

Abstract
Congenital malformations represent approximately 3 in 100 live births within the human population. Understanding their pathogenesis and ultimately formulating effective treatments is underpinned by knowledge of the events and factors that regulate normal embryonic development. Studies in model organisms, primarily in the mouse, the most prominent genetically tractable mammalian model, have equipped us with a rudimentary understanding of mammalian development from early lineage commitment to morphogenetic processes. In this way information provided by studies in the mouse can, in some cases, be used to draw parallels with other mammals, including human. Here we provide an overview of our current understanding of the general sequence of developmental events from early cell cleavages to gastrulation and axis extension occurring in human embryos. We will also review some of the rare birth defects occurring at these stages, in particular those resulting in conjoined twinning or caudal dysgenesis.
Keywords: embryo, development, human, mouse, twinning, conjoined twinning gastrulation, axis extension, primitive streak, tailbud, caudal dysgenesis, sirenomelia
Embryonic development is a long journey that in humans takes approximately 9 months. Each step needs to be carefully regulated to ensure normal development to term. In its first month, the human embryo develops from a single-cell zygote to an embryo of approximately 10mm in size. During this period the primordia of almost all the internal organs are established. Fundamental features of the body plan such as the formation of the three primary germ layers (ectoderm, mesoderm and endoderm), the establishment and elaboration of the axes (anterior-posterior, dorsal-ventral and left-right), as well as the morphogenetic events that will shape the embryo have to be properly executed and coordinated.
About 3% of all live born infants exhibit an obvious congenital anomaly. Half of them are detectable at birth, and most of the rest will become apparent during the first postnatal year. The major causes of abnormal development are genetic factors, such as mutations or chromosomal aberrations, but additional factors including environmental agents, such as drugs, viruses or hypoxia also have an effect. Unfortunately, we still do not know the etiology of more than half of the observed human congenital abnormalities, with up to a quarter likely to be caused by a combination of environmental and genetic factors (referred to as multifactorial inheritance). Severely malformed embryos are usually spontaneously aborted during the first weeks of development, whereas, live born infants affected by congenital anomalies may experience a challenging life. As such, a detailed understanding of the cellular and molecular events that regulate embryonic development is critical for unraveling the pathogenesis of these anomalies with the aim of offering treatment, either therapeutic or supportive.
Due to ethical and religious issues, as well as legal constraints surrounding experimentation with human embryos, very little is known about the mechanistic aspects that underlie early human development. Studies in different model organisms, primarily the mouse, the most prominent mammalian model, have helped formulate an understanding of early development, from lineage segregation to the morphogenetic processes that occur during these initial steps. Here we will review the sequence of events from early cleavage stages to early organogenesis in the human embryo. This description will be accompanied by the current genetic knowledge provided by studies in the mouse and will overview some of the rare birth defects occurring at these stages. It is worth pointing out that defects due to impaired gastrulation are exceedingly rare given the fundamental importance of this process to embryonic development as a whole. Moreover, even though we will discuss caudal dysgenesis and sirenomelia as congenital defects occuring during gastrulation and axial elongation, their etiology remains controversial.
PRE-IMPLANTATION EMBRYO DEVELOPMENT
Soon after fertilization, the zygote engages in a series of mitotic cell divisions, which are referred to as cleavages (Fig. 1B). Daughter cells, which are referred to as blastomeres and are encapsulated in a glycoproteinaceous egg shell, the zona pellucida, decrease in size at each division (Fig. 1C). The first cell divisions are near synchronous, whereas later on they become increasingly asynchronous. These events take place as the embryo travels from the oviduct, the site of fertilization, to the uterus. Since the embryo has yet to implant into the maternal uterus, this period of embryonic development is referred to as pre-implantation. The morula (derived from the Latin morus meaning mulberry) stage embryo, which comprises approximately 12–30 cells, arrives at the uterine cavity around the fourth day of development. At this time, the initially loosely attached blastomeres begin to reinforce their cell-cell junctions and increase their cell-cell contacts. This results in an increase in their adhesion such that blastomeres become tightly aligned against one another in a process called compaction. This change in adhesive properties is accompanied by the segregation and distinction of centrally located blastomeres, which are now referred to as the inner cell mass, from the external cells, which will form the trophoblast.
FIGURE 1. Events occurring the first two weeks of human development.

(A) Zygote. (B) Two-cell stage embryo. (C) Morula before compaction. (D) Blastocyst forms after compaction and formation of the blastocoel. The embryo segregates into epiblast and hypoblast. (E) Implantation starts at the end of the first week. The syncytiotrophoblast begins to invade the uterine wall. (F) The amniotic cavity appears. (G) Hypoblast cells invade the lining of the primary yolk sac. Extraembryonic reticulum forms between the hypoblast and cytotrophoblast. Extraembryonic mesoderm colonizes the reticulum. (H) The reticulum cavitates leaving the chorionic cavity. The extraembryonic mesoderm and underlying cytotrophoblast form the chorion.
At this point fluid from the uterine cavity enters the compacted morula through the zona pellucida and accumulates between the blastomeres of the inner cell mass. This fluid accumulation creates a cavity, at which point the embryo is referred to as a blastocyst (Fig. 1D). The inner cell mass (ICM), now compacted at one side of the cavity is surrounded by the outer cell mass or trophoblast, a single-cell epithelium. The ICM will give rise to the embryo-proper and some extra-embryonic tissues. Human embryonic stem cells or ES cells are derived from the pre-implatation ICM. Due to their therapeutic potential these highly proliferative pluripotent cells are currently the focus of intense research1, 2. On the other hand, the trophoblast will give rise mainly to the embryonic part of the placenta. Lineage allocation in the blastocyst has been extensively studied in the mouse embryo. It is believed that the main genetic regulators of the cell-lineage decisions such as Cdx2 and Pou5f1 (formerly known as Oct4) are also conserved in human embryos although working under a different timing3, 4.
By day 5 after fertilization, the zona pellucida has degenerated and the blastocyst hatches through areas of fracture. Shedding of the zona allows the blastocyst to grow by incorporating nutrients exuded from the uterine glands, and prepares it for implantation into the maternal uterus.
Right before implantation, cells in the inner cell mass segregate in two layers, the epiblast formed by tall columnar epithelial cells and the hypoblast or primary endoderm, composed by a cuboidal epithelium (Fig. 1D). This lineage specification occurs in the mouse embryo also prior to implantation. According to a three-step model of lineage segregation, first in the murine ICM, markers of both epiblast and primitive endoderm are coexpressed in the ICM cells. Later, mutually exclusive expression of the markers indicates lineage commitment. In mouse embryos, FGF signaling controls the process of lineage specification, whereas in human it is not the case5–7. The last step involves the physical segregation of both lineages, whereby the primitive endoderm cells end up on the blastocoelic surface, whereas the epiblast cells remain enclosed. Cell sorting involves multiple cell behaviors that are not fully understood8. In humans however, kinetics of lineage segregation seems to be different.
EMBRYO IMPLANTATION
At the end of the first week of development the embryo attaches to the endometrium, the uterine mucous membrane. As soon as the trophoblast adheres to the uterine wall it starts to proliferate. Only trophoblast cells that form the wall of the blastocyst retain their epithelial structure forming the cytotrophoblast (Fig. 1E). The rest undergo rapid division and through loss of their cellular membranes create a protoplasmatic mass called the syncytiotrophoblast. This highly invasive syncytium secretes enzymes that digest the surrounding tissue providing nutrients to the embryo and allowing it to embed further into the endometrium. By the 12th day of development, the syncytiotrophoblast has enclosed the blastocyst such that the conceptus is completely embedded in the endometrium (Fig. 1G). Proper crosstalk between maternal and embryonic tissues is required to achieve successful implantation and continued embryonic development. A variety of hormones, cytokines and growth factors are involved in this process (reviewed in 9).
At day 8 a cavity forms between cells of the epiblast, and is referred to as the amniotic cavity (Fig. 1F). The cell layer separating the new cavity from the cytotrophoblast and forming the lining of the cavity becomes the amniotic membrane. The amnion will expand steadily and ultimately will enclose the entire embryo and provide protection later in development (Fig. 1G–H). At the floor of the amniotic cavity, the disc-shaped embryo or embryonic disc, composed of two epithelia, epiblast and hypoblast, separates the amniotic cavity from the former blastocoel cavity, which is now referred to as the exocoelomic cavity or primary yolk sac (Fig. 1F). At the same time cells from the hypoblast start to migrate out to colonize the lining of the primary yolk sac. Once formed, a thick layer of extracellular matrix, the extraembryonic reticulum, is secreted between these endoderm cells and the cytotrophoblast (Fig. 1G). Later on, cells from the hypoblast and epiblast, forming the extraembryonic mesoderm, migrate and colonize the reticulum. At some point this reticulum breaks out leaving a fluid-filled cavity surrounding the amnion, embryonic disc and yolk sac that will become the chorionic cavity (Fig. 1H). The extraembryonic mesoderm and the underlying trophoblast layers form the chorion.
