

Abstract
Background: Although accumulating data has implicated a role for the Apelinergic system in cirrhosis, the role of Apelin during different stages of fibrogenesis has not been clarified, whereas no studies have been conducted on its expression in human hepatocellular carcinoma (HCC). This study aimed to elucidate its role in hepatic remodelling and carcinogenesis. Methods: Immunolocalization of Apelin was compared during different stages of HCV-induced liver disease (n = 123). Results: Apelin level in hepatic stellate cells (HSC), portal fibroblast and endothelial cells was significantly elevated in F3 stage .In cirrhosis, the expression was markedly striking and extended as linear staining in the cirrhotic septa and proliferated capillaries. In liver cirrhosis with high grade dysplastic nodule (HGDN) group and liver cirrhosis with HCC group, Apelin was constantly expressed in the hepatocytes with the exemption of non-parenchymatous cells. Apelin gradually increased in HGDN, HCC grade-I and HCC grade II (P < 0.0001). In contrast, Apelin gradually decreased in the cirrhosis adjacent to HGDN, HCC grade-I and HCC grade II (P < 0.0001). The gradual incline in Apelin expression in dysplastic and malignant cells was paralleled by a decline in their adjacent cirrhotic liver (P = 0.013). Conclusion: In HCV chronic hepatitis, Apelin seems to manipulate the differentiation of HSC ending in hepatic remodelling. The uptake of Apelin from the stromal components by the epithelial cells may initiate the transformation of adjacent epithelial cells and supports the evolution and progression of dysplastic nodules and hepatocellular carcinoma. These findings could have both prognostic and therapeutic applications.
Keywords: Apelin, HCV, cirrhosis, hepatocellular carcinoma, high grade dysplasia
Introduction
Apelin is the cognate ligand of the G protein-coupled receptor APJ [1]. Apelin and APJ mRNA are expressed in numerous rodent and human tissues [2] in the central nervous system and peripheral tissues [3,4]. Consensus supports a putative role for the Apelin-APJ system in fostering several physiological processes, pathological conditions and neoplastic growth. It is involved in the physiological regulation of the cardiovascular system, blood pressure, fluid homeostasis, vessel formation, hypothalamic-pituitary-adrenal axis and bone physiology. On the other hand, the apelinergic system shares in the pathophysiology of heart failure, hypertension, obesity, glucose intolerance, diabetes mellitus type 2, diabetes insipidus, gastric ulcer and osteoporosis [2,5,6]. Moreover, Apelin was found to be expressed in one-third of human tumours, such as colonic, skin and pancreatic cancers, malignant gliomas [7,8] as well as oral squamous cell carcinoma [9]. Its overexpression escalates tumour growth in a murine breast-cancer model [7].
Recently, accumulating data has implicated a role for the Apelinergic system in cirrhosis in rats [10] and human livers [11,12]. It has been linked to the hemodynamic disturbance and its complications in cirrhosis [10,11]. However, the role of Apelin during different stages of fibrogenesis has not been clarified whereas no studies have analysed its expression in human hepatocellular carcinoma (HCC). Thus, this study was designed to elucidate its role in hepatic remodelling and carcinogensis. Chronic hepatitis-C virus (HCV) patients pose the highest incidence toward evolution of HCC [13,14], especially in cirrhosis [15]. Thereby, HCV patients are the best model to study the fibrosis-carcinoma sequence. Accordingly, this study immunolocalized Apelin in the different stages of chronic HCV liver disease.
Materials and methods
Tissue samples
A total of 123 hepatic tissue samples of HCV cases were selected from the archives of Ain Shams University Hospitals and Ain Shams Specialized Hospital, Cairo, Egypt. Selection was done based on tissue availability (i.e. biopsies longer than 15 mm with at least 6 portal tracts and sufficient amount of tumour as well as adjacent nontumorous tissue with no/minimal necrosis). All selected cases, as indicated in their medical records, were HCV RNA positive, genotype 4. Cases with the following conditions were excluded from the study: the presence of other liver diseases, hepatitis B virus and/or HIV co-infection, alcohol consumption and radiofrequency ablation or trans-arterial chemoembolization of HCC. The studied cohort were segregated into four groups: Group I: chronic hepatitis (n = 43); Group II: liver cirrhosis (n = 18); Group III: high grade dysplastic nodules (HGDN) with adjacent liver cirrhosis (n = 12) and Group IV: hepatocellular carcinoma (HCC) with adjacent liver cirrhosis (n = 50). Five normal liver biopsies served as control. Sample collection was performed according to the guidelines of the Ethics committee of the Pathology Department, Ain Shams University.
Histopathological evaluation
All available slides (H&E and Masson trichrome stained) of each case were reviewed. Histological evaluation was independently performed by two well-trained liver pathologists. Fibrosis stage and necroinflammatory activity were assessed according to the METAVIR system [16]. Accordingly, fibrosis was staged on a scale of F0 to F4 (F0 = no fibrosis, F1 = portal fibrosis without septa, F2 = few septa, F3 = numerous septa without cirrhosis, and F4 = cirrhosis). Activity was graded on a scale of A0 to A3 (A0 = no activity, A1 = minimal activity, A2 = moderate activity, A3 = severe activity). Presence or absence of hepatic steatosis was also assessed. The criteria for diagnosis of HGDN and HCC were referenced from the World Health Organization and International Consensus Group for Hepatocellular Neoplasia guidelines [17,18]. Accordingly, HCC grading was assigned well-differentiated (G1) and moderately to poorly differentiated (G2/G3).
