

Abstract
Marfan syndrome (MFS) is a genetic disorder of the connective tissue which rarely manifests in the neonatal period and has an ominous prognosis. A case of a first female offspring of healthy parents is described here. The pregnancy was uneventful and the mother had a term caesarean delivery. At birth, some dysmorphic signs became apparent, such as loose redundant skin, dolichocephaly, frontal bossing, deeply sunken eyes, micrognathia, contractures of the elbows, arachnodactyly and hip dysplasia. The echocardiogram showed a mitral and tricuspid valve regurgitation and a long aortic arch. The diagnosis of neonatal MFS came forward and genetic studies revealed a de novo mutation in the fibrillin 1 (FBN1) gene. At 6 months, due to a progressive worsening of the cardiac pathology, she was submitted to mitral valvuloplasty. She is now 2 years and 10 months old, which is a remarkable feat for a child suffering from this condition.
Background
The Marfan syndrome (MFS) is rarely present in the neonatal period, nevertheless, when so, it is usually associated with a very poor prognosis with the majority of affected infants dying from heart failure in the first 2 years of life. Although this severe form shares some features with the classic presentation there are some particular ones, such as severe skeletal problems with flexion contractures and arachnodactyly, congenital emphysema, severe cardiovascular disease (mitral's and/or tricuspid valve's relentlessly progressive regurgitation leading to congestive heart failure and often an enlarged aortic root) and loose redundant skin with characteristic ‘senile’ facial appearance. These features, manifested in the neonatal period, should raise the suspicion for MFS. It is a severe form of the disease with an ominous prognosis so, early diagnosis of this condition is essential for attempted treatment planning and prognosis modification.