Twinning
Twins that originate from two zygotes are referred to as dizygotic or fraternal twins. Twins that arise from the splitting of a single zygote are referred to as monozygotic or identical twins. In human pregnancies, twins occur with an approximate frequency of 1 in 90 births10. In general, the number of multiple births in a population can be estimated using Hellin’s hypothesis, whereby twins occur once in every 89 births, triplets occur once in every 892, quadruplets once in every 893 and so forth11. However in recent decades, the overall rate of twinning in the Western world has increased considerably. This has been attributed mainly to the increase in the age of women having children, as well as the increasing use of fertility-enhancing therapies12.
The rate of dizygotic twins varies among human populations and has been linked to genetic factors13–15. On the other hand, the incidence of monozygotic twinning is constant throughout the world and is not influenced by race, nutrition or other factors. Even though there is no known cause for monozygotic twinning, there is general agreement that a teratogenic event occurring at a critical stage of pregnancy could induce splitting of the embryo16–19.
Dizygotic twins implant separately and develop separate fetal membranes, whereas monozygotic twins are genetically identical and may share one, some or all their fetal membranes. Dizygotic twins always have a dichorionic, diamniotic placenta, since each zygote produces its own trophoblast and ICM. The placentae can be separated or fused along a ridge of membranes. These membranes consist of four layers, two outer amnions and two layers of chorion in the middle and have no vascular connections across them.
Placentation of monozygotic twins depends on the time of embryo spliting. Division of the morula or the zona-intact blastocyst before the inner cell mass forms generates two genetically identical embryos that would implant separately. In such cases placentation will be dichorionic diamniotic (Fig. 2A). This group accounts for up to a third of monozygotic twins. If the inner cell mass within the blastocyst divides between the third and eighth day, the shared trophoblast will give rise to a single placenta and each ICM will generate an amnion of its own. This placentation will be monochorionic, diamniotic and represents up to two thirds of all monozygotic twins (Fig. 2B). If division occurs later than the eighth day, when the bilaminar disc and amnion have formed, twins will share a common amniotic sac in a monochorionic, monoamniotic placentation (Fig. 2C). This last group represents only a 2%–4% of monozygotic pregnancies. Splitting occurring beyond the 13th day of development results in incomplete separation of the embryo forming conjoined twins.
FIGURE 2. Uterine disposition of monozygotic twins.

(A) Dichorionic diamniotic twins develop from a single zygote divided before the inner cell mass forms. (B) Monochorionic diamniotic twins form when the inner cell mass divides between the third and eighth day of development. (C) Monochorionic monoamniotic twins develop from the division of the bilaminar disc.
Monochorionic placentation is associated with a higher rate of morbidity and perinatal loss than dichorionic pregnancies. Malformations in monochorionic twins would fall into three categories; first, being structural defects related to the twinning process; second, vascular disruptions caused by abnormal vascularization and third, constriction deformations16. Several conditions are unique for monochorionic pregnancies20. Twin-twin transfusion syndrome (TTTS) occurs in 15% to 25% MC pregnancies. It is related to the unequal distribution of blood flow due to the vascular connections of the placenta. As a result, one twin is overperfused, while the second is underperfused. The in utero death of a twin can also endanger the survival of the co-twin. Moreover, associated only to monoamniotic placentations, umbilical cord entanglements place a risk for the twins21. Thus, even though twinning does not represent a congenital condition per se, monochoryonic twinning may result in a higher risk of developmental defects and pregnancy complications.
Early and accurate determination of chorionicity is crucial for successful management of pregnancy. Determination of zygosity can be done presumptively by sex determination and examination of the number of placentas. Different imaging techniques such as sonography, initially, MRI later in development and CT to a lesser extent play an important role in the diagnosis and supervision of twin pregnancies22, 23
Conjoined twinning
Conjoined twinning is estimated to occur in 1:50,000 pregnancies. However, since many die in utero, are terminated or are stillborn, the final incidence is about 1:250,000 live births. Interestingly, there is a female predominance 3:1, although there is no proposed explanation for the fact. There are two theories that account for the pathogenesis, the fission and fusion theories. According to the classic and mostly accepted fission view, the splitting of the embryonic axis later than the 13th day of development would lead to two attached embryos. The newer fusion theory proposes the secondary union of two originally separate embryonic discs24.
As discussed previously, accurate determination of chorionicity is critical for pregnancy management and parental counseling. Major ultrasound findings that would suggest conjoined twinning include shared skin coverage between two embryos, fetuses that remain in the same relative positions, fetal scoliosis, extreme neck retroflexion in fetuses facing each other and unusual limb positioning25.
Conjoined twins are classified according to the major site of attachment followed by the suffix pagus (from the Greek, pagos, fixed). A simplified system proposed by Spencer limits the classification to 8 major types which are grouped into dorsal and ventral attachments26 (Table 1 and Fig. 3). Ventral attachments involve the sharing of the yolk sac or abdomen, while dorsal attachments would involve union at the neural tube and exclude the yolk sac and abdomen. Ventral unions comprise cephalopagus, thoracopagus, omphalopagus, ischiopagus and parapagus twins. Dorsal unions encompass craniopagus, pygopagus and rachipagus twins. According to the fusion theory, in ventrally conjoined twins the margins of two embryonic discs would fuse to form one shared umbilicus. Since they arise from a common yolk sac, they also share part of their gastrointestinal system and may involve the thoracic and/or abdominal cavities. Dorsally attached twins would be joined at any site of the neural tube before the neural folds fuse.
TABLE.
A classification of human conjoined twinning.
| Cephalopagus | Have a fused head and often a fused thorax. This category represents approximately 11% of all conjoined twins. Each twin has two arms, two legs and separate lower abdomen and pelvis. According to the fusion theory, they would be united at the oropharyngeal membrane. This category of twins is non-viable, and affected individuals often die in utero. |
| Thoracopagus | Have a fused thorax and shared cardiac anatomy from a single intercardiac vessel to a shared heart. They also display fused sternum, diaphragm, liver and pericardium. They may also have a common small intestine and biliary tree. They represent the major category of conjoined twinning ranging from 20% to 40%. Surgical separation is usually not an option due to complex cardiac anatomy. |
| Omphalopagus | Are fused at the umbilicus and display similar fusion patterns as thoracopagus but without a shared heart. Liver, intestine and biliary tree are shared to different extends. Their incidence ranges from 18% to 33%. From all conjoined twins, omphalopagus have the best chance of survival after surgical separation. |
| Ischiopagus | These twins share a pelvis. They may be oriented face-to-face, with lower abdominal union or end-to-end. They usually exhibit some degree of common genitourinary system. Depending on the number of legs they can be tetrapus (4), tripus (3) or bipus (2) and represent approximately 6–11% of all conjoined twins. According to the fusion theory, these twins are thought to be joined ventrally, at the cloacal membranes. |
| Parapagus | Are joined caudolaterally resulting in a joined abdomen, pelvis and lower limbs and different levels of thoracic and cranial union. They may have 2, 3 o 4 arms and 2 or 3 legs. Dicephalus parapagus have separated heads and faces. Diprosopus parapagus have two faces in a conjoined cranium. Surgical separation is rarely possible and most will die in utero or perinatally. |
| Craniopagus | Are united at any portion of the cranial vault but not including the face, the foramen magnum or the vertebrae. These twins commonly share the cranium, the meninges, the dural venous sinuses and less frequently, the brain itself. They can be subdivided into frontal, parietal, temporoparietal and occipital. Their incidence is 1:10. The extent of the sinuses and brain tissue shared will impact morbidity after surgical separation. Survival usually linked to mental or physical handicap. |
| Pygopagus | Are joined at the sacrum sharing the sacrococcygeal and perineal region, oriented back to back. Half of the cases share anus and rectum but intestines are usually separate. In a third of cases they have fused meninges and spinal cord and a low proportion display shared bladder and urethra. These twins represent an 18–28% of conjoined twins. Evaluation of the degree of spinal cord and sacrococcygeal bony anatomy sharing, and perineal and genitourinary malformations is fundamental before considering surgical separation. |
| Rachipagus | Are fused dorsally at midline, at spine. They lie back-to-back and share variable lengths of the spinal cord and column. The rarest type of twin. Surgical separation would be impossible, but since they are mostly parasites, the parasitic twin can be potentially removed. According to fusion theory, dorsally conjoined embryos derive from two embryonic discs fusing at the neural folds before they fuse. In the case of craniopagus twins, embryonic discs would have fused at the most rostral level; pygopagus would have fused at the caudal neuropore and rachipagus would have resulted from fusion at the midportion of the open neural tube. |
FIGURE 3. Classification of conjoined twins according to Spencer26.