Immunohistochemistry
Apelin primary rabbit polyclonal antibody (Biorhythm, orb77287, 1:200 diluition, Cambridge, UK) was used. Immunohistochemical stains were performed on sections from paraffin blocks using a streptavidin-biotin complex immunoperoxidase technique following the manufacturer’s instruction. Briefly, paraffin embedded tissue sections (5-μm thick) were deparaffinised in xylene and rehydrated through graded alcohol. Sections were immersed in citrate buffer solution (pH 6.0), heated in a pressure cooker and microwaved for 20 minutes. After cooling at room temperature, sections were incubated in 3% hydrogen peroxide for 30 minutes to block endogenous peroxidase activity and followed by normal bovine serum for 30 minutes to block nonspecific antibody binding. The sections were incubated with the primary antibody overnight at 4°C. After repeated wash and incubation with biotin-conjugated secondary antibody, sections reacted with streptavidin-peroxidase (Dako, Carpinteria, California, USA). Finally, sections were incubated with DAB chromogen and counterstained with Mayer’s haematoxylin. To ensure antibody specificity, negative controls were performed by omitting the primary antibody. Moreover, negative controls were used to indicate the level of background staining intensity, when present.
Evaluation of Apelin immunohistochemical results
Distribution pattern and level of expression were separately evaluated in non-parenchymatous cells and hepatocytes. Analysis was independently performed by two pathologists, using computerized Image Analyzing Software (Special SIS starter. version 3.2, Olympus, Germany) connected to an Olympus microscope (model BX51, Olympus Japan). Inter-observer agreement was about 93%. Re-examination of the cases was done in consequence by both pathologists to resolve any disagreement.
Expression of Apelin in non-parenchymatous cells
Non-parenchymatous cells included sinusoidal endothelial cells/hepatic stellate cells (SEC/HSC), portal fibroblast and endothelial cells. The mean percentages of positive cells in five microscopic fields (× 200) was scored, according to the modification of a previously reported method [19], on a scale of 0-4 (0 = negative; 1 = 1-10%; 2 = 11-30%; 3 = 31-70%; 4 ≥ 70%).
Expression of Apelin in hepatocytes
Immunoreactivity for Apelin was separately scored in ballooned hepatocytes in chronic hepatitis and cirrhosis, dysplastic cells in HGDN and malignant cells in HCC. Analysis was modified from a previously reported method [9] according to the extent and relative intensity of staining. Immunoreactivity was graded on a scale of 0 to 4 (0 = negative; 1 = focal & weak, 2 = focal & strong, 3 = diffuse & weak, 4 = diffuse & strong).
Statistical analysis
Continuous variables were expressed as mean and standard deviation (± SD). Categorical variables were expressed as frequencies and percents. Kruskal-Wallis test was used to compare an ordinal variable between more than two study groups. Mann-Whitney test was used to compare an ordinal variable between two study groups. Categorical variables were compared using the chi-square test or Fisher exact test. Linear Correlation and Regression were used as appropriate. P-Value (level of significance) was assigned > 0.05 as non-significant and < 0.05 as Significant. Vassar Statswas used for data analysis and results were confirmed using IBM SPSS statistics (V. 21.0, IBM Corp., USA, 2012).
Results
Clinicopathologic features
This study included 123 cases with hepatitis C virus (HCV) related liver disease. Cases were divided into four groups. Chronic hepatitis group (n = 43, 34.9%) consisted of 40 (93.1%) males and 3 (6.1%) females and their ages ranged from 18 to 64 years old (median 36). Liver cirrhosis group (n = 18, 14.6%) consisted of 15 (83.4%) males and 3 (16.6%) females and their ages ranged from 43 to 66 years old (median 56). HGDN with liver cirrhosis group (n = 12, 9.7%) consisted of 9 (75%) males and 3 (25%) females with ages ranging from 46 to 66 years old (median 57). HCC with liver cirrhosis group (n= 50, 40.6%) consisted of 43 (86%) males and 7 (14%) females with ages ranging from 48 to 66 years old (median 55). The control group (n = 5) consisted of 4 (80%) males and 1 (20%) females and their age ranged from 28 to 49 years old (median 34). There were no significant differences between the studied cohorts with regard to gender. However, significant differences were observed for patients’ age (P < 0.0001).
In chronic hepatitis group, the fibrosis stages were (METAVIR) F1, F2 and F3 in 8 (18.6%), 15 (34.9%) and 20 (46.5%) cases, respectively. Whereas, the cirrhosis group (18 cases) was F4 stage. The necroinflammatory activity grades were A1, A2 and A3 in 9 (14.8%), 22 (36.1%) and 30 (49.1%) cases, respectively. Microvesicular and macrovesicular steatosis was identified in 11/61 (18%) cases, 8/11 (72.7%) cases had chronic hepatitis and 3/11 (27.3%) patients had cirrhosis. The grade of HCC differentiation was grade I and II-III in 16 (32%) and 34 (68%) cases, respectively.
