

Abstract
The integration and fixation preferences of DNA transposons, one of the major classes of eukaryotic transposable elements, have never been evaluated comprehensively on a genome-wide scale. Here, we present a detailed study of the distribution of DNA transposons in the human and bat genomes. We studied three groups of DNA transposons that integrated at different evolutionary times: 1) ancient (>40 My) and currently inactive human elements, 2) younger (<40 My) bat elements, and 3) ex vivo integrations of piggyBat and Sleeping Beauty elements in HeLa cells. Although the distribution of ex vivo elements reflected integration preferences, the distribution of human and (to a lesser extent) bat elements was also affected by selection. We used regression techniques (linear, negative binomial, and logistic regression models with multiple predictors) applied to 20-kb and 1-Mb windows to investigate how the genomic landscape in the vicinity of DNA transposons contributes to their integration and fixation. Our models indicate that genomic landscape explains 16–79% of variability in DNA transposon genome-wide distribution. Importantly, we not only confirmed previously identified predictors (e.g., DNA conformation and recombination hotspots) but also identified several novel predictors (e.g., signatures of double-strand breaks and telomere hexamer). Ex vivo integrations showed a bias toward actively transcribed regions. Older DNA transposons were located in genomic regions scarce in most conserved elements—likely reflecting purifying selection. Our study highlights how DNA transposons are integral to the evolution of bat and human genomes, and has implications for the development of DNA transposon assays for gene therapy and mutagenesis applications.
Keywords: DNA transposons, Myotis lucifugus genome, human genome, integration preferences, multiple linear regression, negative binomial regression, logistic regression
Introduction
Transposable elements (TEs) make up approximately half of the human genome (Lander et al. 2001). Broadly, they are classified as retrotransposons (Class I), which move by means of reverse-transcribed RNA intermediates, and DNA transposons (Class II), which move directly as DNA intermediates by either cut-and-paste (nonreplicative) or copy-and-paste (replicative) mechanism. The cut-and-paste elements occupy up to 3% of the human genome (copy-and-paste elements are absent in human) and fall into at least seven superfamilies (Lander et al. 2001; Pace and Feschotte 2007) of which the most numerous are members of the hAT superfamily (with MER1-Charlie elements occupying 1.39% of the genome) and Tc1/mariner superfamily (with MER2-Tigger elements occupying 1.02% of the genome).
In the human lineage, DNA transposons have not been active for ∼40–50 My (Lander et al. 2001; Pace and Feschotte 2007). In contrast, they are known to have been more recently or still active in several other tetrapod species, for example, in green anole lizard and African clawed frog. In some of these species, DNA transposons were apparently acquired by horizontal gene transfer (Pace et al. 2008). Recently, active DNA transposons have also been found in the genome of the little brown bat (Myotis lucifugus). Ray et al. (2007) discovered six recently active nonautonomous (not encoding all proteins needed for transposition) hAT families in the little brown bat lineage. Subsequently, a more comprehensive characterization of bat TEs was reported (Ray et al. 2008), identifying seven additional DNA transposon families with signs of recent activity (during the last 40 My) and estimating that at least 3.5% of the Myotis genome is derived from DNA transposons. Furthermore, Mitra et al. (2013) showed that the piggyBac1_Ml family (named piggyBat), a member of the cut-and-paste piggyBac superfamily, likely represents the youngest DNA transposon family in the bat genome holding intact coding and cis-acting transposase sequences. Importantly, they demonstrated transpositional activity of this element in bat, human, and yeast cells.
Helitrons, a class of copy-and-paste elements, are abundant in the bat lineage. Pritham and Feschotte (2007) estimated that at least 3% of the M. lucifugus genome is made of Helitron elements. The bulk of Helitrons amplified in the vesper bat (also known as common bat, family Vespertilionidae) lineage 30–36 Ma. The Helitron-encoded replication initiator and replicase protein (RepHel) was reconstructed bioinformatically, and was found to resemble that encoded by plasmids, single-stranded DNA viruses, and bacterial transposons utilizing rolling-circle replication for their amplification (Kapitonov and Jurka 2007b). Therefore, the current model of Helitron transposition is based on bacterial rolling-circle transposons (Kapitonov and Jurka 2007b). Two characteristics of the Helitron integration site have been recognized, that is integration between AT or TT nucleotides at T-rich sites, and no duplication of the host sequences (Kapitonov and Jurka 2001, 2007a).
Several studies of human and mouse cell lines, as well as of transgenic mice, have been conducted to investigate the integration preferences of cut-and-paste DNA transposons in mammalian genomes. These include ex vivo and in vivo assays for three active transposon systems: Sleeping Beauty (SB), a member of the Tc1/mariner superfamily reconstructed from degenerated elements originally isolated from fish (Vigdal et al. 2002; Liu et al. 2005); piggyBac, the founder of the piggyBac superfamily, isolated from and naturally active in the cabbage looper moth (Ding et al. 2005); and Hsmar1, a reconstructed active representative of the Tc1/mariner superfamily (Miskey et al. 2007). SB showed a propensity to integrate near microsatellite repeats (Yant et al. 2005). Several studies also observed the integration of SB and some Tc1 elements in a consensus AT palindrome (Luo et al. 1998; Vigdal et al. 2002; Carlson et al. 2003; Ivics et al. 2004; Liu et al. 2005). This likely reflects transposase sequence recognition specificity or a DNA bendable structure requirement for the integration site (Vigdal et al. 2002).
Another approach to study TE integration/fixation preferences is to analyze the distribution of TEs (stratified by ages or subfamilies) in the genome of a given species together with functional and sequence annotations of this genome. This strategy was recently utilized by Kvikstad and Makova (2010) who focused on LINE and SINE integration preferences in a comparative analysis of primate genomes. Particularly, they used a multiple linear regression (MLR) approach (Kutner et al. 2005), where the response variable was the counts of each category of TEs per genomic window, and the predictors were a diverse list of sequence features measured in the same windows. The resulting models indicated an association of TE counts with, among other predictors, L1 target site sequences, 13-mer genome instability sequences, GC content, and highly conserved elements. This statistical approach effectively unveiled genomic landscapes that were not previously implicated in the integration/fixation of TEs.
In general, most studied TEs have a nonuniform distribution along and among chromosomes (Hua-Van et al. 2011). For instance, some types of TEs are abundant in constitutive heterochromatin (e.g., centromeres and telomeres), and other types show a propensity to colocate with other TEs or to reside in gene-poor regions. One of the best examples of interchromosomal variation is an observation that young Alu elements are enriched in the human Y chromosome (Jurka et al. 2002, 2004). Some of these genomic landscapes have been described in detail for specific DNA transposon subfamilies, for example, integration preference depending on the DNA conformation in the case of SB and P element (Liao et al. 2000; Vigdal et al. 2002; Geurts et al. 2006; Linheiro and Bergman 2008), on the vicinity of transcriptional units for P element in Drosophila (Ryder and Russell 2003) and piggyBac, SPIN and TcBuster studied in HeLa cells (Li et al. 2013), on a particular DNA sequence (i.e., TA repeats for SB, A/TCGG for Ac element [Becker and Kunze 1997], GGGTG or GTGGC for Hobo [Kim et al. 2011]), on nucleosome-free regions in combination with a particular nucleotide sequence for Hermes (Gangadharan et al. 2010), and even on replication origins as exemplified by P element (Spradling et al. 2011).
To the best of our knowledge though, no study so far has produced a comprehensive genome-wide analysis of the integration and fixation preferences of DNA transposons. A study of this kind can clarify many aspects of integration, which could be important for the use of DNA transposons in mutagenesis and genome engineering applications, including DNA delivery for gene therapy (Hackett et al. 2007, 2009; Ivics and Izsvák 2010; Izsvak et al. 2010). Moreover, studying integration preferences in recent events and investigating how the genome adapts to the introduction and further expansion of DNA transposons are important evolutionary questions. Bat genomes provide an excellent opportunity to address these questions; DNA transposon integrations have been hypothesized to be an important influence on the bat lineage diversification into >100 species in a short time frame (Ray et al. 2007).
Here, using the abundant functional annotation of the human genome (Karolchik et al. 2003; Dreszer et al. 2012; Meyer et al. 2013) and performing an extensive genome sequence mining for the little brown bat, we present detailed analyses of the DNA transposon distributions for these two species. In addition, we evaluate two ex vivo data sets—piggyBat (Mitra et al. 2013) and SB (Ammar et al. 2012) integrations in HeLa cells—where we can observe de novo integration preferences essentially in the absence of natural selection. We use statistical regression approaches to investigate whether and how the genomic landscape in the vicinity of DNA transposons contributes to the integration/fixation of these elements. The human case is important because, notwithstanding numerous studies of other TEs (Mager and Medstrand 2005; Cordaux and Batzer 2009; Britten 2010; Kvikstad and Makova 2010; Levy et al. 2010), it remains underexplored from the DNA transposons perspective. Moreover, human DNA transposons have been inactive for the past 40 My, while bat ones remained active; therefore, contrasting human and bat DNA transposon distribution features allows us to evaluate the influence of post-integration selective forces that are expected to be particularly evident in the former but less influential in the latter genome. The bat genome also hosts copy-and-paste elements (the Helitrons), which are absent in the human genome. Our analyses suggest that genomic features play a significant role in determining DNA transposon distributions and allow us to unravel integration mechanisms and/or fixation processes—both in human and in bat.
Results
Human DNA Transposons
The genomic coordinates for the two most abundant human DNA transposon superfamilies, the cut-and-paste Tc1/mariner and hAT superfamilies, were downloaded from the UCSC Genome Browser (Kent et al. 2002; Dreszer et al. 2012; Meyer et al. 2013). Specifically, we considered the Tigger family (∼49,663 primate-specific elements, 64–80 My) from the Tc1/Mariner superfamily, and the Charlie family (∼158,277 eutherian-specific elements only, 80–150 My) from the hAT superfamily (supplementary table S1, Supplementary Material online) (Pace and Feschotte 2007), restricting our attention to elements with lengths above 80 bp, as such elements can be identified with high confidence (Wicker et al. 2007). Table 1 shows the counts of human DNA transposon families analyzed. For subsequent statistical analyses, we partitioned the genome into 1-Mb nonoverlapping windows (2,787 in total), each containing on average 18 (range = 0–53) and 57 (range = 0–317) elements from the Tigger and Charlie data sets, respectively. Substantial variation in counts and coverage by these elements, per window, was observed in the genome (supplementary fig. S1 and table S2, Supplementary Material online).
Table 1.