Other types of anomalous fetuses have been proposed to be variations of abnormal conjoined twins27. This is the case for parasitic twins, fetuses in fetu, acardiacs and teratomas. A conjoined twin parasite is a grossly defective fetus externally fused to a relatively normal fetus (autosite). A fetus in fetu is a fetiform mass enclosed within the body of an autosite. As in parasites, brain and heart are usually vestigial or missing. Acardiacs is a fetiform mass that is entirely separate from the body of the autosite but connected to it by umbilical vessels. Teratomas consist of an unidentifiable mixture of tissues that are rarely differentiated. They are capable of independent growth and might be or become malignant. The origin of teratomas has also proposed to be ectopic primordial germ cells or remnants of the primitive streak.
GASTRULATION & GERM LAYER FORMATION
At the beginning of the third week the embryo adopts an oval shape. Around day 16 a lineal epiblast groove appears at the presumptive caudal part of the embryo, the primitive streak. It constitutes the first break in the symmetry of the embryo. Epiblast cells move along the epithelial sheet, converge at the medial primitive streak and after undergoing an epithelial to mesenchymal transition, they ingress and colonize the space between the epiblast and hypoblast28 (Fig. 4A). Some of the ingressing cells displace the hypoblast and replace it, giving rise to the definitive endoderm. The cells representing the middle layer of the embryo and occupying the space between the endoderm and the epiblast form the mesoderm. Epiblast cells that will remain on the surface at the end of the process will become the ectoderm. Therefore during this process, referred to as gastrulation, in which the three germ (or tissue) layers are established, the bilaminar disc becomes trilaminar. Once gastrulation is complete, the different progenitor cell populations have been allocated, allowing organogenesis to begin. Each germ layer gives rise to specific tissues and organs, even tough, as will be discussed below, lineage boundaries are becoming increasingly unclear. Ectodermal cells predominantly give rise to the epidermis, central and peripheral nervous systems and the retina. The mesoderm gives rise to the cardiovascular system, blood cells, bone marrow, smooth and striated muscles, skeleton, connective tissue, as well as the urogenital tract. The endoderm is the source of the epithelial lining of the respiratory and gastrointestinal tracts, and the glands of associated organs such as the lungs, liver or pancreas.
FIGURE 4. Mesoderm formation in the mammalian embryo.

(A) Cells in the epiblast undergo an epithelial to mesenchymal transition at the primitive streak and colonize the space between the epiblast and hypoblast. (B) Ingressing cells migrate in all directions. Cells ingressing through the node migrate anteriorly forming the notochord. Cells ingressing through the streak and migrating cranially contribute to the head and cardiac mesoderm. Epiblast cells migrating laterally form the mesoderm at each side of the midline. (C) The different mesodermal lineages are established along the mediolateral axis of the embryo.
At the rostral end of the streak, a depression surrounded by a crescent-shaped mound of cells appears forming a structure referred to as the node (Fig. 4B). Cells ingressing through the node and remaining in the midline of the embryo give rise to the notochordal process. The process extends to the rostral extremity of the embryo, the oral membrane, where epiblast and hypoblast are fused. The hollow process fuses with the underlying endoderm creating the notochordal plate. Beginning at the cranial end, notochordal cells proliferate and the plate invaginates to generate the notochord, which then detaches from the endoderm. The notochord, now a solid rod that provides rigidity to the embryo, plays an important role in the induction and specification of the overlying neural tube.
Cells ingressing through the streak migrate in all directions on either side of the midline (Fig. 4B). Mesodermal cells that reach the cranial part of the embryo become the cardiogenic mesoderm where the heart primordium will develop. The two bilateral columns of paraxial mesoderm originate from the mesoderm cells that after ingression settle on both sides of the notochord (Fig. 4C). The paraxial mesoderm will give rise to the somites, temporary rounded structures that will contribute to the dermis (underlying the skin), axial skeleton and musculature. Lateral to the paraxial mesoderm forms the intermediate mesoderm that generates the urinary system and parts of the genital system. The remainder of the lateral mesoderm forms the lateral plate mesoderm, which splits into splachnopleuric and somatopleuric mesoderm. Studies in mouse have shown that mesoderm progenitors in the epiblast, although regionalized in fate, are not restricted in potential 29; the different mesodermal lineages become specified and allocated according to the time and site of ingression at the primitive streak30.
On day 16, the primitive streak spans about half the length of the embryo. As the embryo grows and elongates, the streak regresses caudally and disappears at around day 26. Rarely, remnants of the streak persist in the region and give rise to a sacrococcygeal teratoma. Since they are derived from pluripotent cells from the primitive streak, these tumors contain a variety of tissue types. They are mostly benign and are surgically removed. The caudal eminence, a caudal axial mass of progenitor cells will contribute to axis elongation, once the streak has totally regressed.
Axis elongation
During gastrulation, extension of the anterior-posterior (AP) axis in vertebrate embryos is first achieved by cells derived from ingressing epiblast cells together with a resident stem cell population in the streak and node. However, elongation of the most caudal axis has been a more controversial issue. While some view it as a continuation of the mechanisms of gastrulation, the most accepted view considers elongation of the caudal part of the embryo a slightly different process. This view is supported by the observation that mutations in several genes affect specifically posterior development. Moreover, neurulation has been shown to differ along the AP axis. During primary neurulation, lateral ends of the neural plate elevate and the bilateral neural folds fuse to form the neural tube; whereas during secondary neurulation at the posterior region of the embryo, the neural tube forms by cavitation of the neural rod.
It has been shown in mouse embryos that once overt gastrulation is terminated, descendants of the streak and node progenitors come to reside at the chordoneural hinge (CNH), a structure that is continuous with the caudal end of the notochord and neural tube. This pool of progenitor cells derived from the organizer region serves as a source of cells for the neural tube, notochord and somites31, 32 (Fig. 5B). In human embryos, the CNH might correspond to what human embryologists refer to as caudal eminence. The exact nature of the CNH is poorly understood. To date, clonal lineage analyses in mouse and chick embryos have indicated the presence of long-term axial progenitors intermingled with more fate-restricted precursors in the streak, epiblast and CNH (for review see 33). Interestingly, these multipotent axial progenitors are able to give rise to neural tube, axial and paraxial mesoderm, challenging the established view of early lineage segregation within the embryo34.
FIGURE 5. Signals that regulate axial elongation in the mouse embryo.

(A) Molecular pathways acting at the posterior growth region and contributing to the antagonistic gradients of FGF and RA. (B) Mouse embryo at embryonic day 9. The chordoneural hinge (CNH) is located caudally and lies contiguous with the notochord and neural tube. (C) Noggin expression in the ventral ectodermal ridge (VER) controls the level of BMP signaling required for caudal ventral mesoderm morphogenesis.
Molecular pathways involved in axis elongation
Even though the exact molecular fingerprint of the axial progenitors remains elusive, several signaling pathways have been directly associated with extension of the embryonic AP axis. Primarily, the WNT, FGF and NOTCH pathways, which are believed to work in parallel to promote proliferation and maintain the undifferentiated state on the progenitors, and therefore, regulate axis elongation (Fig. 5A).
Several WNT genes are differentially expressed at the caudal part of the embryo35. Canonical WNT pathway is required for primitive streak formation and mesoderm induction in mouse embryos36. Later in development, the WNT pathway is critical for coordinating mesoderm formation and somitogenesis. It is not only involved in specifying streak identity and regulating mesoderm formation and migration but also governs segmentation of the paraxial mesoderm by controlling the molecular oscillator37. WNT pathway deficient mice exhibit axis truncations at different levels. Wnt3a mutant mice lack posterior somites and tail bud and display an ectopic ventral neural tube38. Wnt5a mutants have a shortened vertebral column with no caudal vertebrae and fused vertebrae39. Moreover, null mutations in Wnt related genes such as Lrp6 and Ctnnb1 also display posterior defects40, 41.