We report a case of a child diagnosed with neonatal MFS (NMFS) during her first month of life and whose evolution has been remarkably positive, considering other paediatric cases described with the same disease.
Case presentation
We describe the case of a first female child of healthy, non-consanguineous young parents (22-year-old mother, height 158 cm; 29-year-old father, height 170 cm). There was no family history of hereditary diseases, including MFS and congenital heart disease.
It was a medically supervised and uneventful pregnancy with normal obstetric ultrasounds.
The child was delivered by caesarean section at 38 weeks due to pelvic position. Her birth weight was 3130 g (25th centile), length 46.5 cm (between 5th and 10th centile) and head circumference 35.5 cm (between 50th and 75th centile). The patient presented an Apgar score of 6 and 8 at 1 and 5 min, respectively. Physical examination revealed several dysmorphic features including frontal bossing, redundant skin folds, prematurely aged appearance, dolichocephaly, deeply set eyes and retromicrognathia (figure 1A, B). Additional findings included limited extension of the elbow joints, arachnodactyly (figure 2A), adduct contracture of thumbs, hindfoot deformity (figure 2B) and hip dysplasia. The chest was symmetric without pectus deformity.
Figure 1.
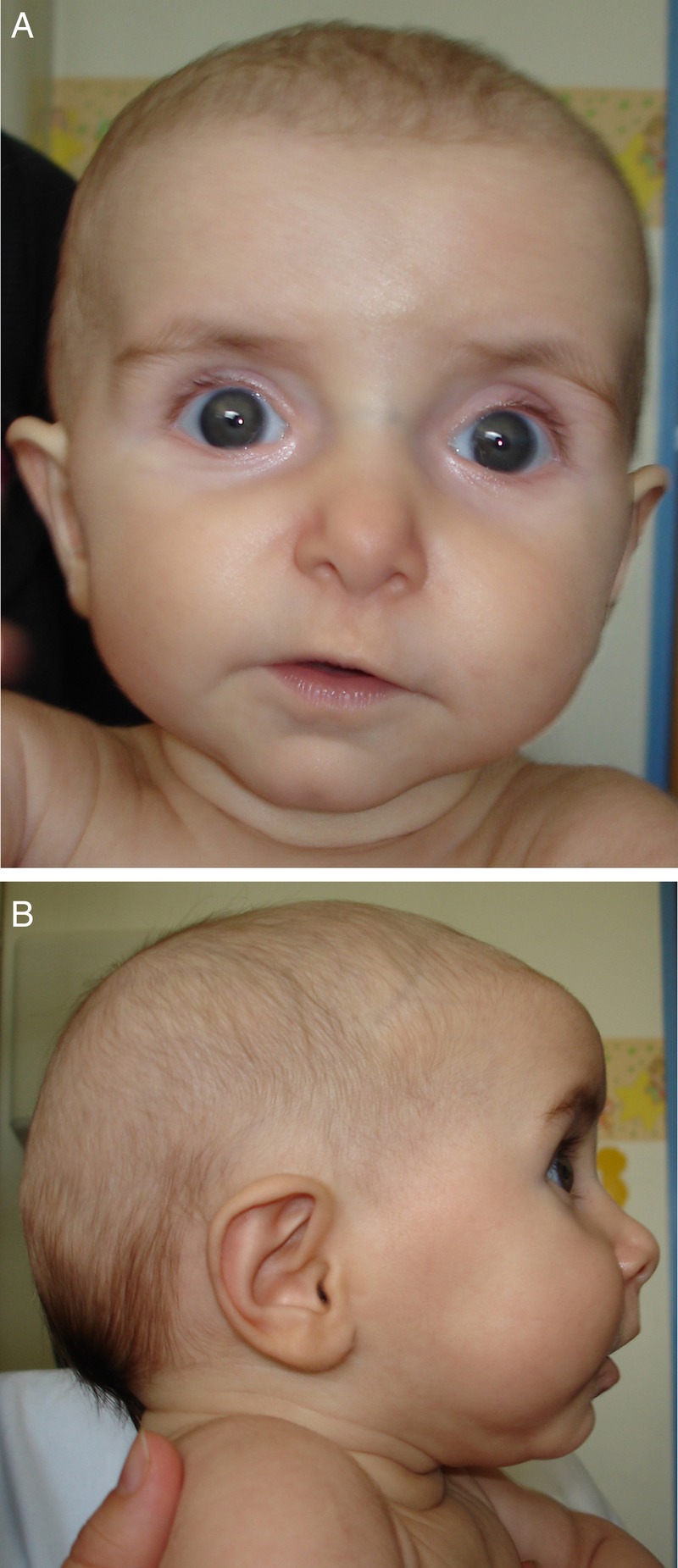
(A and B) Facial features at 4 months old. Note the frontal bossing, redundant skin folds, premature aged appearance, dolichocephaly, deeply set eyes and retromicrognathia.
Figure 2.
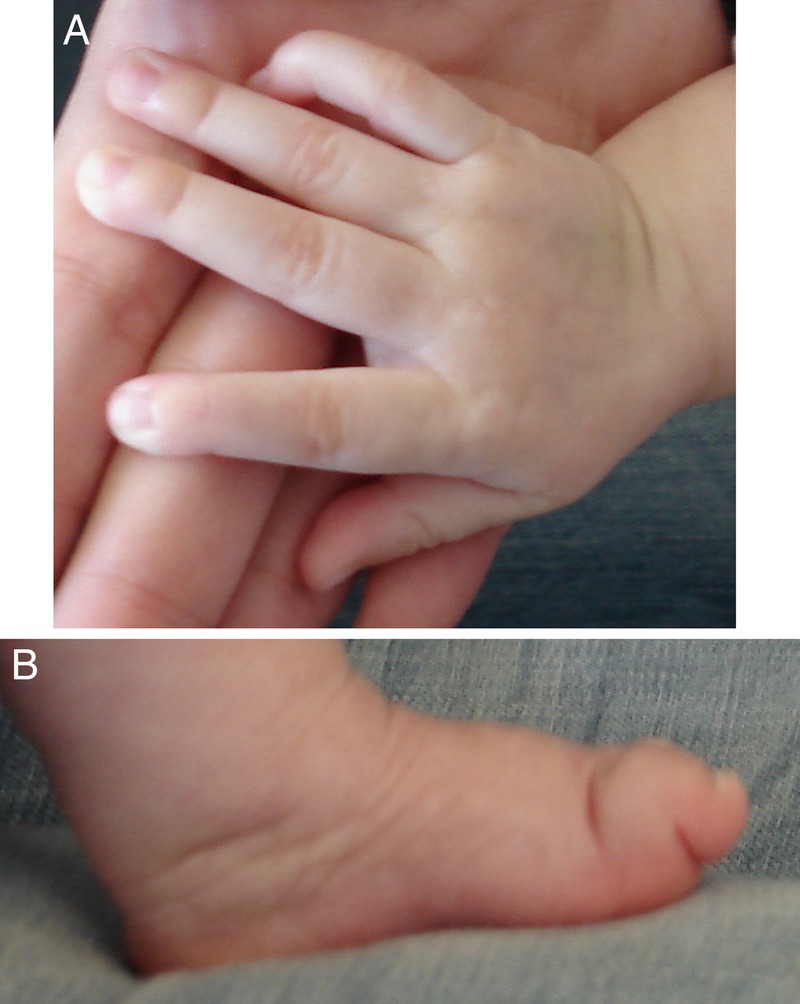
Neonatal Marfan syndrome features in the hand (A) and feet (B) at 4 months of age.