DNA Transposons Identified by Data Set Name and Number of Elements Used in the Regression Analyses after Removing Outliers.
| Data Set | Class/Superfamily | Genome | Number of Elements |
|---|---|---|---|
| Charlie | hAT | Human | 146,171a |
| Tigger | TcMar | Human | 46,177a |
| hAT | hAT | Bat | 90,412a |
| TcMar | TcMar | Bat | 20,215a |
| Helitron | Helitron | Bat | 189,764a |
| piggyBat | piggyBac | Human | 79,711 |
| SB | TcMar | Human | 53,670 |
aThese numbers might represent overestimation because of fragmented elements due to nested integrations.
Human Genomic Features
The premise of our study is that the genomic landscape can affect integration and/or fixation preferences for TEs. Thus, for each 1-Mb window considered, we computed a set of 36 potentially relevant genomic features (supplementary table S3, Supplementary Material online)—some of these were previously shown to be associated with integration of certain types of DNA transposons by experimental assays, and others were implicated in the integration and/or fixation of other TEs abundant in the human genome (see citations in supplementary table S3, Supplementary Material online). Based on the molecular mechanism or sequence/conformation of DNA that could be implicated in their effects, these 36 features can be classified into categories of DNA conformation (non-B structure), DNA sequence, regulation and expression, recombination, chromosome structure, and replication timing (supplementary table S3, Supplementary Material online). Similar to Fungtammasan et al. (2012), we used hierarchical clustering with Spearman’s rank correlation to remove some strongly correlated features, restricting attention to a final group of 28 features (table 2 and supplementary fig. S2, Supplementary Material online). Before running the regression analyses described below, we eliminated windows with outlying values for one or more genomic features based on histograms and scatterplots against transposons coverages or counts; this left us with 2,566 windows (the regression for Tigger coverages actually employed 2,486 windows as additional windows appeared as outliers; supplementary table S2, Supplementary Material online).
Table 2.
Significant Predictors (and Their Sources) in the Regression Analyses of Human, Bat, and Ex Vivo Integrations of DNA Transposons.
| Category | Predictor | Studied in Human, piggyBat and SB | Studied in Bat | Measure (transformation)a | Source | Description |
|---|---|---|---|---|---|---|
| DNA conformation | A-phased repeat | X | X | Content | Cer et al. (2011) | Runs of four or more As or Ts without the flexible TpA step (Rohs et al. 2009) |
| Direct repeat | X | X | Content | Cer et al. (2011) | Two tracts of 10–50 nt separated by 0–5 nt that have the same composition | |
| G-quadruplex repeat | X | X | Count (log10) for human, content for bat | Cer et al. (2011) | Four blocks, with the same number of G bases (from 3 to 7), separated by 1–7 nt | |
| Inverted repeat | X | X | Content | Cer et al. (2011) | Two consecutive DNA sequences (10–100 nt long) separated by 0–100 nt that are palindromic on the same strand. They may fold back and generate double helices | |
| Mirror repeat | X | X | Count | Cer et al. (2011) | Two perfect repeats of 10–100 nt separated by 0–100 nt on the same strand | |
| Triplex motif | X | Count (log10) | Cer et al. (2011) | Similar to Mirror repeat but the repeat can contain only purines or pyrimidines on the same strand, the separation is at most 8 nt. These sequences can form three-stranded isoforms | ||
| Z-DNA repeat | X | X | Content | Cer et al. (2011) | Five or more tandem repeats, each with alternating pyrimidine–purine dinucleotide motif in which the pattern YG is maintained on one of the DNA strands. These motifs can take the Z-DNA conformation | |
| DNA sequence | Mononucleotide STR | X | X | Content | Genome-wide screen | All repeats of one nucleotide with length >9 bp |
| Dinucleotide STR | X | X | Content | Genome-wide screen | All repeats of two nucleotide motifs with length >10 bp | |
| Trinucleotide STR | X | X | Content | Genome-wide screen | All repeats of three nucleotide motifs with length >12 bp | |
| Tetranucleotide STR | X | X | Content | Genome-wide screen | All repeats of four nucleotide motifs with length >16 bp | |
| TRF | X | Content (log10) | Repeats identified by the program Tandem Repeat Finder (Benson 1999) | |||
| SINE | X | X | Count (log10) for human, content for bat | UCSC Genome Browser | Transposable element (Short INterspersed Elements) | |
| LINE | X | X | Count for human, content for bat | UCSC Genome Browser | Transposable element (Long INnterspersed Elements) | |
| L1 target sequence | X | X | Count | Cost et al. (2002) | Sequence associated to target primed reverse transcription which is characteristic of L1 and Alu mobilization | |
| Telomere hexamer sequence | X | X | Count | Morrish et al. (2007); Nergadze et al. (2007) | Sequence associated with DSB and repair by telomerases, and L1 retrotransposition as well | |
| CpG dinucleotides | X | Content | Genome-wide screen | Proportion of CpG dinucleotides in each window | ||
| Gene or exon | X | X | Content | UCSC Genome Browser | All annotated genes or exons in these genomes | |
| Most conserved elements | X | Count | Siepel et al. (2005) | Regions of the genome that contain functional elements identified by comparative genomics | ||
| Expression and regulation | Chromatin accessibility | X | Count (sqrt) | UCSC Genome Browser | Regions of the genome digested by DNAse I reflecting active chromatin | |
| RNA polymerase II occupancy | X | Content | UCSC Genome Browser | Regions of the genome transcribed by RNA polymerase II | ||
| DNA methylation | X | Count (sqrt) | Down et al. (2008) | Epigenetic modification that can modify the chromatin structure and regulate gene expression | ||
| CpG islands | X | X | Content (sqrt) for human, count for bat | UCSC Genome Browser | Regions of the genome rich in GC close to promoters | |
| Recombination | Recombination hotspots | X | Count (log10) | Myers et al. (2005) | Predicted recombination hotspots using SNP data | |
| Position on the chromosome | Distance to telomere | X | Distance in bp (sqrt) | Genome-wide screen | Distance from the tip of the chromosome to each genome window defined here | |
| Distance to centromere | X | Distance in bp (sqrt) | Genome-wide screen | Distance from the centromere to each genome window defined here | ||
| Replication | Replication timing | X | Weighted average (sqrt) | Ryba et al. (2010) | Genome-wide microarray data measuring time in which replication occurs in hESC (human embryonic stem cells) |
aCount is the number of each feature in a particular window. Content is the fraction of a particular window that is occupied by a feature. Weighted average is used when several data intervals overlap within a window border.
Regression Analysis for DNA Transposon Coverage in the Human Genome
Following an approach similar to that in Kvikstad and Makova (2010), we used MLR to study the DNA transposon coverage (response), that is, the proportion of each 1-Mb window occupied by these elements, as a function of the 28 genomic features (predictors) measured in the same 1-Mb windows. The final MLR models explained 42.74% (11 significant predictors) and 31.96% (8 significant predictors) of the response variability for the Charlie and Tigger data sets, respectively (supplementary table S4, Supplementary Material online). Predictor contributions were quantified by means of the Relative Contribution to Variability Explained (RCVE) coefficient (Kelkar et al. 2008) and represented visually through a heatmap (fig. 1 and P values in supplementary fig. S3, Supplementary Material online).
Fig. 1.

Effects of genomic features in various human DNA tranposon models. The intensity of the color is proportional to the RCVE of each predictor in each model, and the color encodes the sign of the effect - positive in red or negative in blue. White are not significant predictors. LR, logistic regression; RS, R-squared; DE, deviance explained.
Predictors with strong positive effects in the Charlie model included SINE count (RCVE 21.60%), L1 target sequence count (RCVE 7.75%) and telomere hexamer sequence count (RCVE 3.84%). Of lesser significance were recombination hotspots count, LINE count, and triplex motif count (supplementary table S4, Supplementary Material online). The Tigger model also included L1 target sequence (RCVE 15.88%) among the strongest positive predictors. In addition, SINE count, LINE count, and RNA polymerase occupancy showed positive effects on Tigger (supplementary table S4, Supplementary Material online).
The predictor with the strongest negative effect in the Charlie model was A-phased repeat content (RCVE 10.09%). Other predictors with negative effects on Charlie were CpG islands count, distance to telomere, inverted repeat content, and TRF content—all with RCVE <1.32% (supplementary table S4, Supplementary Material online). In the case of Tigger, four predictors—most conserved elements count, inverted repeat, and tetranucleotide and trinucleotide content—had negative effects with small RCVEs—all below 2% (supplementary table S4, Supplementary Material online).
We also analyzed alternative regression models for Charlie and Tigger (supplementary table S5, Supplementary Material online), obtained via replacing some of the predictors with highly correlated predictors—based on the hierarchical clustering. For instance, when L1 target was replaced with G-quadruplex repeat content, CpG content, or 13-mer genome instability, the effects of these GC-rich predictors were negative, that is, opposite in sign to the effect of the AT-rich L1 target, as expected. Also, we did not observe notable changes when replacing SINE count with mononucleotide content, or triplex motif with RnYn content (only for Charlie model). Notably, all these alternative models had similar but lower R-squared values (R2) than the main models presented in supplementary table S4, Supplementary Material online.
Regression Analysis for DNA Transposon Counts in the Human Genome
Negative binomial regression (NBR) (Zeileis et al. 2008) was used to study per-window counts of DNA transposons from the Charlie and Tigger data sets (fig. 1 and supplementary table S4, Supplementary Material online). While MLR is appropriate for coverage data, which after log-transformation are approximately Gaussian, NBR was better suited to handle our count data—which could not be easily transformed to approximate a Gaussian distribution. The final NBR models explained 46.10% (Charlie) and 17.18% (Tigger) of the response deviance (supplementary table S4, Supplementary Material online), and by and large revealed predictors and effect signs consistent with those of the MLR models produced above for coverage (fig. 1 and supplementary table S4, Supplementary Material online). Two predictors with negative effects emerged with NBR that were not implicated in the MLR models: dinucleotide microsatellite content (RCVE 4.03% in the Tigger model) and distance to centromere (RCVE 0.49% in the Charlie model). Also, Wn content was a significant positive predictor for the Charlie counts model not implicated in other regression models. Also, here we evaluated alternative NBR models obtained via replacing some of the predictors with highly correlated predictors based on the hierarchical clustering (supplementary table S5, Supplementary Material online), but failed to obtain deviances explained higher than those of the main models presented in supplementary table S4, Supplementary Material online.