Apelin expression in inflammation-fibrosis axis
Pattern of Apelin expression was noted in normal liver, chronic hepatitis group and liver cirrhosis group (Figure 1). In normal liver, Apelin was undetectable. In contrast, Apelin was induced in HCV chronic liver disease. It was localized mainly in non-parenchymatous cells [i.e. sinusoidal endothelial cells/hepatic stellate cells (SEC/HSC), myofibroblasts, endothelial cells]. As HCV-infected livers progressed from chronic hepatitis through cirrhosis, Apelin positive cells extended as linear staining in the cirrhotic fibrous septa and endothelial cells of proliferated capillaries. Notably, this expression on the proliferated capillaries in cirrhosis was absent in all chronic hepatitis and normal livers.
Figure 1.

Apelin expression in chronic hepatitis and liver cirrhosis: (A) Chronic hepatitis showing Apelin expression in hepatic stellate cells (HSC) with exemption of fibrotic septa and hepatocytes, (B) In Metavir F1, Apelin was slightly expressed, (C) In F3, there was significant increase in Apelin positive HSC, (D, E) In cirrhosis, Apelin extended as linear staining in the cirrhotic septa and proliferated capillaries, (F) Apelin expressed in hepatocytes exhibiting steatosis. [Immunohistochemistry, original magnification, (A) × 200; (B, C, E, F) × 400, (D) × 100].
In normal liver, chronic hepatitis and cirrhosis, Apelin was barely expressed in the hepatocytes. However, when expressed, this expression was heterogeneous and was associated with steatosis regardless of the stage of the disease.
In chronic hepatitis group and cirrhosis group, Apelin was expressed in non-parenchymatous cells more frequently than in hepatocytes [35/61 (57.3%) versus 12/61 (19.6%), respectively, P = 0.00001].
Expression in non-parenchymatous cells
Although Apelin expression in non-parenchymatous cells was more frequently noted in F3 stage (11/20, 55%) compared to F1 stage (1/8, 12.5%) and F2 stage (5/15, 33.3%), no statistical difference existed between the three groups (P = 0.093). Overall, Apelin was expressed in 39.5% (17/43) and 100% of chronic hepatitis group and liver cirrhosis, respectively (P = 0.00003). In the positive chronic hepatitis group, non-parenchymatous cells labelled positive were 10% (i.e. score 1) in all cases (17/17, 100%), whereas, in liver cirrhosis group, non-parenchymatous cells labelled positive were > 10% (i.e. score 2 & 3) in 88.8% (16/18) of the cases, (P < 0.0001).
Regarding level of Apelin expression, compared to normal liver, Apelin was significantly induced in HCV chronic hepatitis and cirrhosis. Apelin expression in liver cirrhosis was significantly higher than that in normal liver and chronic hepatitis (Table 1). In chronic hepatitis, the level of expression in SEC/HSC, compared to normal liver, was insignificant in F1 stage and F2 stage. However, this level was significantly elevated in F3 stage. In cirrhosis (i.e. F4), the expression was markedly striking, compared to F3 stages (Table 2). Although Apelin expression increased with necroinflammatory grade A1, A2 and A3 from (0.5 ± 1.01) to (0.9 ± 1.01) to (1.2 ± 1.21) respectively, the increase was statistically insignificant (P = 0.3345).
Table 1.
Apelin expression in normal liver, chronic hepatitis and cirrhosis
| Tissue sample | n | Apelin expression in | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Non-parenchymatous cells | P valuea | Hepatocytes | P valuea | ||||
|
|
|
||||||
| Mean ± SD | Median | Mean ± SD | Median | ||||
| Normal lver | 5 | 0 ± 0.0 | 0 | < 0.001 | 0 ± 0.0 | 0 | 0.58 |
| Chronic hepatitis | 43 | 0.419 ± 0.54 | 0 | 0.23 ± 0.427 | 0 | ||
| Cirrhosis | 18 | 2.5 ± 1.2 | 3 | 0.11 ± 0.32 | 0 | ||
Non-parenchymatous cells: sinusoidal endothelial cells/hepatic stellate cells, myofibroblasts, endothelial cells.
P value presents the differences between the three groups.
Apelin in non-parenchymatous cells & hepatocytes in: Cirrhosis versus normal, P = 0.001 & 0.446, respectively. Cirrhosis versus chronic hepatitis, P < 0.001 & 0.22, respectively. Chronic hepatitis versus normal, P = 0.08 & 0.2, respectively. Significant P-values are in bold.
Table 2.
Apelin expression in normal liver and different stages of chronic HCV liver disease
| Fibrosis stage | n | Apelin expression in | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Non-parenchymatous cells | P valuea | Hepatocytes | P valuea | ||||
|
|
|
||||||
| Mean ± SD | Median | Mean ± SD | Median | ||||
| Normal liver | 5 | 0 ± 0.0 | 0 | < 0.001 | 0 ± 0.0 | 0 | 0.796 |
| F1 | 8 | 0.125 ± 0.35 | 0 | 0.375 ± 0.51 | 0 | ||
| F2 | 15 | 0.33 ± 0.48 | 0 | 0.2 ± 0.41 | 0 | ||
| F3 | 20 | 0.6 ± 0.598 | 1 | 0.2 ± 0.41 | 0 | ||
| F4 | 18 | 2.5 ± 1.2 | 3 | 0.11 ± 0.32 | 0 | ||
Non parenchymatous: sinusoidal endothelial cells/hepatic stellate cells, myofibroblasts, endothelial cells.