FGF signaling also plays a pivotal role in posterior morphogenesis. Fgf8 mutant mice die at early gastrulation42 and Fgfr1 deficient mice fail to undergo gastrulation43, 44. To overcome early lethality and inactivate all FGF acting at the streak, conditional alleles of Fgf4 and Fgf8 were crossed with the pan-mesodermal T-Cre line45. Compound mutant embryos show no expression of markers of undifferentiated mesoderm, whereas markers of nascent somite cell fate expand throughout the tail bud. Thus, deletion of FGF signaling leads to embryonic axis truncation and premature differentiation of the tail bud. Progenitor cells that would contribute to axis elongation differentiate prematurely assuming a somitic fate. Interestingly, restoration of WNT signaling, which is downregulated in these mutants, is not able to rescue premature differentiation and axis truncation.
The third pathway involved in axial progenitors maintenance is the NOTCH pathway. CBF1 (also known as Rbp-jk) knockout mice exhibit posterior defects and growth arrest, a similar but more servere phenotype than that displayed by Notch1 mutant embryos46–48. Resembling the CBF1 mutant phenotype, compound mutants for Presenilin 1/2 exhibit an undulating neural tube and lack of somites49. Moreover, ablation of Lunatic fringe causes axis truncation and posterior vertebral abnormalities50, 51.
Together with WNT, FGF and NOTCH pathways, several genes act to specify the nascent tissues. Usually, their expression is directly regulated by one of the three main pathways. Emergent mesoderm at the streak and node express the transcription factor Brachyury/T, which is downstream of WNT and FGF signaling52. T mutant embryos display axis truncations53. Also under WNT regulation, is the related transcription factor, Tbx6, which is expressed in the paraxial mesoderm. Loss of Tbx6 leads to a failure of paraxial mesoderm specification and concomitant formation of ectopic neural tubes in place of somites54, 55. Indeed, a recent report suggests that mesoderm specification is achieved by neural fate repression at the axial stem cells56.
In contrast to WNT and FGF signaling pathways, which are involved progenitor maintenance, retinoic acid (RA) signaling regulates cellular differentiation. Thus, elongation of the axis is controlled by two opposed gradients, RA and WNT/FGF57 (Fig. 5A). FGF/WNT posterior levels establish the undifferentiated growth zone, whereas anterior levels of RA promote differentiation of the precursors. Levels of RA signaling are established by the balance of its synthesizing and catabolizing enzymes58. Posterior expression of the RA degrading enzyme Cyp26a1, a cytochrome P450 oxidoreductase, which is downstream of FGF8 signaling, allows posterior axial cells to escape the differentiation signaling. Mutations of Cyp26a1, as well as mutations in Por, another cytochrome oxidoreductase, lead to axial truncations59–62. As the tissue elongates, progenitors leave the posterior growth zone and reach the competent zone. Here, inhibition of Aldh1a2, a RA synthesizing enzyme, by FGF8 is no longer effective and cells differentiate upon RA signaling63, 64. Termination of axial elongation is associated with shortening of the undifferentiated mesoderm due to downregulation of FGF/WNT levels at the tail bud around E12.531, 65, 66.
Finally, the role of the ventral ectodermal ridge (VER), an ectoderm thickening positioned on the ventral surface of the tail bud, remains unclear (Fig. 5C). While fate mapping of VER cells has revealed that VER descendants only contribute to the ectoderm and therefore, are not progenitor cells, VER ablation in mice disrupts somite formation and tail outgrowth. Studies suggest a model where VER signaling might sustain Noggin expression in the tail mesoderm thereby facilitating the maintenance of levels of BMP signaling necessary for tail outgrowth67, 68. However, little is currently known about how these various signaling pathways acting in caudal morphogenesis are integrated both temporally and spatially.
Caudal dysgenesis
Caudal dysgenesis (CD), also referred as caudal regression syndrome or sacral agenesis syndrome, is a rare congenital disorder that affects the development of the lower segment. The term of caudal regression syndrome was first coined by Duhamel in 1961 to encompass an array of sacrococcygeal malformations69. CD occurrence numbers range in the literature from 1 per 40,000 to 100,000 pregnancies70, 71. The primary defects of CD are agenesis or incomplete development of the lumbar vertebrae, sacrum and coccyx, hypoplastic lower limbs and anorectal and genitourinary dysgenesis. Depending on the severity of the condition, skeletal malformations affecting the sacrum, coccyx and lumbar vertebrae cause pelvic deformities and hip dislocation. Underdeveloped lower limbs might display malformed knees, absent fibula, equinovarus or calcaneovarus. Infants usually present a “frog-leg” position of the lower extremities. Anorectal malformations often involve displaced or imperforate anus. Urogenital anomalies comprise malformed or fused kidneys, absent bladder and absent or undescended external genitalia. Lower spine defects include myelomeningoceles or open spinal defects. Lower spinal disruption can compromise limb mobility and bladder and bowel continence. Different classifications have been proposed to grade CD according to its severity72, 73. Commonly, sacral agenesis is classified into 4 clinic categories known as the Renshaw classification and each type correlate to ambulatory and other motor function abilities74, 75. Type I involves partial or total unilateral sacral agenesis, whereas Type II involves partial bilateral, symmetrical sacral agenesis. They are usually associated to coccyx agenesis, represent the mild from of sacral agenesis and have a better prognosis. Type III displays total sacral agenesis with variable lumbar anomaly and iliac wings attached to the last lumbar vertebrae. Finally, Type IV involves total sacral agenesis with different levels of lumbar anomaly and iliac wing fissioned behind the last vertebrae, if present. Infants with type III and IV have a worse prognosis and perinatal death is frequent. Infants with CD are usually of normal intelligence. Abnormalities in other systems are rarely associated with this syndrome but might include congenital heart defects and facial clefts76.
Even though the sacrum is not completely ossified at the time, transvaginal ultrasound has allowed in utero diagnosis as early as 11 weeks77. Definitive prenatal diagnosis of CD by ultrasound is possible at 22 weeks of gestation, although sonographic findings are variable and depend on the severity of the defects78–80. Prognosis depends on the severity of spinal and urogenital damage. If vital systems are unaffected, lethality is generally attributed to renal failure resulting from renal malformations. Survivors usually require neurologic, orthopedic and renal interventions as well as long-term attention.
Sirenomelia
Sirenomelia is a very rare disorder, in which fusion of the lower extremities causes the fetus to resemble a mermaid. Other severe defects such as urogenital and gastrointestinal malformations are commonly associated with the disorder. The reported incidence in the population ranges in the literature from 1 per 60,000 to 1 per 100,00081, 82 and has a male to female bias of 2.7 : 1.
Limb fusions exhibit a spectrum of severity. Stocker and Heifetz proposed a classification of sirenomelia into 7 types based on the presence or absence of bony elements83 (Fig. 6). In type I, the mildest form, all bones are present and the fusion only affects superficial tissues. Type II displays fused fibulae, in type III fibulae are absent, type IV involves partially fused femur and fused fibulae and type V, partially fused femurs. Type VI sirenomelia displays single femur and single tibia whereas in type VII, the most severe form, only one bone is present.
FIGURE 6. Classification of sirenomelia by the presence or absence of bones in the lower limb according to Stocker and Heifetz83.
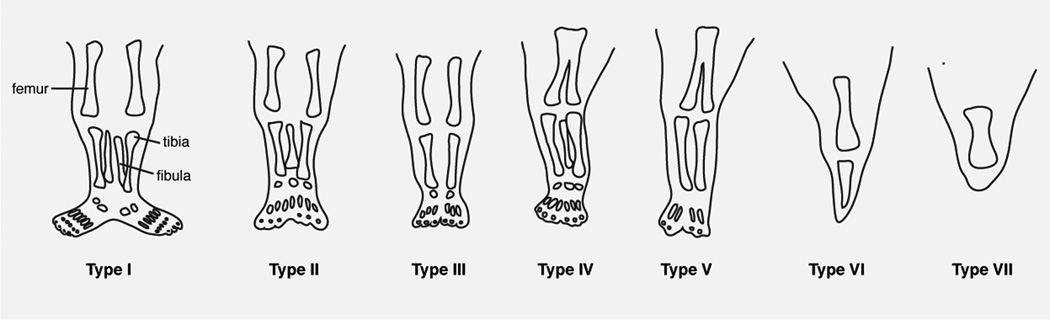
Adapted from 83 with permission.