Myotis DNA Transposons and Genomic Features
A total of ∼1.4 Gb of the Myotis genome (out of the 2.3 Gb estimated genome size, www.broadinstitute.org, last accessed April 25, 2014) was analyzed and partitioned into 1-Mb windows (1,446 windows in total). The bat genome assembly is still fragmented in supercontigs (11,653 supercontigs with N50 = 4,281,594) and contigs (72,784 contigs with N50 = 64,330), and complete chromosomes are yet to be reconstructed. We focused on contigs that were at least 1 Mb in length. We must clarify that nonassembled regions and smaller contigs are possibly rich in TEs, so the results concerning integration/fixation presented here are limited to the regions of the genome we were able to analyze.
Using RepeatMasker 3.3.0 (Smit et al. 1996–2010), we obtained the genomic locations of TEs present in the Myotis genome (draft version with 7× coverage from the Broad Institute). We considered only DNA transposon elements longer than 80 bp, and focused our analysis on two cut-and-paste superfamilies (hAT and TcMar, considered separately), and the copy-and-paste Helitron subclass (Helitron). We found that the cut-and-paste superfamilies have high coverage: the mean 1-Mb window coverages for hAT and TcMar are 1.54% and 0.38%, respectively (supplementary table S6, Supplementary Material online). Moreover, the Helitron class appears to be extremely successful in the bat (mean genome-wide coverage 5.49%) (supplementary table S6 and fig. S4, Supplementary Material online).
Because bat chromosomes are yet to be assembled, we considered only genomic features that could be extracted directly from the nucleotide sequences. For each window we computed: 1) The number of consensus sequences of L1 target insertions (Cost et al. 2002), 2) the content (fraction of a window) of microsatellites based on thresholds previously defined in the literature (mononucleotides >9 bp, dinucleotides >10 bp, trinucleotides >12 bp, and tetranucleotides >16 bp [Ananda et al. 2013]), 3) the number of telomere hexamer sequences, 4) the number of CpG islands, and 5) LINE and (separately) SINE content. In addition, the NonB-DB website was used to predict DNA conformations (Cer et al. 2011), and the exon predictions from the UCSC Genome Browser (Kent et al. 2002; Dreszer et al. 2012) were used to proxy gene content since the bat genome does not have extensive gene annotations. We used 15 out of the 16 predictors selected, as L1 target sequence was highly correlated with inverted repeats (supplementary fig. S5, Supplementary Material online). After filtering out the windows where the computed predictors had outlying values, we were left with 1,372 windows to use in our analyses of the hAT, TcMar, and Helitron groups (table 1 and supplementary table S6, Supplementary Material online).
Regression Analysis for DNA Transposons in the Myotis Genome
Following the approach described above for human DNA transposons, we utilized MLR models to study bat DNA transposon coverage as a function of predictors obtained from the genome sequence. We did not study counts of bat DNA transposons as nested integrations can overestimate unique integrations (Gao et al. 2012). Classifying elements by superfamily and isolating elements specific to the bat lineage allowed us to focus on particular integration and/or fixation preferences by each element group. The final hAT and TcMar models explained 34.23% and 28.48% of the response variability, respectively. The Helitron model was more powerful, with R-squared of 78.54% (supplementary table S7, Supplementary Material online).
The models for the cut-and-paste superfamilies (TcMar and hAT) presented similarities and differences between themselves, and also in comparison to the models built for their human counterparts (see above, RCVEs on figs. 1 and 2, P values in supplementary fig. S6, Supplementary Material online). For instance, similar to all human models, the presence of other TEs had a significant positive effect. LINEs were among the strongest positive predictors for both TcMar (RCVE 10.56%) and hAT (RCVE 8.35%), and SINE content was the strongest positive predictor for hAT (RCVE 11.8%)—although its contribution for TcMar was smaller (RCVE 1.21%). Consistently with the human MLR models (supplementary table S4, Supplementary Material online), L1 target sites had a positive effect on TcMar (RCVE 3.56%; supplementary table S7, Supplementary Material online), whereas telomere hexamer had a positive effect on hAT (RCVE 1.89%). Other predictors with significant but small positive effects on TcMar were CpG islands count (RCVE 0.77%) and direct repeat content (RCVE 0.61%). In the hAT model, Z-DNA repeat content and mononucleotide content had small positive effects (supplementary table S7, Supplementary Material online). Negative predictors for the TcMar model were also detected but their RCVEs were small (<1.47%); these included exon content, Z-DNA content, and trinucleotide content. A stronger predictor with a negative effect on hAT was mirror motif count with an RCVE of 7.52%. We considered replacing L1 target with inverted repeat in the final TcMar model since the two predictors are highly correlated in the hierarchical clustering (supplementary fig. S5, Supplementary Material online), but obtained a model with a slightly lower R-squared (supplementary table S8, Supplementary Material online).
Fig. 2.

Effects of genomic features in various bat DNA transposon models. The intensity of the color is proportional to the RCVE of each predictor in each model, and the color encodes the sign of the effect—positive in red or negative in blue. White are not significant predictors. LR, logistic regression; RS, R-squared; DE, deviance explained.
The powerful model for Helitrons contained 11 predictors (fig. 2). Five were DNA conformation predictors—three with positive effects (direct, G-quadruplex, and mirror repeats; RCVEs 2.89%, 2.93%, and 8.90%, respectively) and two with negative effects (A-phased and Z-DNA repeats; RCVE 0.63% and 21.23%, respectively—the latter is the largest RCVE for this model) (supplementary table S7, Supplementary Material online). Three additional predictors with positive effects were L1 target sequences (RCVE 5.06%), which appear in most models considered, telomere hexamer sequences (RCVE 6.93%), and LINEs (RCVE 8.43%). Finally, three additional predictors had negative effects: SINEs (RCVE 5.78%), trinucleotide repeats (RCVE 1.38%), and exon content (RCVE 2.27%). As for TcMar, we explored an alternative regression model replacing L1 target with inverted repeat. Interestingly, this model was stronger (R2 82.52%) and less complex (nine predictors in total) than the final Helitron model. Inverted repeat had an RCVE of 24.73% in comparison to 5.06% for L1 target (supplementary table S8, Supplementary Material online). In this model, G-quadruplex repeat and trinucleotide content were not significant, but A-phased repeat, direct repeat, LINE, and SINE became stronger predictors (supplementary table S8, Supplementary Material online). We retained the original model in the main text because it is more comparable with the other models presented, however, both models can provide useful information about Helitron biology.
Ex Vivo Integrations of piggyBat and SB in HeLa Cells
Using two ex vivo experimental data sets of piggyBat (Mitra et al. 2013) and SB (Ammar et al. 2012) de novo integrations recovered from HeLa cells, we investigated the genomic landscape of integration for each of these elements using the same regression framework. Regression models here are expected to reflect integration preferences more directly because selection (except for selection against lethals) did not have enough time to act against TE integrations in ex vivo experiments. A total of 85,707 de novo integrations of piggyBat and 57,736 de novo integrations of SB in HeLa cells were localized into 2,766 windows along the human genome (the Y-chromosome was excluded because HeLa cells come from a female; supplementary table S2, Supplementary Material online). Only 28 and 4 of these windows were not targeted by de novo integrations of piggyBat and SB, respectively, indicating a dense distribution of integrations along human chromosomes. Moreover, the count data here, after a square root transformation, did not show overdispersion and in fact appeared approximately Gaussian (supplementary fig. S7 and table S9, Supplementary Material online). We could therefore use MLR (as opposed to NBR) to analyze square root transformed counts as a function of genomic features. After filtering for predictor outliers (see Materials and Methods), we retained 2,553 windows for use in the piggyBat and SB regressions (supplementary table S2, Supplementary Material online).
The piggyBat MLR model for counts had an R2 of 19.4% (supplementary table S10, Supplementary Material online). The relevance of L1 target sequences and SINEs observed for endogenous human and bat cut-and-paste DNA elements was confirmed, with positive effects and high RCVEs (11.39% and 3.46%, respectively). In contrast to the other models, LINE count showed a negative effect (RCVE 0.37%). Non-B DNA conformations—mirror repeat and triplex motif, with negative and positive effects, respectively—were also among the significant predictors but with small RCVEs. Additionally, RNA polymerase II occupancy, which reflects active gene transcription and regulation of expression, showed positive effects and RCVEs of 4.04%. The recombination hotspots (RCVE 1.05%) and telomere hexamer predictors (RCVE 0.51%) showed once again positive effects on the integration of DNA transposons, in this case for the piggyBat superfamily (fig. 1). We must clarify, though, that we could not compare piggyBat de novo models to the endogenous piggyBac due to the low representation of the latter genome-wide in human or bat, making meaningful statistical analysis impossible.
The final SB MLR model for counts had an R2 of 16.75% (fig. 1 and supplementary table S10, Supplementary Material online). This model shared four predictors with positive effects with the piggyBat model (recombination hotspots, RCVE 1.01%; RNApol II, RCVE 1.48%; L1 target sequence, RCVE 1.65%; SINE count, RCVE 1.99%). Other positive predictors were inverted repeats, LINE, replication timing, and most conserved elements—although their effects were relatively small (RCVEs range 0.61–1.65%). Among negative predictors, we found TRF and distance to telomere (RCVE 1.41% and 2.46%, respectively). When we considered alternative piggyBat and SB models obtained replacing L1 target and SINE with highly correlated predictors based on the hierarchical clustering (supplementary table S5, Supplementary Material online), we observed patterns similar to those discussed above for alternative Charlie and Tigger MLR models.
Analyses at Smaller Genomic Scale
To broaden our perspective on how genomic landscape may affect the distribution of DNA transposons, we complemented our 1-Mb analyses with analyses at a much smaller genomic scale. We divided both the human and the bat genomes in 20-kb windows, locating DNA trasposons and recomputing genomic features in such windows, and applied logistic regression using the same set of predictors as in the 1-Mb MLRs and NBRs. Logistic regression was used because in the overwhelming majority of cases 20-kb windows contain only few or no DNA transposons of interest in a regression model—thus we used as binary response capturing presence (1) or absence (0). Importantly, the smaller scale allowed us to draw more consistent comparisons with previous studies, especially for the ex vivo experiments, and to investigate whether the effects of certain genomic features are specific to scale—for example, detectable at small but not large scale, or vice versa.
The human models for Charlie, Tigger, piggyBat, and SB lost most of their explanatory power at the 20-kb scale; the deviances explained ranged from 1.05% to 3.12% (supplementary tables S4 and S10, Supplementary Material online). This may be due to a variety of factors, including smaller “signal-to-noise” ratio and larger autocorrelations across windows at smaller scales (data not shown) as well as the fact that some of the predictors, likewise the response, had to be rendered in binary fashion (i.e., presence or absence of genes, CpG islands, and recombination hotspots in any given window—see Materials and Methods) possibly losing some of their strength. Restricting attention to a subset of the windows (top 10%) with the highest TE coverage or counts did sharpen the signal and reduced some of the autocorrelations, resulting in higher deviances explained—average over 10 replicates ranging from 3.45% to 9.78% (supplementary table S11, Supplementary Material online). Figure 1 shows the predictors shared among all 10 replicates.