Investigations
She was observed by a paediatric orthopaedist, at 9 days of life, and, because of bilateral hip dysplasia with acetabular coverage less than 20%, she was placed on a Pavlik harness.
At her follow-up neonatology consultation, a holosystolic heart murmur (grade II/VI) was heard and at 26 days of life she had a paediatric cardiologist appointment. The ECG was normal and the echocardiogram that revealed an unusually high and long aortic arch and a mitral and tricuspid valve regurgitation with redundant leaflets.
Her first ophthalmological examination showed no lens dislocation. The chest X-ray, the ultrasound study of her kidneys and brain were normal. Owing to the failure to thrive (on caloric supplement intake) and vomiting; and a suspected paraoesophageal hernia (by ultrasound), she had a gastro-oesophageal endoscopy which was normal. This was confirmed on gastroenterology's follow by thoracic MRI. Chromosome study showed 46, XX.
The clinical signs and cardiovascular findings, lead to the suspicion of NMFS.
The genetic study, making use of molecular and sequencing techniques, showed a de novo mutation on the fibrillin 1 (FBN1) gene: c.3458G>A in exon 27 with an amino acid substitution of a cysteine by a tyrosine at position 1153 (p.Cys1153Tyr). Mutation analysis of exon 27 of both parents did not reveal the mutation present in their child.
Outcome and follow-up
At 4 months of age, due to the progressive worsening of fatigue and dyspnoea with failure to thrive and vomiting, she began treatment with losartan and furosemide. Nevertheless, there was a progressive aggravation of the mitral valve's prolapse and regurgitation with great dilation of the left atrium (14.5 cm2), left ventricle (internal diameter of 42 mm) and aortic root that conditioned cardiothoracic surgery (mitral valvuloplasty) at 6 months of age. There were some complications after surgery, including left pneumothorax, pericardial effusion and nosocomial sepsis (Pseudomonas aeruginosa and Staphylococcus aureus). She was discharged 31 days after surgery on furosemide, digoxin, spironolactone, losartan and carvedilol. At 9 months of age there was improvement of heart failure, allowing discontinuation of anticongestive medication while keeping losartan and carvedilol. From 1 to 6 months of life there was a rapid clinical progression with the instalment of subluxation of the lens, slightly blue sclerae, pectus excavatum and severe dorsal lumbar scoliosis. At 11 months of age, the Pavlik harness was removed (X-ray showed a hypoplastic left femoral head ossified nucleus without dislocation) but by 17 months a progressive scoliosis worsening led to the placement of a thoracolumbar plaster cast substituted by a Charleston bending brace due to complicating wounds. At 2.5 years of age, an orthosis for stabilisation of the deformations of the feet and ankles was placed. Follow-up is kept by a multidisciplinary team consisting of paediatric cardiology, paediatric ophthalmology, paediatric orthopaedics, paediatric gastroenterology, physical medicine and rehabilitation, nutrition and medical genetics. Presently, at 2 years and 10 months of age, she is clinically stable although with feeding difficulties, poor appetite and inadequate weight gain (weight 10.3 kg, 5th centile; length 90 cm, 25th centile and head circumference 51 cm, 50th centile; figure 3). She still has some vomiting episodes associated with acute intercurrences. She has an adequate psychomotor development for her age. The last echocardiogram, from June 2013, showed a mild mitral regurgitation, a moderate tricuspid regurgitation, a moderate dilation of all four chambers with good systolic function and a moderate dilation of the aortic root. Currently she is medicated with losartan and carvedilol.
Figure 3.