Urogenital defects comprise renal and urethral dysplasia, with a high incidence of renal agenesis. External genitalia are also frequently absent, whereas gonads seem unaffected. Gastrointestinal defects include colon atresia and imperforate anus83, 84. Lumbosacral and pelvic malformations are usually present. Moreover, affected fetuses often display a single umbilical artery instead of the usual two. This vessel, derived from the vitelline artery, has an abnormal origin high in the abdominal cavity, branching from the aorta, and diverts blood that would normally supply the lower part of the body to the placenta. Sporadically, sirenomelic fetuses display an affected upper body, such as cleft palate, thoracic and cervical vertebral dysplasias, cardiac defects and pulmonary hypoplasia.
Early sonographic diagnostic of sirenomelia may be suggested by bilateral renal agenesis, hypoplastic or fused lower limbs, single umbilical cord and oligohydramnios. Decreased amniotic fluid is a clear sonographic marker of renal malfunction from the second half of the pregnancy onwards85. Extremely poor prognosis is due to the visceral abnormalities, principally to renal failure. Only a few long-term survival cases have been reported owing to an exceptionally functioning kidney86, 87.
Etiology of caudal dysgenesis and sirenomelia
Etiology of CD and sirenomelia are still widely debated. CD has been proposed to be a consequence of abnormal posterior mesoderm development. An embryological insult would take place before the 4th week of development, when the caudal structures of the embryo are forming. The phenotypic variability would depend on the intensity and temporal frame of the insult. Even though genetic factors, maternal diabetes and hypoperfusion have been suggested as probable causes, the nature of the insult remains unclear. CD occurs in up to 1% of pregnancies of diabetic mothers. However, since only 22% of CD patients have diabetic mothers80, 88, CD is not only a diabetes specific condition. Interestingly, sirenomelia is rarely associated with maternal diabetes83.
Moreover, there is no widespread consensus whether sirenomelia and CD are different entities or sirenomelia only represents the most severe form of CD. The presence of the aberrant umbilical vessel in all sirenomelic fetuses supports the idea of a different origin of the disorder. The vascular steal theory suggests that the steal vessel would divert blood from the aorta to the placenta resulting in a hypoplastic aorta, severe hypoperfusion of the lower body and impaired development89, 90. Furthermore, sirenomelia usually involves fusion of the lower limbs, not only hypoplasia as in CD and it is always associated to severe urinary tract malformations. However, an aberrant abdominal artery has also been described in some CD patients91.
The majority of the cases are sporadic, being possible that every case represents an autosomal dominant condition caused by a new spontaneous mutation. In humans, only the Currarino syndrome, a congenital caudal anomaly has been linked to a mutation in the homeobox gene MNX1 (formerly known as HLXB992). The Currarino syndrome is described as the association of a triad of caudal malformations: sacral bony abnormality, presacral mass and anorectal malformation93. However, no link has been found between CD and MNX194.
Even though reports of human sirens are known since ancient times, in the mouse, caudal abnormalities and mermaid-like phenotypes have been described only recently95. In contrast with human cases, mice phenotypes have a known genetic basis. Mice heterozygous for the Brachyury/T locus (short-tail strain) display a short tail and sacral malformations96. In human patients, no clear evidence has been found that directly correlates the T locus to neural tube defects (reviewed in 97). Only recently, new studies argue in favor of an association of the T locus to spina bifida and vertebral malformations98, 99. However, to date no correlation between CD or sirenomelia to the T locus has been reported.
Recent work by van de Ven and colleagues describes the need of combined action of CDX genes, persistent WNT signaling and timely activation of HOX genes for proper posterior development100. Alteration of any of these parameters causes axial growth arrest by affecting the posterior growth zone in mice. Nevertheless, further studies are needed to establish a correlation between this genetic interaction and human CD.
Proper RA signaling is crucial for caudal development. Excess administration of RA to pregnant mice lead to caudal malformations similar to human CD and sirenomelia in most of the survivors101 102. Moreover, maternal diabetes seems to increase the teratogenic effect of RA103. Acting on the same pathway, loss of the RA degrading enzyme Cyp26a1 also leads to axial truncations59, 60, 104. However, a mutational screening of the CYP26A1 gene in CD patients did not provide any correlation between them105.
Another signaling pathway associated with sirenomelia in mouse is the BMP pathway. Bmp7/Twsg double mutants invariably display sirenomelia106. Twsg (twisted gastrulation) encodes for a BMP regulator that either promotes or inhibits BMP signaling. While single mutants are unaffected, compound mutants are embryonic lethal and exhibit sirenomelia; indicating that, whereas loss of Bmp7 might be compensated by other BMP ligands, additional loss of one or two copies of Twsg drops the level of BMP signaling below the threshold necessary for proper morphogenesis. Also Noggin overexpression at the caudal end of chicken embryos occasionally causes a fused limb phenotype as the most severe phenotype67, suggesting that BMP signaling is crucial for caudal ventral mesoderm formation. Bmp7/Twsg compound mutants show a reduction of the posterior Fgf8-expressing region and tail bud mesoderm expressing T. Unpublished data from Otha and colleagues suggest an increase in apoptosis in these mutants. In addition, they also argue for a decrease of the number of ingressing cells through the VER due to reduced levels of BMP as another possible reason for the severe phenotype. Therefore, in the mouse, sirenomelia appears in some cases to be associated with a defect in ventroposterior mesoderm formation. In humans, however, there is no report to date that correlates defective BMP signaling with CD or sirenomelia.
Even though Sparrow and colleagues’ report that the etiology of congenital scoliosis is not directly related to CD, their work suggests a mechanism whereby a genetic risk factor combined with an environmental condition affects the function of the posterior growth zone and somitogenesis causing vertebral malformations107. They show that haplo-insufficiency of NOTCH signaling together with short-term gestational hypoxia, which would disrupt FGF signaling, significantly increases the penetrance and severity of vertebral anomalies in mice. It is tempting to speculate that a similar environmental insult could also affect the phenotypic penetrance of genetically-susceptible embryos in CD.
In conclusion, even though the etiology of CD and sirenomelia remain elusive, the phenotypic similarites between these human congenital anomalies and the above described mouse phenotypes point to similar genetic factors. It is tempting to assume that an early embryonic insult that would interfere specifically with the normal formation of the posterior structures during axial elongation would be sufficient to elicit these caudal malformations.
ACKNOWLEDGEMENTS
We thank Ann Foley and members of our laboratory for discussions and comments on this review. Our work is supported by the National Institutes of Health (RO1-HD052115 and RO1-DK084391), and NYSTEM the New York State Stem Cell Science initiative (C024318).