P-value presents the differences between all groups.
Apelin in non-parenchymatous cells in: Normal versus F1, p = 0.429. Normal versus F2, p = 0.146. Normal versus F3, P = 0.032. F1 versus F2, P = 0.289. F2 versus F3, P = 0.181. F3 versus F4, P < 0.001. Significant P-values are in bold.
Expression in hepatocytes
Apelin expression in hepatocytes did not correlate with the stage of fibrosis (P = 0.796). The expression was more likely to occur in cases with steatosis than those without steatosis [8/11 (72.7%) versus 4/50 (8%), P = 0.00002, odd ratio = 8.36, 95% C.I = 2.128-32.87).
Apelin, in the hepatocytes, was expressed with insignificant negligible levels in chronic hepatitis group (0.23 ± 0.42) and liver cirrhosis (0.11 ± 0.32). The mean Apelin score in hepatocytes was significantly higher in cases with steatosis (0.727 ± 0.467) compared to those without steatosis (0.08 ± 0.27) (P = 0.0004).
Apelin expression in fibrosis-carcinoma axis
The linear staining in the fibrous septa with the exemption of the hepatic parenchyma detected in liver cirrhosis group was reversed in liver cirrhosis with HGDN group and liver cirrhosis with HCC group. In the two latter groups, expression was constantly observed in the hepatocytes with the exemption of non-parenchymatous cells. Accordingly, the data presented in this section represents the results of Apelin expression in the hepatocytes only (Figure 2).
Figure 2.
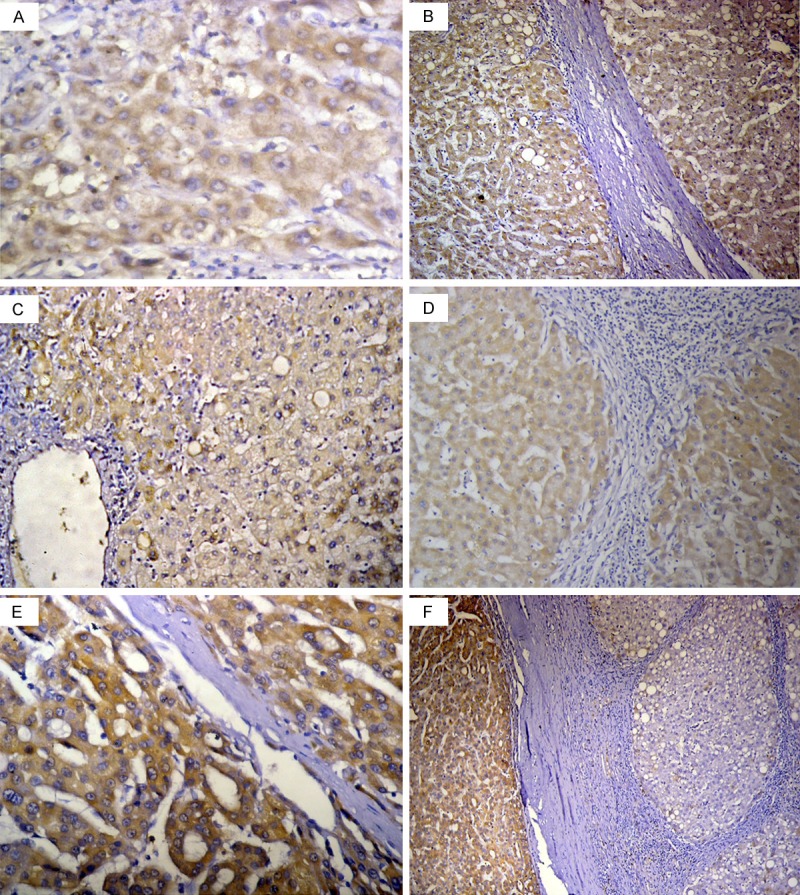
Apelin expression in High grade dysplastic nodule (HGDN), hepatocellular carcinoma (HCC) and their adjacent cirrhosis: Apelin was expressed in dysplastic/neoplastic cells with significant tendency to increase with tumour progression. In the cirrhosis adjacent to HGDN and HCC, Apelin was expressed in hepatocytes and exempted the fibrous septa with a significant tendency to decrease with tumour progression. (A) High grade dysplastic nodule (HGDN), (B) Cirrhosis adjacent to HGDN, (C) HCC grade I, (D) Cirrhosis adjacent to HCC grade I, (E) HCC grade II-III, (F) Cirrhosis adjacent to HCC grade II-III. [Immunohistochemistry, original magnification, (A, E) × 400; (B, F) × 100; (C, D) × 200].
