded molecules rises to 51% (ΔG = −0.02 kcal/mol). Because most diseases are recessive, heterozygotes with one wild-type allele and one null allele (i.e., having 50% functional molecules as in the wild-type) are often phenotypically normal. Hence, a homozygote with the median destabilizing wt-DAR and median compensatory effect, producing 51% of folded molecules, likely has a normal phenotype. In other words, the compensation detected, although small in terms of ΔΔG, may be sufficient in restoring the normal phenotype. The substantial reduction of the fraction of unfolded molecules, which are often cytotoxic, may render the compensation even more important.
That a large fraction of wt-DARs are explainable, at the genomic scale, by the presence of spatially neighboring compensatory residues supports the importance of (intramolecular) epistasis in protein evolution (Breen et al. 2012). The compensatory residues of the DARs identified through our evolutionary analysis may help understand the molecular basis of the involved diseases. Nevertheless, rampart epistasis in protein evolution also means that findings from animal models of human diseases need to be interpreted with care (Liao and Zhang 2008). It is noteworthy that in 5.4% of the cases when a DAR is the wild-type in a species, that species has identical neighboring residues as human. In these cases, whether compensatory residues reside outside the neighborhood defined or other mechanisms are at work remains to be explored.
Materials and Methods
Neighboring Residues
For each residue in a protein, we calculated the number of residues whose spatial distance from this focal residue is between 0 and 0.1 Å, between 0.1 and 0.2 Å, and so on. We then computed the residue density, defined as the number of residues per Å3, for each range of radial distance. We averaged the density across all residues of all nonredundant protein structures from the protein structure database CATH (Sillitoe et al. 2013). The density peaks at 1.4 and 3.3 Å (supplementary fig. S5, Supplementary Material online), representing residue pairs in contact via N–O and hydrogen bonds, respectively. The density drops drastically and appears uniformly distributed at spatial distances above 4 Å. Because the density is contributed by residues that are in contact and residues that are not in contact, the uniformly low density suggests that residues with distances beyond 4 Å tend not to be in contact. Further, proteins are primarily stabilized by electrostatic bonds, hydrogen bonds, and van der Waals interactions, which have distances of ∼3.0, 2.6–3.5, and averaging 3.6 Å between two nonhydrogen atoms, respectively. Therefore, we identify potential compensatory residues within the 4 Å radius.
Protein Structures
Human protein structures were downloaded from PDB (Berman 2008), whereas the SIFTS database (Velankar et al. 2013) was used to map the structures with corresponding proteins in UniProt (UniProt Consortium 2011). Based on the alignments of the structures and their corresponding wild-type sequences, we removed the structures that have point mutations or insertions/deletions (indels) totaling >10% of amino acids in the structures. For the remaining structures that contain point mutations or indels totaling ≤10%, we used them as templates to predict structure models of their corresponding wild-type proteins for the aligned regions, by MODELLER (Eswar et al. 2008). Because the templates and queries have sequence identities ≥90%, the predicted structure models are likely to be highly accurate. These models and native structures formed the structure pool for testing the compensation hypothesis.
We mapped DARs onto the protein structures. When one DAR is mapped to multiple structures, we used the structure containing the highest number of DARs, which reduces structure redundancy in the sample and saves computational time. One-to-one orthologs were obtained from the orthologous matrix database (Altenhoff et al. 2011). Only structure–ortholog alignments with deletion sites <10% of the amino acid residues in the structures were used. From these alignments, we found that 1,077 human DARs appear as the wild-type in at least one nonhuman species. In an alignment between a human protein and one of its orthologs where a DAR appears as the wild-type, if none of the neighboring residues of the DAR site in the human protein corresponds to a gap site in the ortholog and at least one neighboring residue differs between the human protein and the ortholog, the corresponding neighboring residues in the ortholog are considered to be potential compensatory residues for the DAR. A total of 1,008 wt-DARs have at least one set of potential compensatory residues.
Human SAAPs were acquired from UniProt. SAAPs were cross-linked to their single nucleotide polymorphisms (SNPs) in dbSNP where the minor allele frequencies (MAFs) in humans were obtained. Only SAAPs with MAFs ≥ 0.01 were used.
Prediction of ΔΔG
Program “ddg_min” in Rosetta with default parameters was used for energy minimizations of protein structures. Then, “ddg_monomer” was used to predict protein stability reductions upon point mutations. Low Resolution Protocol was set for the prediction using default parameters except for the following changes. We repacked the residues with Cα in 7 Å rather than 8 Å to the site of the point mutation. The 7 Å in Cα distance was chosen because we found it corresponds to 4 Å in heavy atom distance from the structures used in the “neighboring residues” section. The iteration parameter was set to 30 instead of 50 to save computational time. FoldX (Guerois et al. 2002) was used to optimize the neighboring residue side chain orientation in a protein structure upon the replacement of neighboring residues.
Relationship between Fraction of Protein Molecules Folded and Protein Stability
Under the assumption of thermodynamic equilibrium, the fraction of protein molecules folded is given by 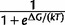 , where ΔG is protein stability, k is Boltzmann constant (1.986 cal/mol/K), and T is absolute temperature (Pakula and Sauer 1989).