Footnotes
RESOURCES
For further reading about early human development
Moore KL, Persaud TVN. The developing human: clinically oriented embryology. Saunders, 2003, 560
http://embryology.med.unsw.edu.au/embryo.htm (accessed November 30, 2011)
REFERENCES
- 1.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.Yu J, Thomson JA. Pluripotent stem cell lines. Genes Dev. 2008;22:1987–1997. doi: 10.1101/gad.1689808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossant J, Tam PP. Blastocyst lineage formation, early embryonic asymmetries and axis patterning in the mouse. Development. 2009;136:701–713. doi: 10.1242/dev.017178. [DOI] [PubMed] [Google Scholar]
- 4.Cockburn K, Rossant J. Making the blastocyst: lessons from the mouse. J Clin Invest. 2010;120:995–1003. doi: 10.1172/JCI41229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lanner F, Rossant J. The role of FGF/Erk signaling in pluripotent cells. Development. 2010;137:3351–3360. doi: 10.1242/dev.050146. [DOI] [PubMed] [Google Scholar]
- 6.Roode M, Blair K, Snell P, Elder K, Marchant S, Smith A, Nichols J. Human hypoblast formation is not dependent on FGF signalling. Dev Biol. 2012;361:358–363. doi: 10.1016/j.ydbio.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuijk EW, van Tol LT, Van de Velde H, Wubbolts R, Welling M, Geijsen N, Roelen BA. The roles of FGF and MAP kinase signaling in the segregation of the epiblast and hypoblast cell lineages in bovine and human embryos. Development. 2012;139:871–882. doi: 10.1242/dev.071688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plusa B, Piliszek A, Frankenberg S, Artus J, Hadjantonakis AK. Distinct sequential cell behaviours direct primitive endoderm formation in the mouse blastocyst. Development. 2008;135:3081–3091. doi: 10.1242/dev.021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh M, Chaudhry P, Asselin E. Bridging endometrial receptivity and implantation: network of hormones, cytokines, and growth factors. J Endocrinol. 2011;210:5–14. doi: 10.1530/JOE-10-0461. [DOI] [PubMed] [Google Scholar]
- 10.Benirschke K, Masliah E. The placenta in multiple pregnancy: outstanding issues. Reprod Fertil Dev. 2001;13:615–622. doi: 10.1071/rd01037. [DOI] [PubMed] [Google Scholar]
- 11.Hellin D. Die Ursache der Multiparität der uniparen Tiere überhaupt und der Zwillingsschwangerschaft beim Menschen insbesondere. München: Seitz und Schauer. 1895 [Google Scholar]
- 12.Martin JA, Park MM. Trends in twin and triplet births: 1980–97. Natl Vital Stat Rep. 1999;47:1–16. [PubMed] [Google Scholar]
- 13.Bulmer M. The biology of twinning in man. Oxford (UK): Claredon Press; 1970. [Google Scholar]
- 14.Nylander PP. The factors that influence twinning rates. Acta Genet Med Gemellol (Roma) 1981;30:189–202. doi: 10.1017/s0001566000007650. [DOI] [PubMed] [Google Scholar]
- 15.Busjahn A, Knoblauch H, Faulhaber HD, Aydin A, Uhlmann R, Tuomilehto J, Kaprio J, Jedrusik P, Januszewicz A, Strelau J, et al. A region on chromosome 3 is linked to dizygotic twinning. Nat Genet. 2000;26:398–399. doi: 10.1038/82515. [DOI] [PubMed] [Google Scholar]
- 16.Schinzel AA, Smith DW, Miller JR. Monozygotic twinning and structural defects. J Pediatr. 1979;95:921–930. doi: 10.1016/s0022-3476(79)80278-4. [DOI] [PubMed] [Google Scholar]
- 17.Kaufman MH, O'Shea KS. Induction of monozygotic twinning in the mouse. Nature. 1978;276:707–708. doi: 10.1038/276707a0. [DOI] [PubMed] [Google Scholar]
- 18.Ferm VH. Congenital malformations in hamster embryos after treatment with vinblastine and vincristine. Science. 1963;141:426. doi: 10.1126/science.141.3579.426. [DOI] [PubMed] [Google Scholar]
- 19.Stockard C. Developmental rate and structural expression: an experimental study of twins, "double monsters" and single deformities, and the interaction among embryonic organs during their origin and development. Am J Anat. 1921;28:115–277. [Google Scholar]
- 20.Trevett T, Johnson A. Monochorionic twin pregnancies. Clin Perinatol. 2005;32:475–494. doi: 10.1016/j.clp.2005.02.007. viii. [DOI] [PubMed] [Google Scholar]
- 21.Dias T, Mahsud-Dornan S, Bhide A, Papageorghiou AT, Thilaganathan B. Cord entanglement and perinatal outcome in monoamniotic twin pregnancies. Ultrasound Obstet Gynecol. 2010;35:201–204. doi: 10.1002/uog.7501. [DOI] [PubMed] [Google Scholar]
- 22.Monteagudo A, Roman AS. Ultrasound in multiple gestations: twins and other multifetal pregnancies. Clin Perinatol. 2005;32:329–354. doi: 10.1016/j.clp.2005.02.006. vi. [DOI] [PubMed] [Google Scholar]
- 23.McHugh K, Kiely EM, Spitz L. Imaging of conjoined twins. Pediatr Radiol. 2006;36:899–910. doi: 10.1007/s00247-006-0121-6. quiz 1002–1003. [DOI] [PubMed] [Google Scholar]
- 24.Spencer R. Theoretical and analytical embryology of conjoined twins: part I: embryogenesis. Clin Anat. 2000;13:36–53. doi: 10.1002/(SICI)1098-2353(2000)13:1<36::AID-CA5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Winkler N, Kennedy A, Byrne J, Woodward P. The imaging spectrum of conjoined twins. Ultrasound Q. 2008;24:249–255. doi: 10.1097/RUQ.0b013e31818c8858. [DOI] [PubMed] [Google Scholar]
- 26.Spencer R. Anatomic description of conjoined twins: a plea for standardized terminology. J Pediatr Surg. 1996;31:941–944. doi: 10.1016/s0022-3468(96)90417-0. [DOI] [PubMed] [Google Scholar]
- 27.Spencer R. Parasitic conjoined twins: external, internal (fetuses in fetu and teratomas), and detached (acardiacs) Clin Anat. 2001;14:428–444. doi: 10.1002/ca.1079. [DOI] [PubMed] [Google Scholar]
- 28.Ferrer-Vaquer A, Viotti M, Hadjantonakis AK. Transitions between epithelial and mesenchymal states and the morphogenesis of the early mouse embryo. Cell Adh Migr. 2010;4:447–457. doi: 10.4161/cam.4.3.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beddington SP. An autoradiographic analysis of the potency of embryonic ectoderm in the 8th day postimplantation mouse embryo. J Embryol Exp Morphol. 1981;64:87–104. [PubMed] [Google Scholar]
- 30.Kinder SJ, Tsang TE, Quinlan GA, Hadjantonakis AK, Nagy A, Tam PP. The orderly allocation of mesodermal cells to the extraembryonic structures and the anteroposterior axis during gastrulation of the mouse embryo. Development. 1999;126:4691–4701. doi: 10.1242/dev.126.21.4691. [DOI] [PubMed] [Google Scholar]
- 31.Cambray N, Wilson V. Two distinct sources for a population of maturing axial progenitors. Development. 2007;134:2829–2840. doi: 10.1242/dev.02877. [DOI] [PubMed] [Google Scholar]
- 32.Cambray N, Wilson V. Axial progenitors with extensive potency are localised to the mouse chordoneural hinge. Development. 2002;129:4855–4866. doi: 10.1242/dev.129.20.4855. [DOI] [PubMed] [Google Scholar]
- 33.Wilson V, Olivera-Martinez I, Storey KG. Stem cells, signals and vertebrate body axis extension. Development. 2009;136:1591–1604. doi: 10.1242/dev.021246. [DOI] [PubMed] [Google Scholar]
- 34.Tzouanacou E, Wegener A, Wymeersch FJ, Wilson V, Nicolas JF. Redefining the progression of lineage segregations during mammalian embryogenesis by clonal analysis. Dev Cell. 2009;17:365–376. doi: 10.1016/j.devcel.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi T. Genetics of Wnt signaling during early mammalian development. Vol. 468. Totowa, NY: Elizabeth Vincan; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A. Requirement for Wnt3 in vertebrate axis formation. Nat Genet. 1999;22:361–365. doi: 10.1038/11932. [DOI] [PubMed] [Google Scholar]
- 37.Dunty WC, Jr, Biris KK, Chalamalasetty RB, Taketo MM, Lewandoski M, Yamaguchi TP. Wnt3a/beta-catenin signaling controls posterior body development by coordinating mesoderm formation and segmentation. Development. 2008;135:85–94. doi: 10.1242/dev.009266. [DOI] [PubMed] [Google Scholar]