In general, we observed that some genomic features remained significant and showed consistent effects at large and small scales. The most relevant were L1 target sequences for all human DNA transposons, SINEs, and RNA pol II for piggyBat and SB, and distance to telomere for Charlie and SB. In addition, for Charlie models, A-phased repeats, Triplex motif, and TRF also remained significant and kept the same effect sign. For Tigger models most conserved elements kept a negative effect, and for SB models LINE and replication timing kept a positive effect (supplementary tables S4 and S10, Supplementary Material online, and fig. 1).
However, as to be expected, moving from 1-Mb to 20-kb windows, several predictors that were significant at large scales but with low RCVEs lost significance—for example, CpG islands, LINE count, and distance to centromere for Charlie (supplementary table S4, Supplementary Material online). Others, in turn, acquired significance. Compared with the 1-Mb model, the 20-kb model for Charlie gained 11 predictors, three of them with high RCVEs: CpG content (5.94%, negative effect), DNA methylation (26%, positive effect), and gene content (2.41%, positive effect). Compared with the 1-Mb model, the 20-kb model for Tigger gained seven predictors, the strongest being G-quadruplex repeat (4.04%, negative effect), DNA methylation (5.98%, negative effect), and gene content (2.32%, positive effect). The models for ex vivo data sets also gained CpG content as a significant negative predictor (RCVEs 9.49% and 11.71% for piggyBat and SB, respectively). Chromatin accessibility (RCVE 10.68%) and replication timing (RCVE 5.10%) were new positive predictors for piggyBat. Finally, mirror repeat (RCVE 4.65%) and distance to centromere (RCVE 4.36%) were also among the strongest newly gained predictors for SB at the 20-kb scale (fig. 1 and supplementary tables S4 and S10, Supplementary Material online). These results must be taken with caution, as the deviances explained for the human DNA transposon models are low.
Unlike the ones for human DNA transposons, the logistic regressions for bat models at 20-kb explained high deviances (40.06% for hAT, 14.37% for TcMar, and 61.06% for Helitron), keeping an explanatory power comparable to that obtained at 1-Mb. Again, moving from the 1-Mb to the 20-kb scale modified the scenario in terms of significant predictors, with more predictors gained than lost—except for TcMar, which lost six predictors. Inverted repeat content was the most significant new predictor with positive influence for hAT (RCVE 7.5%), TcMar (RCVE 15.75%), and Helitron (RCVE 12.93%), confirming observations from alternative models evaluated above at 1 Mb (supplementary table S8, Supplementary Material online). Among the predictors that retained positive influence are LINE count for all bat models, SINE count for hAT and TcMar, telomere hexamer for hAT and Helitron, and Z-DNA and mononucleotide repeats for hAT only. On the other hand, exon content and Z-DNA kept their negative influence for Helitron models, and trinucleotide content for hAT kept its negative effect at both scales.
Discussion
The increasing availability of genome sequences is allowing researchers to unravel the impact of DNA transposons on the evolution of many species (Dooner and Weil 2007; Feschotte and Pritham 2007). Moreover, the utilization of DNA transposons as tools for molecular biology (e.g., the Nextera protocol for Illumina sequencing library construction) and DNA delivery vectors for gene therapy and other biomedical applications is on the rise, especially for SB (Izsvak et al. 2010) and piggyBac (integration of cassettes up to 100 kb [Burnight et al. 2012]). Although a few TEs have been studied in detail ex vivo, the current challenge is to employ bioinformatics and statistical approaches to investigate TE integration/fixation preferences genome-wide. These approaches can also extract important information from ex vivo integration data sets. In the present study, we included a comprehensive list of predictors that describe the genomic landscape of large, genome-wide collections of DNA transposons in human (∼192,000 elements) and bat (∼300,400 elements). Our data sets allowed us to compare and contrast DNA transposons at three distinct evolutionary time scales: Ancient inactive elements (>40 My, in human), more recently active elements (<40 My, in bat), and de novo insertion events of piggyBat and SB recovered in human cell culture. The features discovered in the latter data reflect predominantly intrinsic transposon integration preferences rather than the action of natural selection to fix elements differentially in the genome (similar to Wagstaff et al. [2012] for Alu integrations).
In line with previous studies, our models implicate a group of genomic landscape features as significant modulators of the integration/fixation patterns of DNA transposons in the genome (e.g., recombination hotspots, the presence of other TEs, features linked to expression, and regulation). In addition to these, we were able to identify novel predictors related to the integration/fixation of bat and human DNA transposons, such as diverse non-B DNA conformations (e.g., Z-DNA, inverted, G-quadruplex, and mirror repeats), L1 target sequences, and subtelomeric regions. Comparing human and bat models, we observed similar roles for some predictors associated with DNA conformation, presence of other TEs (Feschotte and Pritham 2007; Levy et al. 2010; Gao et al. 2012), and telomere hexamer sequences. The models for ex vivo PiggyBat and SB data also revealed integration preferences toward active transcriptional regions—as evidenced by RNApol II occupancy and other predictors (Jiang and Wessler 2001).
Non-B DNA Conformation Predictors
Our models identify several DNA conformation features as positive predictors at the 1-Mb scale and thus suggest that they play a significant role in shaping the genomic distribution of various DNA transposons we studied in human (e.g., triplex motif for Charlie and PiggyBac, and Inverted repeat for SB) and in bat (e.g., Z-DNA repeat for hAT, direct repeats for TcMar and Helitron elements, and mirror and G-quadruplex repeat for Helitrons). Triplex motifs can stall replication forks and cause double-strand breaks (DSBs) (Zhao et al. 2010), and these perturbations of the normal DNA structure might be employed by a transposase to facilitate new integration events in the human genome. Indeed, several DNA transposons are known to target replication forks during transposition, for example, Ac in maize (Ros and Kunze 2001), P element in Drosophila (Spradling et al. 2011), Tn7 and IS200/IS605 members in bacteria (Parks et al. 2009; Ton-Hoang et al. 2010). The conformation features significant in our bat models have been studied extensively in human cells. While direct and mirror repeats cause hairpins/cruciforms and overlap with chromosome regions undergoing somatic and germline rearrangements (Zhao et al. 2010), G-quadruplex repeats play a role in homologous recombination (Sen and Gilbert 1988) and telomere maintenance (Zhao et al. 2010). Also, Z-DNA sequences have been associated with genomic instability causing DSBs in mammalian cells (Wang et al. 2006; Zhao et al. 2010). It is conceivable that these DNA conformations and the associated processes are utilized by bat DNA transposons opportunistically to facilitate their integration and amplification in the genome.
In contrast, some other DNA conformation features appear as negative predictors in our 1-Mb models and thus may impede DNA transposon integrations. The most significant are A-phased repeats in Charlie, Z-DNA in Helitron, and G-quadruplex in the alternative models for Charlie, Tigger, PiggyBat, and SB. A-phased repeats, or adenine tracts, cause a narrowing of minor grooves of DNA, which can be recognized by some proteins such as transcriptional activators (Barbic et al. 2003). Z-DNA repeats, which are frequently located in close proximity to transcription start sites and are stabilized during transcription, are associated with transcribed regions of the genome (Zhao et al. 2010). G-quadruplex sequences are associated with ∼42.7% of human promoters and also with 3′-UTRs collaborating in transcription termination (Huppert and Balasubramanian 2007; Zhao et al. 2010). It is therefore plausible that selection might work against maintaining DNA transposons in regions of the genome rich in A-phased repeats, Z-DNA, and G-quadruplex. Moreover, we identified negative effects for mirror repeats in the bat hAT and piggyBat models, and for inverted repeats in Charlie and Tigger models. Some of these repeats promote repair of DSBs by homologous recombination, for example, inverted Ty elements in yeast (Downing et al. 2008), which is counterproductive for DNA transposon expansion (see below) and might explain their negative effects. An exception here might be the ex vivo integrations; inverted repeats had positive effect in the SB model—possibly due to lack of strong selection on these recent integrations (in human and to a lesser extent in bat, our observations reflect both integration and fixation preferences).
The complementary analysis conducted using 20-kb windows revealed that the most important predictors for Charlie (i.e., A-phased and inverted repeats), hAT (i.e., Z-DNA repeat), and Helitron (i.e., mirror and Z-DNA repeat) models preserved their effects at this scale, though their RCVEs were smaller than at the 1-Mb scale. Therefore, we have evidence that these DNA structures are essential for integration of the corresponding DNA transposon superfamilies or families, with effects that remain detectable and consistent when observed at various scales. Moving to a smaller genomic scale, we also observed changes in the sign of the effects for a few predictors in the hAT, Helitron, and Tigger models—but the RCVEs of these predictors decreased substantially in comparison to the 1-Mb analysis. Important new predictors detected as significant at the 20-kb scales were G-quadruplex in all human models and in the TcMar bat model, and inverted repeats in all bat models. At a small genomic scale, G-quadruplex repeats appear to be important structures that impede integration of elements from the above mentioned families, while inverted repeats appear to foment integration in the bat genome.
Our results support the importance of non-B DNA conformations to the binding and/or catalytic activity of transposases (Geurts et al. 2006). In fact, there is evidence that DNA conformation characteristics contribute to the identification of potential integration sites for P elements (Liao et al. 2000; Bergman and Quesneville 2007; Linheiro and Bergman 2008) and SB transposons (Liu et al. 2005). In vivo, non-B DNA conformations are known to form during replication, transcription, repair, and recombination, explaining the relevance of such conformations to disease (Cer et al. 2011). Based on the effects we were able to ascertain for de novo integrations of piggyBat and SB, we conjecture that their involvement is also plausible for DNA transposon integrations. We note that methodologies like the one developed to study transposition catalyzed by RAG recombinase (Posey et al. 2006) can be applied in future studies to experimentally validate the effects of diverse DNA conformations on integrations, as suggested by our models. At the same time, the role of non-B DNA conformations for older events is likely to also reflect selection.