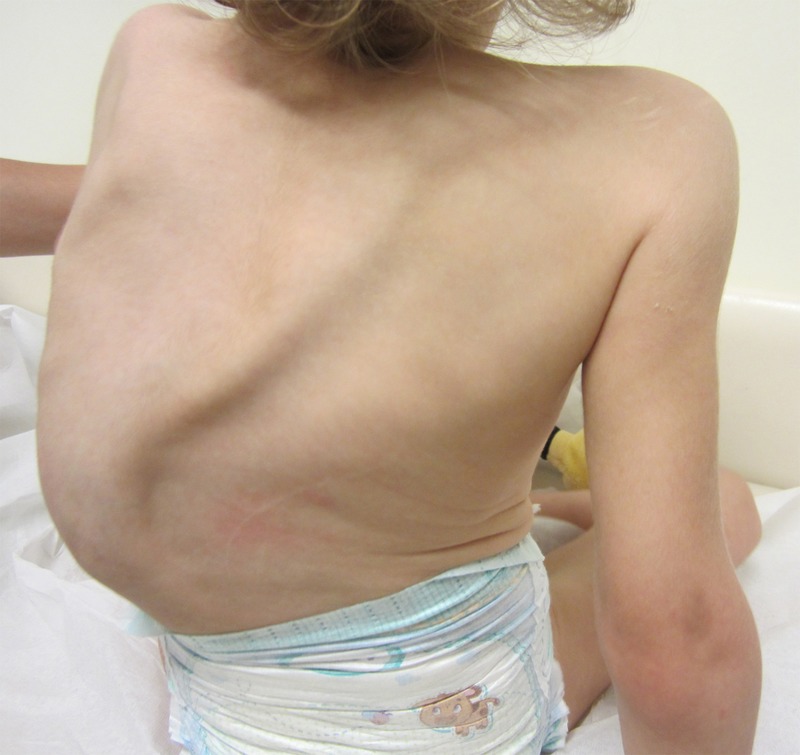
Scoliosis at 2 years and 10 months of age.
Discussion
MFS (MIM 154700), first described in 1896 by the French paediatrician Antoine Marfan, is an inherited autosomal dominant connective tissue disorder that shows significant pleiotropism and marked clinical variability. The three foremost systems affected by this condition are the cardiovascular, ocular and skeletal, being the cardinal manifestations, the aortic aneurysm with dissection, ectopia lentis and long bone overgrowth. This disease occurs at about 1 in 5000–10 000 individuals. Family history may be positive, but nearly 25% of cases arise from a de novo mutation.1 This syndrome is notable for its variable expression, namely in the severity of clinical manifestations, age at onset and targeted tissues, among and within affected families. It can range from the severest phenotype manifested in the neonatal period (NMFS) to a very mild clinical form. The diagnosis may be difficult and is based on typical clinical features and, when available, in a positive family history. Specific diagnostic criteria for the disorder have been established, being first promulgated in 1988 (the Berlin criteria) and updated in 1996 (the Ghent criteria).2 3 Owing to the penetrance of some features being age dependent, the Ghent criteria must be used with caution in children. MFS is caused by mutations in the FBN1 gene mapped on chromosome 15q21.1. This gene encodes the FBN1, a 320 kDa glycoprotein that is a main component of microfibrils in the extracellular matrix. To date, more than 2000 mutations have been identified in the FBN1 gene, most of which are unique to individual families. The same mutation can be associated with different phenotypes besides classic MFS, including isolated ectopia lentis (MIM 129 600), isolated ascending aortic aneurysm and dissection, isolated skeletal features and NMFS.4 Presently, no clear genotype–phenotype correlation has been established with the exception of the so-called ‘neonatal’ region in FBN1 exons 24–32 where mutations are associated with more severe and complete phenotypes, with poorer prognosis when compared with patients with a mutation located elsewhere. However, despite mutations in this location being associated with NMFS and atypically severe phenotypes in non-neonatal cases, mutations associated with classic MFS can also occur in this region. Although the location of the mutations in exon 24–32 appears to be a relevant factor for the severity of the phenotype in these patients, others such as the type of mutation or the involvement of modifier gene can also be important.5
The NMFS is a rare form of this disease having the severest phenotype and the worst prognosis. It may be manifested in the prenatal period and is characterised, at birth, by some other features beyond the ones it shares with the classic form. These include: flexion contractures, characteristic facial dysmorphology (crumpled ears, loose redundant skin and a characteristic ‘senile’ facial appearance), pulmonary emphysema and severe cardiovascular disease, rapidly progressive, which often includes mitral or tricuspid regurgitation and dilated aortic root. Skeletal manifestations such as arachnodactyly, dolichostenomelia and pectus deformities are typically present.6
The majority of these clinical manifestations are not unique to NMFS and may also be seen in the severe/lethal form of Beals-Hecht syndrome or congenital contractural arachnodactyly (CCA; MIM 121050). This form of CCA, in addition to the typical features of classical CCA (a connective tissue disorder characterised by multiple contractures, arachnodactyly, kyphoscoliosis, dolichostenomelia, crumpled ears and, less commonly, craniofacial abnormalities as dolichocephaly, micrognathia and frontal bossing) has cardiovascular and gastrointestinal abnormalities. Besides those overlapping features with NMFS there are some important differences that may be useful in the final differentiation of these severe forms of the two syndromes, namely: cardiovascular (mitral and tricuspid valve anomalies and aortic root dilation mainly in NMFS and congenital structural defects in CCA); skeletal abnormalities (scoliosis and vertebral anomalies mostly in CCA); gastrointestinal abnormalities (oesophageal atresia, duodenal atresia and intestinal malrotation in CCA) and finally the genetic test (mutation on the FBN2 gene located on chromosome 5q23-q31 in CCA). The correct diagnosis between both has important prognostic significance.7