, where ΔG is protein stability, k is Boltzmann constant (1.986 cal/mol/K), and T is absolute temperature (Pakula and Sauer 1989).
Supplementary Material
Supplementary figures S1–S6 and table S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/) is valid.
Acknowledgments
The authors thank Wei-Chin Ho, Jian-Rong Yang, and three anonymous reviewers for valuable comments. This work was supported in part by research grant R01GM103232 from the U.S. National Institutes of Health to J.Z. All data used in this study can be obtained at http://www.umich.edu/∼zhanglab/download/Jinrui_MBE_Suppl/index.htm.
References
- Altenhoff AM, Schneider A, Gonnet GH, Dessimoz C. OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 2011;39:D289–D294. doi: 10.1093/nar/gkq1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baresic A, Hopcroft LE, Rogers HH, Hurst JM, Martin AC. Compensated pathogenic deviations: analysis of structural effects. J Mol Biol. 2010;396:19–30. doi: 10.1016/j.jmb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Berman HM. The Protein Data Bank: a historical perspective. Acta Crystallogr A. 2008;64:88–95. doi: 10.1107/S0108767307035623. [DOI] [PubMed] [Google Scholar]
- Breen MS, Kemena C, Vlasov PK, Notredame C, Kondrashov FA. Epistasis as the primary factor in molecular evolution. Nature. 2012;490:535–538. doi: 10.1038/nature11510. [DOI] [PubMed] [Google Scholar]
- Davis BH, Poon AF, Whitlock MC. Compensatory mutations are repeatable and clustered within proteins. Proc Biol Sci. 2009;276:1823–1827. doi: 10.1098/rspb.2008.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayhoff MO, Schwartz R, Orcutt BC. A model of evolutionary change in proteins. In: Dathoff MO, editor. Atlas of protein sequence and structure. Silver Spring (MD): National Biomedical Research Foundation; 1978. pp. 345–352. [Google Scholar]
- Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- Ferrer-Costa C, Orozco M, de la Cruz X. Characterization of compensated mutations in terms of structural and physico-chemical properties. J Mol Biol. 2007;365:249–256. doi: 10.1016/j.jmb.2006.09.053. [DOI] [PubMed] [Google Scholar]
- Gao L, Zhang J. Why are some human disease-associated mutations fixed in mice? Trends Genet. 2003;19:678–681. doi: 10.1016/j.tig.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins. 2011;79:830–838. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Vihinen M. Performance of protein stability predictors. Hum Mutat. 2010;31:675–684. doi: 10.1002/humu.21242. [DOI] [PubMed] [Google Scholar]
- Kondrashov AS, Sunyaev S, Kondrashov FA. Dobzhansky-Muller incompatibilities in protein evolution. Proc Natl Acad Sci U S A. 2002;99:14878–14883. doi: 10.1073/pnas.232565499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulathinal RJ, Bettencourt BR, Hartl DL. Compensated deleterious mutations in insect genomes. Science. 2004;306:1553–1554. doi: 10.1126/science.1100522. [DOI] [PubMed] [Google Scholar]
- Liao BY, Zhang J. Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc Natl Acad Sci U S A. 2008;105:6987–6992. doi: 10.1073/pnas.0800387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Miyazawa S, Yasunaga T. Two types of amino acid substitutions in protein evolution. J Mol Evol. 1979;12:219–236. doi: 10.1007/BF01732340. [DOI] [PubMed] [Google Scholar]
- Pakula AA, Sauer RT. Genetic analysis of protein stability and function. Annu Rev Genet. 1989;23:289–310. doi: 10.1146/annurev.ge.23.120189.001445. [DOI] [PubMed] [Google Scholar]
- Poon A, Davis BH, Chao L. The coupon collector and the suppressor mutation: estimating the number of compensatory mutations by maximum likelihood. Genetics. 2005;170:1323–1332. doi: 10.1534/genetics.104.037259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe I, Cuff AL, Dessailly BH, Dawson NL, Furnham N, Lee D, Lees JG, Lewis TE, Studer RA, Rentzsch R, et al. New functional families (FunFams) in CATH to improve the mapping of conserved functional sites to 3D structures. Nucleic Acids Res. 2013;41:D490–D498. doi: 10.1093/nar/gks1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- Thiltgen G, Goldstein RA. Assessing predictors of changes in protein stability upon mutation using self-consistency. PLoS One. 2012;7:e46084. doi: 10.1371/journal.pone.0046084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N, Tawfik DS. Stability effects of mutations and protein evolvability. Curr Opin Struct Biol. 2009;19:596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2011;39:D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar S, Dana JM, Jacobsen J, van Ginkel G, Gane PJ, Luo J, Oldfield TJ, O’Donovan C, Martin MJ, Kleywegt GJ. SIFTS: Structure Integration with Function, Taxonomy and Sequences resource. Nucleic Acids Res. 2013;41:D483–D489. doi: 10.1093/nar/gks1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol. 2005;353:459–473. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Zhang J. Rates of conservative and radical nonsynonymous nucleotide substitutions in mammalian nuclear genes. J Mol Evol. 2000;50:56–68. doi: 10.1007/s002399910007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.