Apelin expression in dysplastic cells of HGDN and malignant cells of HCC
In the HGDN group and HCC group, Apelin was diffusely expressed (i. e. score 3 or 4) in 91.6% (11/12) of dysplastic nodules and in 100% (50/50) of HCCs. The expression was more frequently strong (i.e. score 4) in the latter group (38/50, 76%) compared to the former group (1/11, 9%), (P = 0.0006). Strong Apelin expression (i.e. score 4) was more frequent in HCC grade II-III (30/34, 88.2%) than in HCC grade I (8/16, 50%) (P = 0.005).
There was a significant difference in the level of Apelin in the various stages of hepatocarcinogenesis. There was a significant tendency to increase with tumour progression. The expression in HCC grade II-III was higher than that in HCC-grade I. Likewise, the expression in HCC grade I was higher than that in HGDN (Table 3).
Table 3.
Apelin expression in high grade dysplastic nodule (HGDN), hepatocellular carcinoma (HCC) and their adjacent cirrhosis
| Tissue sample | Apelin expression in | P valuea | ||||
|---|---|---|---|---|---|---|
| n | Dysplastic/malignant cells | Hepatocytes of adjacent cirrhosis | ||||
| Mean ± SD | Median | Mean ± SD | Median | |||
| HGDN | 12 | 3 ± 0.426 | 3 | 3.166 ± 0.389 | 3 | 0.27 |
| HCC grade I | 16 | 3.5 ± 0.516 | 3.5 | 2.5 ± 0.73 | 3 | 0.008 |
| HCC grade II-III | 34 | 3.882 ± 0.327 | 4 | 1.35 ± 1.151 | 1 | < 0.0001 |
P-value presents the differences between Apelin in HGDN/HCC and their adjacent cirrhosis.
Apelin in hepatocytes in: HGDN versus HCC grade-I versus HCC grade-II, P < 0.0001. HGDN versus HCC grade-I, P = 0.021. HCC grade-I versus HCC grade-II, P = 0.0158. Cirrhosis adjacent to HGDN versus adjacent to HCC grade-I versus adjacent to HCC grade-II, p < 0.0001. Cirrhosis adjacent to HGDN versus adjacent to HCC grade-I, P = 0.017. Cirrhosis adjacent to HCC grade-I versus adjacent to HCC grade-II, P = 0.0012. Significant P-values are in bold.
Apelin expression in cirrhosis adjacent to HGDN and cirrhosis adjacent to HCC
In cirrhosis adjacent to HGDN (n = 12), Apelin was universally expressed. The expression was diffuse-weak (i.e. score 3) to diffuse-strong (i.e. score 4) in 83.3% (10/12) and 16.6% (2/12), respectively. In cirrhosis adjacent to HCC grade I, Apelin expression was focal-weak (i.e. score 1) in 12.5% (2/16), focal-strong (i.e. score 2) in 25% (4/16) and diffuse-weak (i.e. score 3) in 62.5% (10/16). In cirrhosis adjacent to HCC grades II-III, Apelin was weakly expressed either diffuse-weak (i.e. score 3) or focal-weak (i.e. score 1) in 29.4% (10/34) and 47% (16/34), respectively. Apelin was not expressed in 23.5% (8/34). Diffuse Apelin expression was more frequent in cirrhosis adjacent to HGDN (12/12, 100%) compared to cirrhosis adjacent to HCC (20/50, 40%) (P = 0.0001). Focal to absent (scores 0, 1) Apelin expression was more frequent in cirrhosis adjacent to HCC grade II-III (24/34, 70.6%) compared to cirrhosis adjacent HCC grade I (6/16, 37%) (P = 0.027) and cirrhosis adjacent to HGDN (0/12) (P < 0.00001).
Apelin was negligibly expressed in the hepatic parenchyma in liver cirrhosis group (0.11 ± 0.32). On the contrary, it was constantly and significantly expressed in the hepatocytes in cirrhosis adjacent to HGDN and cirrhosis adjacent to HCC. There was a significant tendency to decrease with tumour progression. The expression in cirrhosis adjacent to HGDN was higher than cirrhosis adjacent to HCC grade I and cirrhosis adjacent to HCC-grade I was higher than that adjacent to HCC grade II-III (Table 3).
Apelin expression in HGDN and HCC compared to their adjacent cirrhosis
In HGDN group, the staining intensity in 75% (9/12) of the dysplastic nodules was similar to their adjacent cirrhosis with insignificant difference (P = 0.7). In HCC grade I, diffuse-strong (i.e. score 4) to diffuse-weak (i.e. score 3) Apelin expression in the neoplastic cells was universal in all cases (16/16, 100%). This diffuse expression was less frequent in their adjacent cirrhosis (10/16, 62.5%) (P = 0.008). Likewise, in HCC grade II-III, diffuse-strong (score 4) to diffuse-weak (score 3) Apelin expression in the neoplastic cells was universal in all cases (34/34, 100%). This diffuse expression was less frequent in their adjacent cirrhosis (10/34, 29.4%) (P < 0.0001).
Regarding the mean staining scores of Apelin, in HGDN it was close to that of the corresponding adjacent cirrhotic tissue. On the contrary, the level of Apelin in HCC-grade I and HCC grade II-III were consistently and significantly higher as compared with that of their corresponding adjacent cirrhotic tissue (Table 3).