The average life span of the patients with NMFS is severely decreased and is mostly related to the severity of cardiovascular disease. The primary cause of death is congestive heart failure secondary to mitral and tricuspid regurgitation, usually in the first year of life, in contrast to the classic MFS, where the mortality is more related to complications associated with aortic root disease (aortic dilation with aortic aneurism or dissection). The vast majority of FBN1 mutations in patients with NMFS are sporadic and located in exons 24–32.8
The confirmation of the syndrome and its mutation are important for genetic counseling, and prenatal diagnosis. During counselling the variability of the disease should be emphasised, particularly if there is an affected parent (the child may be more or less affected than the affected parent). In the sporadic cases, although neither parent carries this mutation, it should be stated that there is an estimated recurrence risk of 3–5% based on the possibility of some degree of germline mosaicism that was not detected by mutation screening on peripheral blood analysis.8
Learning points.
The neonatal form is a severe and rare presentation of Marfan syndrome (MFS) usually associated with a very poor prognosis.
Beyond the few features it shares with the classic presentation, it has some particular ones, such as severe skeletal problems with flexion contractures and arachnodactyly, congenital emphysema, severe cardiovascular disease (mitral's and/or tricuspid valve's relentlessly progressive regurgitation leading to congestive heart failure and often an enlarged aortic root) and loose redundant skin with characteristic ‘senile’ facial features. The presence of these features at birth should raise the suspicion of neonatal MFS.
In situations of suspected neonatal MFS it is important to differentiate from severe/lethal form of congenital contractural arachnodactyly as these two entities share overlapping features.
Early diagnosis of this severe form of the MFS allows a timely approach and a multidisciplinary follow-up. This way it is possible to identify early complications (as scoliosis, lens dislocation and cardiac abnormalities) with subsequent improvement of an ominous prognosis with high likelihood to cause mortality in infancy or early childhood, particularly by cardiac failure secondary to severe mitral regurgitation.
The genetic counseling should emphasise the clinical variability of the disease and the estimated recurrence risk of 3–5% on sporadic cases.
Footnotes
Contributors: MA was involved in the literature research and writing of the manuscript. MAC, RF and TL contributed to the review of the manuscript.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Stheneur C, Laffond C, Rioux S, et al. Recent progress in Marfan syndrome. Arch Pediatr 2012;19:551–5 [DOI] [PubMed] [Google Scholar]
- 2.Beighton P, de Paepe A, Danks D, et al. International nosology of heritable disorders of connective tissue. Am J Med Genet 1988;29:581–94 [DOI] [PubMed] [Google Scholar]
- 3.De Paepe A, Devereux RB, Dietz HC, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996;62:417–26 [DOI] [PubMed] [Google Scholar]
- 4.Faivre L, Collod-Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 2007;81:454–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faivre L, Collod-Beroud G, Callewaert B, et al. Clinical and mutation-type analysis from an international series of 198 probands with a pathogenic FBN1 exons 24–32 mutation. Eur J Hum Genet 2009;17:491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghandi Y, Zanjani KS, Mazhari-Mousavi SE, et al. Neonatal Marfan syndrome: report of two cases. Iran J Pediatr 2013;23:113–17 [PMC free article] [PubMed] [Google Scholar]
- 7.Jurko A, Jr, Krsiakova J, Minarik M, et al. Congenital contractural arachnodactyly (Beals-Hecht syndrome): a rare connective tissue disorder. Wien Klin Wochenschr 2013;125:288–90 [DOI] [PubMed] [Google Scholar]
- 8.Sutherell J, Zarate Y, Tinkle BT, et al. Novel fibrillin 1 mutation in a case of neonatal Marfan syndrome: the increasing importance of early recognition. Congenit Heart Dis 2007;2:342–6 [DOI] [PubMed] [Google Scholar]