Sequence Predictors: Telomere Hexamer Sequences and CpG Content
We observed a positive effect of the telomere hexamer sequence predictor in the Charlie, hAT, Helitron, and piggyBat models, with the strongest effect in the Helitron model at the 1-Mb scale. The analysis at 20-kb scale confirmed a positive effect in these models, detected a positive effect also for Tigger and a small negative effect for TcMar in the bat. Telomere hexamer repeats, (TTAGGG)n, are the characteristic of DSB repair by telomerases at chromosome ends and internally (i.e., at interstitial telomeric sequences—ITS), as well as of telomerase-dependent RNA retrotransposition and of non-canonical L1 integrations (Morrish et al. 2007; Nergadze et al. 2007). Short ITSs are distributed throughout mammalian chromosomes (Lin and Yan 2008) and have been implicated as hotspots for chromosome breakage, recombination, DNA repair, and even regulation of gene expression (Lin and Yan 2008). The significance of telomere hexamer in our models could reflect retrotransposition of L1s (as this sequence is associated with noncanonical L1 integrations) and colocalization of DNA transposons with them (see below).
CpG content was a highly significant negative predictor in the 1-Mb alternative models analyzed for Charlie, Tigger, and PiggyBat. This observation was confirmed at the 20-kb scale. Therefore, regions with fewer CpGs presented more DNA transposons, consistent with positive effects of AT-rich predictors (e.g., L1 target sequence).
Recombination
Recombination hotspots were a positive predictor in human Charlie, piggyBat, and SB models at the 1-Mb scale, and also at 20-kb scale for Charlie. Even though the RCVEs were low (0.48–1.70%), this suggests a potential involvement of recombination in the expansion of DNA transposons—echoing findings by Myers et al. (2005) who discovered a strong overrepresentation of two retrovirus-like retrotransposons (THE1A and THE1B) in genomic regions enriched in recombination hotspots. In Caenorhabditis elegans, DNA transposons were associated with high recombination chromosomal regions which could be explained by requirements of the transposition mechanism (Duret et al. 2000). It has been shown that DNA transposons can make use of homologous recombination to increase their copy number (e.g., P element in Drosophila, Tc1 in C. elegans, Ds element and MuDR in maize) (Engels et al. 1990; Plasterk 1991; Hsia and Schnable 1996; Rubin and Levy 1997). Therefore, this predictor might indeed reflect integration preferences, especially for Charlie elements in the human genome. Lack of data prevented us from evaluating this predictor in the bat models.
Location on the Chromosome
Our models indicate that regions of the human genome located in the proximity of telomeres contain more Charlie elements and ex vivo SB integrations at both large and small genomic scales. We also detected a weak negative effect of the distance to centromere for Charlie at the 1-Mb scale. To the best of our knowledge, this is the first report of chromosomal position bias for human DNA transposons, although positional preference was well documented in Dictyostelium discoideum (20% of total centromere length) (Glockner and Heidel 2009).
Hua-Van et al. (2011) proposed that subtelomeric and peri-centromeric regions are enriched in TEs in a great variety of species, potentially due to relaxed selection on these regions. Some examples include the telomeres of chromosome 3 (500 Mb) and 4 (400 Mb) from the short-tailed opossum that are almost completely comprised of ERV and LTR sequences (Gentles et al. 2007). A contrasting case is evidenced by Drosophila, where telomeric retrotransposons have taken over the function of telomere maintenance (Villasante et al. 2007). A more recent example of specialized chromosomal localization in the primate lineage is represented by the LAVA element that has expanded (600–1,200 copies) in the centromere of the hoolock gibbon (Carbone et al. 2012). Therefore, TEs may occupy certain portions of the chromosome due to selection and may influence chromosome structure.
LINEs and SINEs
We found that most human and bat DNA transposons are located in regions with high numbers of LINEs and SINEs (piggyBat elements were positively associated only with SINEs, and Helitrons with LINEs but not with SINEs). These effects were mostly consistent at large and small genomic scales. For the vast majority of human DNA transposons, the integration explosion happened before the expansion of LINE and SINE elements (Batzer and Deininger 2002; Pace and Feschotte 2007; Cordaux and Batzer 2009), therefore the effects captured in our models reflect mostly co-localization and not causation of integration. Unfortunately the bat genome lacks extensive annotation and dating of retrotransposons, preventing us from drawing sensible age-related interpretations. Nevertheless, for a few endogenous elements and ex vivo data in human (i.e., the SB integrations) and for the younger elements we consider in the bat, our models may be detecting different mechanisms in action. One could be the mechanism of nested integrations—an important aspect of TE biology (Levy et al. 2010). Gao et al. (2012) observed that DNA transposons tend to be the preferred insertion sites for other TEs, except for Helitrons, which experience fewer nesting events and nested less into other TEs. Yang and Bennetzen (2009), on the other hand, observed that Helitrons have a preference to integrate within themselves. An investigation of human TEs by Levy et al. (2010) indicated that L1s suffer the most integrations into themselves, and that AluSxs integrate preferentially in DNA transposons of the Charlie family. Gentles et al. (2007) found that genome regions hosting nested integrations may be protected from deletion, with further integrations seldomly removed or selected against. These regions may also be “safe havens” where TE integrations do not damage the host genome (Levin and Moran 2011).
In all of our models (except hAT) L1 target sites had significant positive effects. These sites are a signature of LINE and SINE integrations, as they are characteristic of the target primed reverse transcription mechanism used by such elements to retrotranspose (Cost et al. 2002). The significance of L1 target sites in our models partially reflects the colocalization of DNA transposons with LINEs and SINEs. In addition, the L1 target sequence (i.e., TTTTAA) contains the motif that has been previously associated with insertion site preferences for different TcMar superfamily members—including piggyBac and hAT elements in bats (Feschotte and Pritham 2007; Ray et al. 2007; Li et al. 2013), as well as piggyBat (Mitra et al. 2013). The positive effects of LINEs, SINEs, and L1 targets on de novo and old integration events in our analyses support a bias for integration close to other TEs in both the human and bat genomes.
The Potential Involvement of DSBs
Some predictors that may be involved in producing or fixing DSBs, caused either by special DNA conformations (e.g., triplex motif, G-quadruplex repeats, and Z-DNA) or enzymatic reactions (telomere hexamer repeats and L1 target sequence) are prominent in our models—particularly for the most prolific Helitrons. DSBs that recruit the host machinery to be repaired might also stimulate integration of DNA transposons. This was observed for Tn7 (a bacterial DNA transposon), in which induction of DSB incited new integrations in adjacent chromosomal areas (Peters and Craig 2000). According to Peters and Craig (2000), to localize integration sites, Tn7 transposase recognizes either DSB directly or some repair components on the DSB. Another line of evidence comes from the interaction of Drosophila Ku70 and BLM (helicase) proteins to resolve DSBs, through nonhomologous end joining caused by P element excision (Min et al. 2004). Moreover, it is known that one of SB DSB repair mechanisms needs direct interaction of Ku protein with the transposase (Izsvák et al. 2004). L1 endonucleases can induce DSB frequently and cause genome instability (Hedges and Deininger 2007), which could also be associated with DNA transposon expansion. The evidence we gathered with our analyses therefore warrants confirmation in ex vivo assays, as it suggests a novel mechanism not previously investigated for eukaryotic DNA transposon mobilization.
DNA Methylation
We observed a strong positive effect of DNA methylation at the 20-kb scale for Charlie, piggyBat, and SB, but negative effect for Tigger. It is known that methylation at CpG dinucleotides is used by host genomes to control expansion of TEs (Oliver and Greene 2009) and this might be the effect we are capturing for the old elements. However, DNA methylation is also important in transcriptional start sites and exonic regions which showed to be relevant for the ex vivo data sets (Down et al. 2008).
Replication Timing
Replication timing was a significant predictor for ex vivo integrations at the 20-kb scale, and also at the 1-Mb scale for SB. This suggests that late replicating areas of the genome are targeted for integration. It was observed for P element in Drosophila that movement of the elements is coordinated with replication sites as this increases the copy number of the element through homologous repair of the excision site (Spradling et al. 2011). It is also known that early replicating regions of the genome are GC-rich, gene dense (Woodfine et al. 2004) and have high transcription levels (Ryba et al. 2010). A word of caution must be raised here since replication timing differs among cell types—in our data ex vivo integrations were studied in HeLa cells, while replication timing measures were generated in hESC (Ryba et al. 2010).
Potential Selection
Most conserved elements were a significant negative predictor at both large and small genomic scales for Tigger and at the 20-kb scale for Charlie—similar to our previous findings concerning L1s (Kvikstad and Makova 2010). The opposite was observed at 1-Mb scale for SB. This suggests a role of natural selection in the localization of TEs avoiding their fixation into or close to cis-regulatory sequences (Siepel et al. 2005), but not for recent integrations. Indeed, it was previously demonstrated that conserved sequences have a negative association with the fixation frequencies of some TEs (including hATs) in the human genome (Sironi et al. 2005; Sironi et al. 2006).
We used CpG islands as a proxy for genic and expressed regions of the genome because they occur near or within proximal promoters (Ioshikhes and Zhang 2000). In the Charlie models, CpG islands had a significant negative effect at the 1-Mb scale. This is consistent with other studies (Li et al. 2013) where DNA transposons were shown to avoid integration or be less likely to become fixed in genic and actively transcribed areas of the genome. In contrast, CpG islands had a small positive effect on TcMar at the 1-Mb scale and on piggyBat and SB ex vivo at the 20-kb scale. This is consistent with observations that some DNA transposons have in vivo integration preference for gene-rich regions (Wilson et al. 2007; Bellen et al. 2011). Additional positive predictors were RNA polymerase II occupancy (which localizes at actively transcribed genes) for Tigger, piggyBat, and SB at both scales; chromatin accessibility (which reflects active regions of the genome) for Charlie and piggyBat models at the 20-kb scale; and gene content for all models except TcMar and Helitron (not significant for SB). Consistent with Ammar et al. (2012) we did not find any bias of SB integrations toward genes or open chromatin. Similar to Mitra et al. (2013), we also observed a peculiar integration preference toward actively transcribed genes for piggyBat—reflected by the RNA polymerase II occupancy. Such pattern was reported previously for piggyBac (Handler 2002) and P element—which integrate in promoter regions of genes actively transcribed in the germline (Ryder and Russell 2003; Spradling et al. 2011).
Mammalian DNA transposons are difficult to study in vivo due to the fact that in most mammals these elements are less active (and even dead) compared with other TEs. Nevertheless, by applying rigorous statistical methods, we were able to extract valuable information that improves our understanding of the biology of these intriguing elements. A better knowledge of site integration preferences can aid in the design of gene therapy strategies and insertional mutagenesis assays. Moreover, our analysis pipeline can be reproduced for other genomes, such as those made available by the Genome 10K Project (Genome 10K Community of Scientists 2009)—further refining the evidence and perspectives presented in this article.