Apelin expression in HGDN, HCC grade-I and HCC grade II inversely correlated with their adjacent cirrhosis (r = -0.31). Apelin gradually increased in HGDN, HCC grade-I and HCC grade II. In contrast, Apelin gradually decreased in the cirrhosis adjacent to HGDN, HCC grade-I and HCC grade II. The gradual incline in Apelin expression in dysplastic and malignant cells was paralleled by a decline in its adjacent cirrhotic liver (P = 0.013).
Discussion
Recently accumulating data has implicated an emerging role for the Apelinergic system in hepatic remodelling [10]. In the current study, Apelin, in healthy liver, was undetectable but it was expressed in the liver of HCV patients with either late fibrosis (F3) or cirrhosis. This expression was significantly different among the two groups being higher in the latter. This is in line with previous investigations demonstrating that patients with cirrhosis showed significant increase in apelin circulating levels [10,20,21]. This finding, in accordance to previous studies [12], suggests that Apelin is induced in the course of liver disease and links this induction to the process of fibrogenesis.
Recent studies have demonstrated that the signalling of Apelin and its receptor APJ is weakly expressed in the liver [12]. Its expression has been confirmed in the sinusoidal endothelial cells/hepatic stellate cells (SEC/ HSC) in the cirrhotic liver of human and rats [10,11,23] and in proliferative hepatic arterial capillaries in human cirrhotic liver [11,23]. This study has, for the first time, analysed the pattern of Apelin expression in different stages of human chronic liver disease. At the early stage of hepatic fibrosis (i.e. F1 & F2), Apelin was almost undetectable. With progression of fibrosis (i.e. F3), Apelin positive cells were located in SEC/HSC. In cirrhosis, Apelin positive cells shifted to the fibrotic septa and extended as linear staining in the septa and on the proliferated capillary endothelial cells. APJ has been detected in the hepatocytes [10], SEC/HSC, myofibroblastic cells and capillary endothelial cells in the cirrhotic fibrous septa [11]. The co-localization of the APJ receptor and its ligand (Apelin) in SEC/HSC, myofibroblastic cells and capillary endothelial cells, observed in this study, supports the suggestion of Kälin et al, for the presence of a possible autocrine or paracrine signalling pathway [8]. It is noteworthy to mention, Apelin, in cirrhotic liver, did not bind to its receptor (APJ) in hepatocytes.
The selective activation of the apelinergic system on the HSC and the exemption of the hepatic parenchyma imply some insights to the possible mechanism of the aplinergic system in the liver. The former finding suggests that Apelin might be primarily induced in the HSC. Although, the gene of Apelin receptor is up-regulated in response to acute and repeated stress [24], recent evidence showed that Apelin, within the gastrointestinal tract, regulates the expression of its receptor [25]. Accordingly, early in the course of the disease, Apelin induced in the HSC did not bind to its receptor in the hepatocytes. Whatever the source of Apelin is, binding of Apelin to its receptor on HSC is involved in the pathway of collagen production ending in hepatic remodelling. This remodelling is independent from the apelinergic system present in the hepatocytes.
In the current study, the expression of Apelin in the fibrotic liver was different from that in the cirrhotic liver. In the fibrotic liver it was solely localized on SEC/ HSC. In cirrhotic liver, it was localized on the myofibroblasts and in proliferated capillaries in the septa. A recent study has identified a special type of cell that poses the potential to differentiate into both mesenchymal stromal cells and endothelial cells. This cell is named mesenchymoangioblasts. Interestingly, these mesenchymoangioblasts were found to express Apelin receptors. Undifferentiated mesenchymoangioblasts were homogeneously Apelin receptor negative. On day 2-3 of differentiation, Apelin receptor was induced and up-regulated simultaneously with mesodermal colony-forming activity [26]. Apelin, in other studies, induced profibrogenic genes in the HSC [12,21] and contributed to its in vitro platelet-derived growth factor-induced proliferation [11]. Taken the published data together with the results of the current study might suggest that HSC has a mesenchymoangioblasts-like behaviour. The binding of Apelin to its receptor on HSC, in fibrotic liver, manipulates its differentiation towards stromal cells and endothelial cells in the cirrhotic septa.
This study has, for the first time, analysed the pattern of Apelin expression during neoplastic transformation. With the initiation of carcinogenesis, a paradox shift occurred where Apelin unbound its receptor in the fibrous septa and proliferated capillaries and became bound to its receptors in hepatocytes in the cirrhosis adjacent to high grade dysplastic nodule (HGDN) and hepatocellular carcinoma (HCC). This paradox shift printed a primary impression that hepatocytes have scavenged Apelin from the fibrous tissue. This impression was ensured when we found that gradual decline in Apelin expression in the hepatocytes in cirrhosis adjacent to high grade dysplastic nodule (HGDN) and hepatocellular carcinoma (HCC) was paralleled by an incline in its expression in the HGDN and HCC. Apelin may represent a material that is taken up rather than produced de novo. The significant incline in Apelin expression in HGDN, HCC grade-I and HCC grade-II suggests that Apelin-APJ pathway may promote HCV-induced liver cell dysplasia and progression of carcinoma.