- 38.Takada S, Stark KL, Shea MJ, Vassileva G, McMahon JA, McMahon AP. Wnt-3a regulates somite and tailbud formation in the mouse embryo. Genes Dev. 1994;8:174–189. doi: 10.1101/gad.8.2.174. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi TP, Bradley A, McMahon AP, Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–1223. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- 40.Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- 41.Aulehla A, Wiegraebe W, Baubet V, Wahl MB, Deng C, Taketo M, Lewandoski M, Pourquie O. A beta-catenin gradient links the clock and wavefront systems in mouse embryo segmentation. Nat Cell Biol. 2008;10:186–193. doi: 10.1038/ncb1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999;13:1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- 44.Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev. 1994;8:3032–3044. doi: 10.1101/gad.8.24.3032. [DOI] [PubMed] [Google Scholar]
- 45.Naiche LA, Holder N, Lewandoski M. FGF4 and FGF8 comprise the wavefront activity that controls somitogenesis. Proc Natl Acad Sci U S A. 2011;108:4018–4023. doi: 10.1073/pnas.1007417108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oka C, Nakano T, Wakeham A, de la Pompa JL, Mori C, Sakai T, Okazaki S, Kawaichi M, Shiota K, Mak TW, et al. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 1995;121:3291–3301. doi: 10.1242/dev.121.10.3291. [DOI] [PubMed] [Google Scholar]
- 47.de la Pompa JL, Wakeham A, Correia KM, Samper E, Brown S, Aguilera RJ, Nakano T, Honjo T, Mak TW, Rossant J, et al. Conservation of the Notch signalling pathway in mammalian neurogenesis. Development. 1997;124:1139–1148. doi: 10.1242/dev.124.6.1139. [DOI] [PubMed] [Google Scholar]
- 48.Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–1545. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- 49.Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang N, Gridley T. Defects in somite formation in lunatic fringe-deficient mice. Nature. 1998;394:374–377. doi: 10.1038/28625. [DOI] [PubMed] [Google Scholar]
- 51.Evrard YA, Lun Y, Aulehla A, Gan L, Johnson RL. lunatic fringe is an essential mediator of somite segmentation and patterning. Nature. 1998;394:377–381. doi: 10.1038/28632. [DOI] [PubMed] [Google Scholar]
- 52.Yamaguchi TP, Takada S, Yoshikawa Y, Wu N, McMahon AP. T (Brachyury) is a direct target of Wnt3a during paraxial mesoderm specification. Genes Dev. 1999;13:3185–3190. doi: 10.1101/gad.13.24.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herrmann BG, Labeit S, Poustka A, King TR, Lehrach H. Cloning of the T gene required in mesoderm formation in the mouse. Nature. 1990;343:617–622. doi: 10.1038/343617a0. [DOI] [PubMed] [Google Scholar]
- 54.Chapman DL, Papaioannou VE. Three neural tubes in mouse embryos with mutations in the T-box gene Tbx6. Nature. 1998;391:695–697. doi: 10.1038/35624. [DOI] [PubMed] [Google Scholar]
- 55.Hofmann M, Schuster-Gossler K, Watabe-Rudolph M, Aulehla A, Herrmann BG, Gossler A. WNT signaling, in synergy with T/TBX6, controls Notch signaling by regulating Dll1 expression in the presomitic mesoderm of mouse embryos. Genes Dev. 2004;18:2712–2717. doi: 10.1101/gad.1248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takemoto T, Uchikawa M, Yoshida M, Bell DM, Lovell-Badge R, Papaioannou VE, Kondoh H. Tbx6-dependent Sox2 regulation determines neural or mesodermal fate in axial stem cells. Nature. 2011;470:394–398. doi: 10.1038/nature09729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diez del Corral R, Olivera-Martinez I, Goriely A, Gale E, Maden M, Storey K. Opposing FGF and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extension. Neuron. 2003;40:65–79. doi: 10.1016/s0896-6273(03)00565-8. [DOI] [PubMed] [Google Scholar]
- 58.Niederreither K, Dolle P. Retinoic acid in development: towards an integrated view. Nat Rev Genet. 2008;9:541–553. doi: 10.1038/nrg2340. [DOI] [PubMed] [Google Scholar]
- 59.Sakai Y, Meno C, Fujii H, Nishino J, Shiratori H, Saijoh Y, Rossant J, Hamada H. The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001;15:213–225. doi: 10.1101/gad.851501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abu-Abed S, Dolle P, Metzger D, Beckett B, Chambon P, Petkovich M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001;15:226–240. doi: 10.1101/gad.855001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ribes V, Otto DM, Dickmann L, Schmidt K, Schuhbaur B, Henderson C, Blomhoff R, Wolf CR, Tickle C, Dolle P. Rescue of cytochrome P450 oxidoreductase (Por) mouse mutants reveals functions in vasculogenesis, brain and limb patterning linked to retinoic acid homeostasis. Dev Biol. 2007;303:66–81. doi: 10.1016/j.ydbio.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 62.Otto DM, Henderson CJ, Carrie D, Davey M, Gundersen TE, Blomhoff R, Adams RH, Tickle C, Wolf CR. Identification of novel roles of the cytochrome p450 system in early embryogenesis: effects on vasculogenesis and retinoic Acid homeostasis. Mol Cell Biol. 2003;23:6103–6116. doi: 10.1128/MCB.23.17.6103-6116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Molotkova N, Molotkov A, Sirbu IO, Duester G. Requirement of mesodermal retinoic acid generated by Raldh2 for posterior neural transformation. Mech Dev. 2005;122:145–155. doi: 10.1016/j.mod.2004.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shum AS, Poon LL, Tang WW, Koide T, Chan BW, Leung YC, Shiroishi T, Copp AJ. Retinoic acid induces down-regulation of Wnt-3a, apoptosis and diversion of tail bud cells to a neural fate in the mouse embryo. Mech Dev. 1999;84:17–30. doi: 10.1016/s0925-4773(99)00059-3. [DOI] [PubMed] [Google Scholar]
- 65.Gomez C, Ozbudak EM, Wunderlich J, Baumann D, Lewis J, Pourquie O. Control of segment number in vertebrate embryos. Nature. 2008;454:335–339. doi: 10.1038/nature07020. [DOI] [PubMed] [Google Scholar]
- 66.Tenin G, Wright D, Ferjentsik Z, Bone R, McGrew MJ, Maroto M. The chick somitogenesis oscillator is arrested before all paraxial mesoderm is segmented into somites. BMC Dev Biol. 2010;10:24. doi: 10.1186/1471-213X-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ohta S, Suzuki K, Tachibana K, Tanaka H, Yamada G. Cessation of gastrulation is mediated by suppression of epithelial-mesenchymal transition at the ventral ectodermal ridge. Development. 2007;134:4315–4324. doi: 10.1242/dev.008151. [DOI] [PubMed] [Google Scholar]
- 68.Goldman DC, Martin GR, Tam PP. Fate and function of the ventral ectodermal ridge during mouse tail development. Development. 2000;127:2113–2123. doi: 10.1242/dev.127.10.2113. [DOI] [PubMed] [Google Scholar]
- 69.Duhamel B. From the Mermaid to Anal Imperforation: The Syndrome of Caudal Regression. Arch Dis Child. 1961;36:152–155. doi: 10.1136/adc.36.186.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Entezami M, Albig M, Knoll U, Gasiorek-Wiens A, Becker R. Ultrasound diagnosis of fetal anomalies: Thieme. 2003 [Google Scholar]
- 71.Boulas MM. Recognition of caudal regression syndrome. Adv Neonatal Care. 2009;9:61–69. doi: 10.1097/ANC.0b013e31819de44f. quiz 70–61. [DOI] [PubMed] [Google Scholar]
- 72.Welch JP, Aterman K. The syndrome of caudal dysplasia: a review, including etiologic considerations and evidence of heterogeneity. Pediatr Pathol. 1984;2:313–327. doi: 10.3109/15513818409022263. [DOI] [PubMed] [Google Scholar]
- 73.Cama A, Palmieri A, Capra V, Piatelli GL, Ravegnani M, Fondelli P. Multidisciplinary management of caudal regression syndrome (26 cases) Eur J Pediatr Surg. 1996;6(Suppl 1):44–45. [PubMed] [Google Scholar]
- 74.Renshaw TS. Sacral agenesis. J Bone Joint Surg Am. 1978;60:373–383. [PubMed] [Google Scholar]
- 75.Van Buskirk CS, Ritterbusch JF. Natural history of distal spinal agenesis. J Pediatr Orthop B. 1997;6:146–152. doi: 10.1097/01202412-199704000-00011. [DOI] [PubMed] [Google Scholar]
- 76.Singh SK, Singh RD, Sharma A. Caudal regression syndrome--case report and review of literature. Pediatr Surg Int. 2005;21:578–581. doi: 10.1007/s00383-005-1451-4. [DOI] [PubMed] [Google Scholar]
- 77.Cullier F, Charpentier A, M'Lamali H, Colbert R. Jarcho-Levin syndrome with caudal regression. Available at: http://www.thefetus.net.