Materials and Methods
DNA Transposons Data in Human
We obtained the location of every DNA transposon in the human genome version hg19 from the UCSC Genome Browser, which contains the most recent masking information available. These were then lifted-over to hg18 with Galaxy (Giardine et al. 2005; Blankenberg et al. 2010; Goecks et al. 2010) because most genomic features and annotations are available on this version of the genome. Since the best-described elements are the members of the superfamilies TcMar and hAT, we focused on them, further restricting our attention to ∼208,000 elements in the superfamilies longer than 80 bp (Wicker et al. 2007). These were analyzed as counts (number of elements) and coverages (fraction of a window) based on a subdivision of the genome into nonoverlapping 1-Mb windows (a total of 2,787 including sex chromosomes). Some elements of these classes were accurately dated (Pace and Feschotte 2007) and are considered to be primate- and eutherian-specific. Thus, these groups of elements allowed us to evaluate whether some genomic features are important for a particular timing of integration and family. Supplementary table S1, Supplementary Material online, provides a complete list of the elements under consideration (nomenclature from Repeat Masker) and their total counts.
Genomic Features in Human
A total of 36 genomic features (supplementary table S3, Supplementary Material online) were considered as potentially affecting the process of integration and/or fixation of DNA transposons. Each was obtained from the UCSC Genome Browser (Kent et al. 2002; Dreszer et al. 2012) or from previous publications using the hg18 version of the human genome. All features were evaluated for normality, and transformed to approximate it where necessary (table 2). To remove outliers, we generated scatterplots of each predictor against the response variable and identified extreme data points. In addition, to improve the efficacy of model selection for our regression analyses and reduce the chances of multicollinearity, we implemented a preselection of the predictors (Fungtammasan et al. 2012). We ran hierarchical clustering based on Spearman’s rank correlation and identified clusters of predictors using a threshold of 80% (supplementary fig. S2, Supplementary Material online). From each such cluster, we took one “representative” feature, thus restricting attention to 28 (out of 36) predictors characterized by relatively low linear dependencies (table 2).
Regression Analyses in Human
For the MLRs in each of the classes under consideration, we started by evaluating transformations that would approximate normality of the coverage response. For Charlie and Tigger data sets, we applied a logarithmic transformation (base 10) after shifting coverage by a very small amount (+0.0001) in every window (some windows contained no Charlie and/or Tigger DNA transposons, thus having a coverage of 0). To select predictors, we applied the best subset procedure based on the Akaike Information Criterion (AIC). Variance inflation factors were checked and influential outliers (detected with Cook’s distance) were removed at each stage—rendering data sets of 2,566 or more windows (92% of the initial data set). The R2 was used to capture the explanatory power of each model considered, and various diagnostic plots (residuals, case influence, Q-Q, and spread level [Hoaglin et al. 1983; Fox 1997, 2008; Fox and Weisberg 2011]) were used to evaluate the performance of the models at each step of the process. We also ran models substituting some of the predictors with other predictors highly correlated with them (in other words, we changed some of the “representative” features for the clusters described before). We found that our main results were robust to these “swaps.”
We also utilized NBRs to model count responses as a function of the genomic features. The Negative Binomial framework (Zeileis et al. 2008), in addition to allowing zero response values (which do occur for DNA transposons counts in 1-Mb windows), can deal with over-dispersion (variance exceeding mean) that we do observe in our data sets (supplementary table S10, Supplementary Material online). The procedure for model selection was the same as for the MLRs used for coverage. Here, the deviance explained replaces the R2 as a measure of the explanatory power of each model considered.
For each final MLR model, the contribution of individual predictors was computed as the relative contribution to variance explained (RCVE), which is given by the formula: 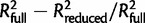 , where
, where 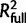 represents the share of explained variability by all the predictors in the final model, whereas
represents the share of explained variability by all the predictors in the final model, whereas  represents the explained variability without the specific predictor whose contribution is being evaluated (Kelkar et al. 2008). For each final NBR model, the contribution of individual predictors was computed with the formula, [(D0 − Dfull) − (D0 − Dreduced)]/(D0 − Dfull), where D0 is the null deviance, Dfull is the residual deviance of the full model, and Dreduced is the deviance of the model without the predictor under evaluation (Fungtammasan et al. 2012).
represents the explained variability without the specific predictor whose contribution is being evaluated (Kelkar et al. 2008). For each final NBR model, the contribution of individual predictors was computed with the formula, [(D0 − Dfull) − (D0 − Dreduced)]/(D0 − Dfull), where D0 is the null deviance, Dfull is the residual deviance of the full model, and Dreduced is the deviance of the model without the predictor under evaluation (Fungtammasan et al. 2012).
All statistical analyses were performed in the R environment (Team 2011), using the packages MASS (Venables and Ripley 2002), BiodiversityR (Kindt and Coe 2005), bestglm (McLeod and Xu 2010), and car (Fox and Weisberg 2011). RCVEs and P values (fig. 1 and supplementary fig. S3, Supplementary Material online) were represented graphically in heatmaps, using the package pheatmap (Kolde 2013).
Data and Analyses for the M. lucifugus
The second version (7× coverage) of the little brown bat genome (M. lucifugus) was released in 2010 (www.broadinstitute.org). Using RepeatMasker 3.3.0 (Smit et al. 1996–2010), we were able to extract the genomic locations of all TEs known in the bat plus additional annotations from previous publications (Pace et al. 2008; Ray et al. 2007, 2008; Gilbert et al. 2010; Mitra et al. 2013; Zhuo et al. 2013). As described above for human, we restricted attention to elements larger than 80 bp and apportioned them to 1-Mb nonoverlapping windows along the bat genome. We focused on superfamilies highly represented in this species, as well as on elements that are considered to be bat-specific and have experienced a recent explosion of integration. The classes considered were: hAT (∼94,048 fragments/elements), TcMar (∼21,400 fragments/elements), and Helitron (∼203,205 fragments/elements; supplementary fig. S4, Supplementary Material online, shows their distributions). In terms of predictors, we computed 16 sequence-related genomic features (table 2) using our own scripts, by computational prediction using NonB-DB website (Cer et al. 2011), or downloading the information from Ensembl (Hubbard et al. 2007) and UCSC (Dreszer et al. 2012; Kent et al. 2002). As for human, we filtered out windows with outlying predictors values, thus restricting ourselves to 1,372 windows (∼95% of all 1-Mb windows). We then ran only the MLR for coverage in every class of elements, since counts could be overestimated by nesting events (Gao et al. 2012).
Ex Vivo Integrations of piggyBat and SB
PiggyBat and SB integrations in HeLa cells were obtained from Mitra et al. (2013) and Ammar et al. (2012), respectively. These data sets correspond to the control assays in their experiments. The genomic coordinates of all de novo integrations were used to compute count responses in 1-Mb windows of hg18. We removed windows from the Y-chromosome, as HeLa cells are derived from a female. Few windows did not experience integrations (28 for piggyBat, and 4 for SB). Moreover, both data sets did not show overdispersion, and in fact were approximately normally distributed after square root transformation. Therefore, we applied to transformed counts in the ex vivo data sets the MLR workflow described above. A total of 85,707 integrations for piggyBat, and 57,736 for SB were studied in 2,766 windows of hg18 (supplementary fig. S7, Supplementary Material online, shows their distribution). As for the other regressions, we removed windows with predictor outliers based on scatterplots thus 2,553 windows were used in the analyses.
Analyses at the 20-kb Scale
We complemented our 1-Mb analyses with analyses conducted on much smaller, 20-kb windows—142,630 on the human genome and 69,151 in the bat genome. We used a logistic regression approach because at the 20-kb scale almost half of all windows contained no DNA transposons, and the other half contained only one or a few elements—making a binary response appropriate (0 = absence, 1 = presence). We used the same predictors as in the 1-Mb analyses but predictor transformations (to regularize their distributions) were not necessary here—except for some predictors that showed dramatically bimodal distributions. We dealt with these through binarization like we did for the response. For the human genome, we encoded in binary format (presence/absence) gene content, CpG islands, and recombination hotspots; for the bat—exon content and CpG islands. We utilized the same preprocessing pipeline, performing hierarchical clustering to select a subset of nonredundant predictors (supplementary figs. S8 and S9, Supplementary Material online) and removing outliers—which led to retaining 130,815 and 66,433 windows for the human and bat analyses, respectively. Best subset selection was again performed to identify the best predictors for each model. Deviance explained and RCVEs were obtained for each of the models (supplementary tables S4, S7, and S10, Supplementary Material online, and figs. 1 and 2, P values in supplementary figs. S3 and S6, Supplementary Material online). To refine our human models, we collected the top 10% windows with most counts or highest content of elements, and contrasted them to the same number of empty windows picked up at random and replicated 10 times (supplementary table S11, Supplementary Material online). We followed the same workflow mentioned before to generate the best logistic regression models.
Supplementary Material
Supplementary tables S1–S11 and figures S1–S9 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Guruprasad Ananda and Arkarachai Fungtammassan for their advice and help with the computation of genomic features used in our analyses, and Erika Kvikstad for her help with the initial data set of genomic predictors. They also thank Jainy Thomas for providing Myotis lucifugus Helitron element library prior to publication. Nancy Craig provided published data for piggyBat, and Zsuzsanna Izsvak provided published data for Sleeping Beauty. They declare no conflict of interests related to the publication of this research. This work was supported by the National Science Foundation grant number DBI-0965596 to K.D.M.; the National Institute of General Medical Sciences grant number GM087472 to K.D.M. and Kristin Eckert, and GM077582 to C.F.; the Penn State Clinical and Translational Science Institute and the Pennsylvania Department of Health using Tobacco Settlement Funds (the Department specifically disclaims responsibility for any analyses, interpretations, or conclusions).