The data presented in this study together with data published provide strong evidence for a cross-talk link between the apelinergic system and transforming growth factor-β (TGF-β) pathway in the liver. First, TGF-β, similar to Apelin, is a major modulator of cellular responses involved in aberrant wound healing in the liver [27]. Second, published data points that during liver injury TGF-β cytokine, likewise Apelin, is primarily induced in HSC and exerts an autocrine regulation on it ending in collagen deposition [27,28]. Furthermore, sustained hypoxia plays a major role in the activation of both pathways; the apelinergic system [29] and TGF-beta1 pathways [30]. Third, analogous to our results of Apelin, during hepatic carcinogenesis, the loss of TGF-β signalling in the stromal components and its shift to the adjacent hepatocytes “activates the microenvironment” which, consequently, initiates and supports the neoplastic transformation of adjacent epithelial cells [31]. Fourth, TGF-β is produced as a precursor that contains latency associated peptide (LAP). The LAP-TGF-β complex is sequestered in a bond by a large protein termed latent TGF-β-binding protein (LTBP). The latter anchors the extracellular matrix components [32,27]. Accordingly, LTBP in the extracellular matrix sequester TGF-β cytokine forming a reservoir for it without the need for de novo synthesis [27,33]. In the current study, the gradual transferral of Apelin from the fibrotic septa to the cirrhosis adjacent to HGDN and HCC then to the HGDN and HCC implies that Apelin, similar to TGF-β cytokine, has been taken up rather than synthesized de novo. In this scenario, the extracellular matrix can potentially share in modulating the hepatic apelinergic system. Fifth, in this study, Apelin, in chronic hepatitis and cirrhosis, was almost unexpressed in the hepatocytes. Nevertheless, when it was expressed, the expression was heterogeneous with the highest levels associated with steatosis, regardless of the stage of the disease. This finding may suggest that Apelin may chip in steatohepatitis similar to the TGF-β cytokine [34].
The data in the current study shows that Apelin initiates and supports hepatocarcinogensis. If the Apelinergic system is one of the “other pathways” [35,36] suggested to cross-talk with TGF-β family protein, this cross-talk may be one of the factors that help the hepatocytes to escape “the growth-inhibitory signals of TGF-β” [37] and consequently shift the signalling of TGF-β from tumour suppression to oncogenesis. Advancing the knowledge detailing the hepatic aplinergic pathway may provide further comprehension for a probable cross-talk with TGF-β pathway.
Steatosis is a complication of HCV infection and entails an increased risk for the development of hepatocellular carcinoma [38]. The data in the current study supports and provides an insight to the possible underlying mechanism for this associated risk. In chronic hepatitis, activation of the apelinergic system in the hepatocytes with steatosis was significantly more likely to occur (with high odd ratio) than without steatosis. Consequently, this activation, as formerly discussed, triggers microenvironment activation which in turn drives the neoplastic transformation. This finding warrants further detailed analysis.
By analogy, the results in this study could provide rationale for the use of apelinergic system in anti-fibrogenic and anti-carcinogenic strategies. Especially that, although blockage of the TGF-β pathway was an effective antifibrotic [27] and anti-carcinogenic [39] approach, other alternative pathways were suggested to compensate to this blockage [27]. Overall, the data points that targeting the hepatic apelinergic system, whether dependent or in dependent on the TGF-β signalling pathway, can modify the course of liver disease in HCV patients.
In conclusion, Apelin is expressed in the liver of HCV patients compared to healthy individuals and is involved in the disease progression. It may be implicated in multiple facets in the fibrosis-carcinoma axis. In chronic hepatitis, Apelin binds to its receptor on the HSC. HSC may have a mesenchymoangioblasts-like behaviour. Activation of apelinergic system on HSC seems to manipulate the differentiation of HSC ending in hepatic remodelling. This remodelling is independent from the apelinergic system in the hepatocytes. Extra-cellular matrix may act as a reservoir pouring Apelin, rather than its de novo synthesis. The uptake of Apelin from the stromal components by the epithelial cells may result in an ‘activated’ microenvironment. This microenvironment initiates the transformation of adjacent epithelial cells and supports the evolution and progression of dysplastic nodules and hepatocellular carcinoma. In addition, the data point to interrelated events between the apelinergic pathway and TGF-β signalling. These findings imply prognostic and therapeutic implications which could halt the progression of fibrosis and evolution of hepatocellular carcinoma in HCV patients.
Disclosure of conflict of interest
None.