- 78.Gonzalez-Quintero VH, Tolaymat L, Martin D, Romaguera RL, Rodriguez MM, Izquierdo LA. Sonographic diagnosis of caudal regression in the first trimester of pregnancy. J Ultrasound Med. 2002;21:1175–1178. doi: 10.7863/jum.2002.21.10.1175. [DOI] [PubMed] [Google Scholar]
- 79.Nagy GR, Csapo Z, Barakonyi E, Nagy B, Rigo J., Jr Prenatal diagnosis and fetopathological investigation of dorsolumbosacral agenesis. Pathol Res Pract. 2009;205:490–493. doi: 10.1016/j.prp.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 80.Stroustrup Smith A, Grable I, Levine D. Case 66: caudal regression syndrome in the fetus of a diabetic mother. Radiology. 2004;230:229–233. doi: 10.1148/radiol.2301020942. [DOI] [PubMed] [Google Scholar]
- 81.Martinez-Frias ML, Cucalon F, Urioste M. New case of limb body-wall complex associated with sirenomelia sequence. Am J Med Genet. 1992;44:583–585. doi: 10.1002/ajmg.1320440510. [DOI] [PubMed] [Google Scholar]
- 82.Johnson B. Sirenomelia (Mermaid Fetus). Br. J. Clin. Pract. 1966;20:12. [Google Scholar]
- 83.Stocker JT, Heifetz SA. Sirenomelia. A morphological study of 33 cases and review of the literature. Perspect Pediatr Pathol. 1987;10:7–50. [PubMed] [Google Scholar]
- 84.Thottungal AD, Charles AK, Dickinson JE, Bower C. Caudal dysgenesis and sirenomelia-single centre experience suggests common pathogenic basis. Am J Med Genet A. 2010;152A:2578–2587. doi: 10.1002/ajmg.a.33599. [DOI] [PubMed] [Google Scholar]
- 85.van Zalen-Sprock MM, van Vugt JM, van der Harten JJ, van Geijn HP. Early second-trimester diagnosis of sirenomelia. Prenat Diagn. 1995;15:171–177. doi: 10.1002/pd.1970150211. [DOI] [PubMed] [Google Scholar]
- 86.Pinette MG, Hand M, Hunt RC, Blackstone J, Wax JR, Cartin A. Surviving sirenomelia. J Ultrasound Med. 2005;24:1555–1559. doi: 10.7863/jum.2005.24.11.1555. [DOI] [PubMed] [Google Scholar]
- 87.Murphy JJ, Fraser GC, Blair GK. Sirenomelia: case of the surviving mermaid. J Pediatr Surg. 1992;27:1265–1268. doi: 10.1016/0022-3468(92)90270-h. [DOI] [PubMed] [Google Scholar]
- 88.Zaw W, Stone DG. Caudal Regression Syndrome in twin pregnancy with type II diabetes. J Perinatol. 2002;22:171–174. doi: 10.1038/sj.jp.7210614. [DOI] [PubMed] [Google Scholar]
- 89.Stevenson RE, Jones KL, Phelan MC, Jones MC, Barr M, Jr, Clericuzio C, Harley RA, Benirschke K. Vascular steal: the pathogenetic mechanism producing sirenomelia and associated defects of the viscera and soft tissues. Pediatrics. 1986;78:451–457. [PubMed] [Google Scholar]
- 90.Twickler D, Budorick N, Pretorius D, Grafe M, Currarino G. Caudal regression versus sirenomelia: sonographic clues. J Ultrasound Med. 1993;12:323–330. doi: 10.7863/jum.1993.12.6.323. [DOI] [PubMed] [Google Scholar]
- 91.Duesterhoeft SM, Ernst LM, Siebert JR, Kapur RP. Five cases of caudal regression with an aberrant abdominal umbilical artery: Further support for a caudal regression-sirenomelia spectrum. Am J Med Genet A. 2007;143A:3175–3184. doi: 10.1002/ajmg.a.32028. [DOI] [PubMed] [Google Scholar]
- 92.Ross AJ, Ruiz-Perez V, Wang Y, Hagan DM, Scherer S, Lynch SA, Lindsay S, Custard E, Belloni E, Wilson DI, et al. A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet. 1998;20:358–361. doi: 10.1038/3828. [DOI] [PubMed] [Google Scholar]
- 93.Currarino G, Coln D, Votteler T. Triad of anorectal, sacral, and presacral anomalies. AJR Am J Roentgenol. 1981;137:395–398. doi: 10.2214/ajr.137.2.395. [DOI] [PubMed] [Google Scholar]
- 94.Merello E, De Marco P, Mascelli S, Raso A, Calevo MG, Torre M, Cama A, Lerone M, Martucciello G, Capra V. HLXB9 homeobox gene and caudal regression syndrome. Birth Defects Res A Clin Mol Teratol. 2006;76:205–209. doi: 10.1002/bdra.20234. [DOI] [PubMed] [Google Scholar]
- 95.Gluecksohn-Schoenheimer S, Dunn L. Sirens, aprosopi and intestinal abnormalities in the mouse mouse. Anat Rec. 1945;92:13. [Google Scholar]
- 96.Gluecksohn-Schoenheimer S. The Development of Two Tailless Mutants in the House Mouse. Genetics. 1938;23:573–584. doi: 10.1093/genetics/23.6.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boyles AL, Hammock P, Speer MC. Candidate gene analysis in human neural tube defects. Am J Med Genet C Semin Med Genet. 2005;135C:9–23. doi: 10.1002/ajmg.c.30048. [DOI] [PubMed] [Google Scholar]
- 98.Ghebranious N, Blank RD, Raggio CL, Staubli J, McPherson E, Ivacic L, Rasmussen K, Jacobsen FS, Faciszewski T, Burmester JK, et al. A missense T (Brachyury) mutation contributes to vertebral malformations. J Bone Miner Res. 2008;23:1576–1583. doi: 10.1359/jbmr.080503. [DOI] [PubMed] [Google Scholar]
- 99.Carter TC, Pangilinan F, Troendle JF, Molloy AM, VanderMeer J, Mitchell A, Kirke PN, Conley MR, Shane B, Scott JM, et al. Evaluation of 64 candidate single nucleotide polymorphisms as risk factors for neural tube defects in a large Irish study population. Am J Med Genet A. 2011;155A:14–21. doi: 10.1002/ajmg.a.33755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.van de Ven C, Bialecka M, Neijts R, Young T, Rowland JE, Stringer EJ, Van Rooijen C, Meijlink F, Novoa A, Freund JN, et al. Concerted involvement of Cdx/Hox genes and Wnt signaling in morphogenesis of the caudal neural tube and cloacal derivatives from the posterior growth zone. Development. 2011;138:3451–3462. doi: 10.1242/dev.066118. [DOI] [PubMed] [Google Scholar]
- 101.Padmanabhan R. Retinoic acid-induced caudal regression syndrome in the mouse fetus. Reprod Toxicol. 1998;12:139–151. doi: 10.1016/s0890-6238(97)00153-6. [DOI] [PubMed] [Google Scholar]
- 102.Kessel M. Respecification of vertebral identities by retinoic acid. Development. 1992;115:487–501. doi: 10.1242/dev.115.2.487. [DOI] [PubMed] [Google Scholar]
- 103.Chan BW, Chan KS, Koide T, Yeung SM, Leung MB, Copp AJ, Loeken MR, Shiroishi T, Shum AS. Maternal diabetes increases the risk of caudal regression caused by retinoic acid. Diabetes. 2002;51:2811–2816. doi: 10.2337/diabetes.51.9.2811. [DOI] [PubMed] [Google Scholar]
- 104.Pennimpede T, Cameron DA, MacLean GA, Li H, Abu-Abed S, Petkovich M. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res A Clin Mol Teratol. 2010;88:883–894. doi: 10.1002/bdra.20709. [DOI] [PubMed] [Google Scholar]
- 105.De Marco P, Merello E, Mascelli S, Raso A, Santamaria A, Ottaviano C, Calevo MG, Cama A, Capra V. Mutational screening of the CYP26A1 gene in patients with caudal regression syndrome. Birth Defects Res A Clin Mol Teratol. 2006;76:86–95. doi: 10.1002/bdra.20225. [DOI] [PubMed] [Google Scholar]
- 106.Zakin L, Reversade B, Kuroda H, Lyons KM, De Robertis EM. Sirenomelia in Bmp7 and Tsg compound mutant mice: requirement for Bmp signaling in the development of ventral posterior mesoderm. Development. 2005;132:2489–2499. doi: 10.1242/dev.01822. [DOI] [PubMed] [Google Scholar]
- 107.Sparrow DB, Chapman G, Smith AJ, Mattar MZ, Major JA, O'Reilly VC, Saga Y, Zackai EH, Dormans JP, Alman BA, et al. A mechanism for gene-environment interaction in the etiology of congenital scoliosis. Cell. 2012;149:295–306. doi: 10.1016/j.cell.2012.02.054. [DOI] [PubMed] [Google Scholar]