References
- Ammar I, Gogol-Doring A, Miskey C, Chen W, Cathomen T, Izsvak Z, Ivics Z. Retargeting transposon insertions by the adeno-associated virus Rep protein. Nucleic Acids Res. 2012;40:6693–6712. doi: 10.1093/nar/gks317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananda G, Walsh E, Jacob KD, Krasilnikova M, Eckert KA, Chiaromonte F, Makova KD. Distinct mutational behaviors differentiate short tandem repeats from microsatellites in the human genome. Genome Biol Evol. 2013;5:606–620. doi: 10.1093/gbe/evs116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbic A, Zimmer DP, Crothers DM. Structural origins of adenine-tract bending. Proc Natl Acad Sci U S A. 2003;100:2369–2373. doi: 10.1073/pnas.0437877100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- Becker HA, Kunze R. Maize activator transposase has a bipartite DNA binding domain that recognizes subterminal sequences and the terminal inverted repeats. Mol Gen Genet. 1997;254:219–230. doi: 10.1007/s004380050410. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Levis RW, He YC, Carlson JW, Evans-Holm M, Bae E, Kim J, Metaxakis A, Savakis C, Schulze KL, et al. The Drosophila Gene Disruption Project: progress using transposons with distinctive site specificities. Genetics. 2011;188:731–743. doi: 10.1534/genetics.111.126995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman CM, Quesneville H. Discovering and detecting transposable elements in genome sequences. Brief Bioinform. 2007;8:382–392. doi: 10.1093/bib/bbm048. [DOI] [PubMed] [Google Scholar]
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Chapter 19. Curr Protoc Mol Biol. 2010 doi: 10.1002/0471142727.mb1910s89. Unit 19.10:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ. Transposable element insertions have strongly affected human evolution. Proc Natl Acad Sci U S A. 2010;107:19945–19948. doi: 10.1073/pnas.1014330107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnight ER, Staber JM, Korsakov P, Li X, Brett BT, Scheetz TE, Craig NL, McCray PB., Jr A hyperactive transposase promotes persistent gene transfer of a piggyBac DNA transposon. Mol Ther Nucleic Acids. 2012;1:e50. doi: 10.1038/mtna.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone L, Harris RA, Mootnick AR, Milosavljevic A, Martin DIK, Rocchi M, Capozzi O, Archidiacono N, Konkel MK, Walker JA, et al. Centromere remodeling in hoolock leuconedys (hylobatidae) by a new transposable element unique to the gibbons. Genome Biol Evol. 2012;4:648–658. doi: 10.1093/gbe/evs048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CM, Dupuy AJ, Fritz S, Roberg-Perez KJ, Fletcher CF, Largaespada DA. Transposon mutagenesis of the mouse germline. Genetics. 2003;165:243–256. doi: 10.1093/genetics/165.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cer RZ, Bruce KH, Mudunuri US, Yi M, Volfovsky N, Luke BT, Bacolla A, Collins JR, Stephens RM. Non-B DB: a database of predicted non-B DNA-forming motifs in mammalian genomes. Nucleic Acids Res. 2011;39:D383–D391. doi: 10.1093/nar/gkq1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost GJ, Feng QH, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002;21:5899–5910. doi: 10.1093/emboj/cdf592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Wu XH, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac resource (PB) transposon in mammalian cells and mice. Cell. 2005;122:473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Dooner HK, Weil CF. Give-and-take: interactions between DNA transposons and their host plant genomes. Curr Opin Genet Dev. 2007;17:486–492. doi: 10.1016/j.gde.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Gräf S, Johnson N, Herrero J, Tomazou EM, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008;26:779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing B, Morgan R, VanHulle K, Deem A, Malkova A. Large inverted repeats in the vicinity of a single double-strand break strongly affect repair in yeast diploids lacking Rad51. Mutat Res. 2008;645:9–18. doi: 10.1016/j.mrfmmm.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreszer TR, Karolchik D, Zweig AS, Hinrichs AS, Raney BJ, Kuhn RM, Meyer LR, Wong M, Sloan CA, Rosenbloom KR, et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res. 2012;40:D918–D923. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Marais G, Biemont C. Transposons but not retrotransposons are located preferentially in regions of high recombination rate in Caenorhabditis elegans. Genetics. 2000;156:1661–1669. doi: 10.1093/genetics/156.4.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels WR, Johnsonschlitz DM, Eggleston WB, Sved J. High-frequency P-element loss in Drosophila is homolog dependent. Cell. 1990;62:515–525. doi: 10.1016/0092-8674(90)90016-8. [DOI] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J. Applied regression, linear models, and related methods. Thousand Oaks (CA): Sage; 1997. [Google Scholar]
- Fox J. Applied regression analysis and generalized linear models. Los Angeles (CA): Sage; 2008. [Google Scholar]
- Fox J, Weisberg S. An {R} companion to applied regression. Thousand Oaks (CA): Sage; 2011. [Google Scholar]
- Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, Makova KD. A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res. 2012;22:993–1005. doi: 10.1101/gr.134395.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangadharan S, Mularoni L, Fain-Thornton J, Wheelan SJ, Craig NL. DNA transposon Hermes inserts into DNA in nucleosome-free regions in vivo. Proc Natl Acad Sci U S A. 2010;107:21966–21972. doi: 10.1073/pnas.1016382107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao CH, Xiao ML, Ren XD, Hayward A, Yin JM, Wu LK, Fu DH, Li JN. Characterization and functional annotation of nested transposable elements in eukaryotic genomes. Genomics. 2012;100:222–230. doi: 10.1016/j.ygeno.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Genome 10K Community of Scientists. Genome 10K: a proposal to obtain whole-genome sequence for 10 000 vertebrate species. J Hered. 2009;100:659–674. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentles AJ, Wakefield MJ, Kohany O, Gu W, Batzer MA, Pollock DD, Jurka J. Evolutionary dynamics of transposable elements in the short-tailed opossum Monodelphis domestica. Genome Res. 2007;17:992–1004. doi: 10.1101/gr.6070707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts AM, Hackett CS, Bell JB, Bergemann TL, Collier LS, Carlson CM, Largaespada DA, Hackett PB. Structure-based prediction of insertion-site preferences of transposons into chromosomes. Nucleic Acids Res. 2006;34:2803–2811. doi: 10.1093/nar/gkl301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, Schaack S, Pace JK, Brindley PJ, Feschotte C. A role for host–parasite interactions in the horizontal transfer of transposons across phyla. Nature. 2010;464:1347–1350. doi: 10.1038/nature08939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glockner G, Heidel AJ. Centromere sequence and dynamics in Dictyostelium discoideum. Nucleic Acids Res. 2009;37:1809–1816. doi: 10.1093/nar/gkp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J, Galaxy T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett CS, Geurts AM, Hackett PB. Predicting preferential DNA vector insertion sites: implications for functional genomics and gene therapy. Genome Biol. 2007;8:S12. doi: 10.1186/gb-2007-8-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett PB, Largaespada DA, Cooper LJ. A transposon and transposase system for human application. Mol Ther. 2009;18:674–683. doi: 10.1038/mt.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler AM. Use of the piggyBac transposon for germ-line transformation of insects. Insect Biochem Mol Biol. 2002;32:1211–1220. doi: 10.1016/s0965-1748(02)00084-x. [DOI] [PubMed] [Google Scholar]
- Hedges DJ, Deininger PL. Inviting instability: transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat Res. 2007;616:46–59. doi: 10.1016/j.mrfmmm.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoaglin DC, Mosteller F, Tukey JW. Understanding robust and exploratory data analysis. New York: Wiley; 1983. [Google Scholar]
- Hsia AP, Schnable PS. DNA sequence analyses support the role of interrupted gap repair in the origin of internal deletions of the maize transposon, MuDR. Genetics. 1996;142:603–618. doi: 10.1093/genetics/142.2.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua-Van A, Le Rouzic A, Boutin TS, Filee J, Capy P. The struggle for life of the genome’s selfish architects. Biol Direct. 2011;6:19. doi: 10.1186/1745-6150-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard TJP, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T, et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S. G-quadruplexes in promoters throughout the human genome (vol. 35, pg 406, 2006) Nucleic Acids Res. 2007;35:2105–2105. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioshikhes IP, Zhang MQ. Large-scale human promoter mapping using CpG islands. Nat Genet. 2000;26:61–63. doi: 10.1038/79189. [DOI] [PubMed] [Google Scholar]
- Ivics Z, Izsvák Z. The expanding universe of transposon technologies for gene and cell engineering. Mob DNA. 2010;1:25. doi: 10.1186/1759-8753-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Kaufman CD, Zayed H, Miskey C, Walisko O, Izsvak Z. The Sleeping Beauty transposable element: evolution, regulation and genetic applications. Curr Issues Mol Biol. 2004;6:43–55. [PubMed] [Google Scholar]
- Izsvak Z, Hackett PB, Cooper LJ, Ivics Z. Translating sleeping beauty transposition into cellular therapies: victories and challenges. Bioessays. 2010;32:756–767. doi: 10.1002/bies.201000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izsvák Z, Stüwe EE, Fiedler D, Katzer A, Jeggo PA, Ivics Z. Healing the wounds inflicted by sleeping beauty transposition by double-strand break repair in mammalian somatic cells. Mol Cell. 2004;13:279–290. doi: 10.1016/s1097-2765(03)00524-0. [DOI] [PubMed] [Google Scholar]
- Jiang N, Wessler SR. Insertion preference of maize and rice miniature inverted repeat transposable elements as revealed by the analysis of nested elements. Plant Cell. 2001;13:2553–2564. doi: 10.1105/tpc.010235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kohany O, Pavlicek A, Kapitonov VV, Jurka MV. Duplication, coclustering, and selection of human Alu retrotransposons. Proc Natl Acad Sci U S A. 2004;101:1268–1272. doi: 10.1073/pnas.0308084100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Krnjajic M, Kapitonov VV, Stenger JE, Kokhanyy O. Active Alu elements are passed primarily through paternal germlines. Theor Popul Biol. 2002;61:519–530. doi: 10.1006/tpbi.2002.1602. [DOI] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci U S A. 2001;98:8714–8719. doi: 10.1073/pnas.151269298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Helitrons in fruit flies. Repbase Rep. 2007a;7:127–132. [Google Scholar]
- Kapitonov VV, Jurka J. Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet. 2007b;23:521–529. doi: 10.1016/j.tig.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelkar YD, Tyekucheva S, Chiaromonte F, Makova KD. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Res. 2008;18:30–38. doi: 10.1101/gr.7113408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Hice RH, O’Brochta DA, Atkinson PW. DNA sequence requirements for hobo transposable element transposition in Drosophila melanogaster. Genetica. 2011;139:985–997. doi: 10.1007/s10709-011-9600-2. [DOI] [PubMed] [Google Scholar]
- Kindt R, Coe R. Tree diversity analysis. A manual and software for common statistical methods for ecological and biodiversity studies. 2005 Nairobi: World Agroforestry Centre (ICRAF) [Google Scholar]
- Kolde R, editor. 2013. pheatmap: Pretty Heatmaps. R package version 0.7.7. Available from: http://CRAN.R-project.org/package=pheatmap. [Google Scholar]
- Kutner MH, Nachtsheim CJ, Neter J, Li W. Applied linear statistical models. New York: McGraw-Hill; 2005. [Google Scholar]
- Kvikstad EM, Makova KD. The (r)evolution of SINE versus LINE distributions in primate genomes: sex chromosomes are important. Genome Res. 2010;20:600–613. doi: 10.1101/gr.099044.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Levin HL, Moran JV. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet. 2011;12:615–627. doi: 10.1038/nrg3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy A, Schwartz S, Ast G. Large-scale discovery of insertion hotspots and preferential integration sites of human transposed elements. Nucleic Acids Res. 2010;38:1515–1530. doi: 10.1093/nar/gkp1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ewis H, Hice RH, Malani N, Parker N, Zhou L, Feschotte C, Bushman FD, Atkinson PW, Craig NL. A resurrected mammalian hAT transposable element and a closely related insect element are highly active in human cell culture. Proc Natl Acad Sci U S A. 2013;110:E478–E487. doi: 10.1073/pnas.1121543109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao GC, Rehm EJ, Rubin GM. Insertion site preferences of the P transposable element in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2000;97:3347–3351. doi: 10.1073/pnas.050017397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KW, Yan J. Endings in the middle: current knowledge of interstitial telomeric sequences. Mutat Res. 2008;658:95–110. doi: 10.1016/j.mrrev.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Linheiro RS, Bergman CM. Testing the palindromic target site model for DNA transposon insertion using the Drosophila melanogaster P-element. Nucleic Acids Res. 2008;36:6199–6208. doi: 10.1093/nar/gkn563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, Geurts AM, Yae K, Srinivasan AR, Fahrenkrug SC, Largaespada DA, Takeda J, Horie K, Olson WK, Hackett PB. Target-site preferences of Sleeping Beauty transposons. J Mol Biol. 2005;346:161–173. doi: 10.1016/j.jmb.2004.09.086. [DOI] [PubMed] [Google Scholar]
- Luo GB, Ivics Z, Izsvak Z, Bradley A. Chromosomal transposition of a Tc1/mariner-like element in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 1998;95:10769–10773. doi: 10.1073/pnas.95.18.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager DL, Medstrand P. Encyclopedia of life sciences. NJ: John Wiley & Sons, Ltd. 2005. Retroviral repeat sequences; p. 7. [Google Scholar]
- McLeod AI, Xu C, editors. 2011. bestglm: Best Subset GLM. R package version 0.33. Available from: http://CRAN.R-project.org/package=bestglm. [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min B, Weinert BT, Rio DC. Interplay between Drosophila Bloom’s syndrome helicase and Ku autoantigen during nonhomologous end joining repair of P element-induced DNA breaks. Proc Natl Acad Sci U S A. 2004;101:8906–8911. doi: 10.1073/pnas.0403000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskey C, Papp B, Mates L, Sinzelle L, Keller H, Izsvak Z, Ivics Z. The ancient mariner sails again: transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Mol Cell Biol. 2007;27:4589–4600. doi: 10.1128/MCB.02027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra R, Li XH, Kapusta A, Mayhew D, Mitra RD, Feschotte C, Craig NL. Functional characterization of piggyBat from the bat Myotis lucifugus unveils an active mammalian DNA transposon. Proc Natl Acad Sci U S A. 2013;110:234–239. doi: 10.1073/pnas.1217548110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrish TA, Garcia-Perez JL, Stamato TD, Taccioli GE, Sekiguchi J, Moran JV. Endonuclease-independent LINE-1 retrotransposition at mammalian telomeres. Nature. 2007;446:208–212. doi: 10.1038/nature05560. [DOI] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- Nergadze SG, Santagostino MA, Salzano A, Mondello C, Giulotto E. Contribution of telomerase RNA retrotranscription to DNA double-strand break repair during mammalian genome evolution. Genome Biol. 2007;8:R260. doi: 10.1186/gb-2007-8-12-r260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver KR, Greene WK. Transposable elements: powerful facilitators of evolution. Bioessays. 2009;31:703–714. doi: 10.1002/bies.200800219. [DOI] [PubMed] [Google Scholar]
- Pace JK, Feschotte C. The evolutionary history of human DNA transposons: evidence for intense activity in the primate lineage. Genome Res. 2007;17:422–432. doi: 10.1101/gr.5826307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace JK, Gilbert C, Clark MS, Feschotte C. Repeated horizontal transfer of a DNA transposon in mammals and other tetrapods. Proc Natl Acad Sci U S A. 2008;105:17023–17028. doi: 10.1073/pnas.0806548105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AR, Li Z, Shi Q, Owens RM, Jin MM, Peters JE. Transposition into replicating DNA occurs through interaction with the processivity factor. Cell. 2009;138:685–695. doi: 10.1016/j.cell.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, Craig NL. Tn7 transposes proximal to DNA double-strand breaks and into regions where chromosomal DNA replication terminates. Mol Cell. 2000;6:573–582. doi: 10.1016/s1097-2765(00)00056-3. [DOI] [PubMed] [Google Scholar]
- Plasterk RHA. The origin of footprints of the Tc1 transposon of Caenorhabditis elegans. EMBO J. 1991;10:1919–1925. doi: 10.1002/j.1460-2075.1991.tb07718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey JE, Pytlos MJ, Sinden RR, Roth DB. Target DNA structure plays a critical role in RAG transposition. PLoS Biol. 2006;4:e350. doi: 10.1371/journal.pbio.0040350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritham EJ, Feschotte C. Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc Natl Acad Sci U S A. 2007;104:1895–1900. doi: 10.1073/pnas.0609601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray DA, Feschotte C, Pagan HJT, Smith JD, Pritham EJ, Arensburger P, Atkinson PW, Craig NL. Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Res. 2008;18:717–728. doi: 10.1101/gr.071886.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray DA, Pagan HJT, Thompson ML, Stevens RD. Bats with hATs: evidence for recent DNA transposon activity in genus Myotis. Mol Biol Evol. 2007;24:632–639. doi: 10.1093/molbev/msl192. [DOI] [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. The role of DNA shape in protein-DNA recognition. Nature. 2009;461:1248–U81. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros F, Kunze R. Regulation of activator/dissociation transposition by replication and DNA methylation. Genetics. 2001;157:1723–1733. doi: 10.1093/genetics/157.4.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin E, Levy AA. Abortive gap repair: underlying mechanism for Ds element formation. Mol Cell Biol. 1997;17:6294–6302. doi: 10.1128/mcb.17.11.6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Lu JJ, Itoh M, Kulik M, Zhang JF, Schulz TC, Robins AJ, Dalton S, Gilbert DM. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761–770. doi: 10.1101/gr.099655.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder E, Russell S. Transposable elements as tools for genomics and genetics in Drosophila. Brief Funct Genomic Proteomic. 2003;2:57–71. doi: 10.1093/bfgp/2.1.57. [DOI] [PubMed] [Google Scholar]
- Sen D, Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou MM, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi M, Menozzi G, Comi GP, Bresolin N, Cagliani R, Pozzoli U. Fixation of conserved sequences shapes human intron size and influences transposon-insertion dynamics. Trends Genet. 2005;21:484–488. doi: 10.1016/j.tig.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Sironi M, Menozzi G, Comi GP, Cereda M, Cagliani R, Bresolin N, Pozzoli U. Gene function and expression level influence the insertion/fixation dynamics of distinct transposon families in mammalian introns. Genome Biol. 2006;7:R120. doi: 10.1186/gb-2006-7-12-r120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. RepeatMasker Open-3.0. 1996–2010 Available from: http://www.repeatmasker.org. [Google Scholar]
- Spradling AC, Bellen HJ, Hoskins RA. Drosophila P elements preferentially transpose to replication origins. Proc Natl Acad Sci U S A. 2011;108:15948–15953. doi: 10.1073/pnas.1112960108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RDC. Vienna (Austria): R Foundation for Statistical Computing; 2011. R: a language and environment for statistical computing. [Google Scholar]
- Ton-Hoang B, Pasternak C, Siguier P, Guynet C, Hickman AB, Dyda F, Sommer S, Chandler M. Single-stranded DNA transposition is coupled to host replication. Cell. 2010;142:398–408. doi: 10.1016/j.cell.2010.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables WN, Ripley BD. Modern applied statistics with S. New York: Springer; 2002. [Google Scholar]
- Vigdal TJ, Kaufman CD, Izsvak Z, Voytas DF, Ivics Z. Common physical properties of DNA affecting target site selection of Sleeping Beauty and other Tc1/mariner transposable elements. J Mol Biol. 2002;323:441–452. doi: 10.1016/s0022-2836(02)00991-9. [DOI] [PubMed] [Google Scholar]
- Villasante A, Abad JP, Planello R, Mendez-Lago M, Celniker SE, de Pablos B. Drosophila telomeric retrotransposons derived from an ancestral element that was recruited to replace telomerase. Genome Res. 2007;17:1909–1918. doi: 10.1101/gr.6365107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff BJ, Hedges DJ, Derbes RS, Campos Sanchez R, Chiaromonte F, Makova KD, Roy-Engel AM. Rescuing Alu: recovery of new inserts shows LINE-1 preserves Alu activity through A-tail expansion. PLoS Genet. 2012;8:e1002842. doi: 10.1371/journal.pgen.1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Christensen LA, Vasquez KM. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc Natl Acad Sci U S A. 2006;103:2677–2682. doi: 10.1073/pnas.0511084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Wilson MH, Coates CJ, George AL. PiggyBac transposon-mediated gene transfer in human cells. Mol Ther. 2007;15:139–145. doi: 10.1038/sj.mt.6300028. [DOI] [PubMed] [Google Scholar]
- Woodfine K, Fiegler H, Beare DM, Collins JE, McCann OT, Young BD, Debernardi S, Mott R, Dunham I, Carter NP. Replication timing of the human genome. Hum Mol Genet. 2004;13:191–202. doi: 10.1093/hmg/ddh016. [DOI] [PubMed] [Google Scholar]
- Yang L, Bennetzen JL. Structure-based discovery and description of plant and animal Helitrons. Proc Natl Acad Sci U S A. 2009;106:12832–12837. doi: 10.1073/pnas.0905563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant SR, Wu XL, Huang Y, Garrison B, Burgess SM, Kay MA. High-resolution genome-wide mapping of transposon integration in mammals. Mol Cell Biol. 2005;25:2085–2094. doi: 10.1128/MCB.25.6.2085-2094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeileis A, Kleiber C, Jackman S. Regression models for count data in R. J Stat Softw. 2008;27:1–25. [Google Scholar]
- Zhao J, Bacolla A, Wang G, Vasquez K. Non-B DNA structure-induced genetic instability and evolution. Cell Mol Life Sci. 2010;67:43–105. doi: 10.1007/s00018-009-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo X, Rho M, Feschotte C. Genome-wide characterization of endogenous retroviruses in the bat Myotis lucifugus reveals recent and diverse infections. J Virol. 2013;87:8493–8501. doi: 10.1128/JVI.00892-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.