References
- 1.Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251:471–76. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- 2.Kleinz MJ, Davenport AP. Emerging roles of apelin in biology and medicine. Pharmacol Ther. 2005;107:198–211. doi: 10.1016/j.pharmthera.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, Liu Y, Osmond DH, George SR, O’Dowd BF. Characterization of apelin, the ligand for the APJ receptor. J Neurochem. 2000;74:34–41. doi: 10.1046/j.1471-4159.2000.0740034.x. [DOI] [PubMed] [Google Scholar]
- 4.Medhurst AD, Jennings CA, Robbins J, Davis RP, Ellis C, Winborn KY, Lawrie KW, Hervieu G, Riley G, Bolaky JE, Herrity NC, Murdock P, Darker JG. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J Neurochem. 2003;84:1162–1172. doi: 10.1046/j.1471-4159.2003.01587.x. [DOI] [PubMed] [Google Scholar]
- 5.Falcao-Pires I, Leite-Moreira AF. Apelin: a novel neurohumoral modulator of the cardiovascular system: pathophysiologic importance and potential use as a therapeutic target. Rev Port Cardiol. 2005;24:1263–76. [PubMed] [Google Scholar]
- 6.O’Carroll AM, Lolait SJ, Harris LE, Pope GR. The apelin receptor APJ: journey from an orphan to a multifaceted regulator of homeostasis. J Endocrinol. 2013;219:R13–35. doi: 10.1530/JOE-13-0227. [DOI] [PubMed] [Google Scholar]
- 7.Sorli SC, Le Gonidec S, Knibiehler B, Audigier Y. Apelin is a potent activator of tumour neoangiogenesis. Oncogene. 2007;26:7692–9. doi: 10.1038/sj.onc.1210573. [DOI] [PubMed] [Google Scholar]
- 8.Kalin RE, Kretz MP, Meyer AM, Kispert A, Heppner FL, Brandli AW. Paracrine and autocrine mechanisms of apelin signaling govern embryonic and tumour angiogenesis. Dev Biol. 2007;305:599–614. doi: 10.1016/j.ydbio.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Heo K, Kim YH, Sung HJ, Li HY, Yoo CW, Kim JY, Park JY, Lee UL, Nam BH, Kim EO, Kim SY, Lee SH, Park JB, Choi SW. Hypoxia-induced up-regulation of apelin is associated with a poor prognosis in oral squamous cell carcinoma patients. Oral Oncol. 2012;48:500–6. doi: 10.1016/j.oraloncology.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 10.Principe A, Melgar-Lesmes P, Fernández-Varo G, delArbol LR, Ros J, Morales-Ruiz M, Bernardi M, Arroyo V, Jiménez W. The hepatic apelin system: a new therapeutic target for liver disease. Hepatology. 2008;48:1193–201. doi: 10.1002/hep.22467. [DOI] [PubMed] [Google Scholar]
- 11.Yokomori H, Oda M, Yoshimura K, Machida S, Kaneko F, Hibi T. Overexpression of apelin receptor (APJ/AGTRL1) on hepatic stellate cells and sinusoidal angiogenesis in human cirrhotic liver. J Gastroenterol. 2011;46:222–31. doi: 10.1007/s00535-010-0296-3. [DOI] [PubMed] [Google Scholar]
- 12.Melgar-Lesmes P, Casals G, Pauta M, Ros J, Reichenbach V, Bataller R, Morales-Ruiz M, Jimenez W. Apelin mediates the induction of profibrogenic genes in human hepatic stellate cells. Endocrinology. 2010;151:5306–14. doi: 10.1210/en.2010-0754. [DOI] [PubMed] [Google Scholar]
- 13.Raza SA, Clifford GM, Franceschi S. Worldwide variation in the relative importance of hepatitis B and hepatitis C viruses in hepatocellular carcinoma: a systematic review. Br J Cancer. 2007;96:1127–34. doi: 10.1038/sj.bjc.6603649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hui KM. Human hepatocellular carcinoma: expression profiles-based molecular interpretations and clinical applications. Cancer Lett. 2009;286:96–102. doi: 10.1016/j.canlet.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Wright JH, Johnson MM, Shimizu-Albergine M, Bauer RL, Hayes BJ, Surapisitchat J, Hudkins KL, Riehle KJ, Johnson SC, Yeh MM, Bammler TK, Beyer RP, Gilbertson DG, Alpers CE, Fausto N, Campbell JS. Paracrine activation of hepatic stellate cells in platelet-derived growth factor C transgenic mice: Evidence for stromal induction of hepatocellular carcinoma. Int J Cancer. 2014;134:778–88. doi: 10.1002/ijc.28421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The French METAVIR Cooperative Study Group. Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. Hepatology. 1994;20:15–20. [PubMed] [Google Scholar]
- 17.Hamilton SR, Aaltonen LA, Hirohashi S, Ishak KG, Kojiro M. Hepatocellular carcinoma. In: Hamilton SR, Aaltonen LA, editors. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Digestive System. Lyon, France: IARC Press; 2000. pp. 159–172. [Google Scholar]
- 18.International Consensus Group for Hepatocellular Neoplasia. Pathologic diagnosis of early hepatocellular carcinoma: a report of the International Consensus Group for Hepatocellular Neoplasia. Hepatology. 2009;49:658–664. doi: 10.1002/hep.22709. [DOI] [PubMed] [Google Scholar]
- 19.Schmitt-Gräff A, Krüger S, Bochard F, Gabbiani G, Denk H. Modulation of alpha smooth muscle actin and desmin expression in perisinusoidal cells of normal and diseased human livers. Am J Pathol. 1991;138:1233–42. [PMC free article] [PubMed] [Google Scholar]
- 20.Barnias G, Zouboulis-Vafiadis I, Nikolaou P, Siakavellas SI, Perrea D, Ladas SD. Increased serum levels of apelin in patients with cirrhosis. Gastroenterology. 2009;136:A416. [Google Scholar]
- 21.El-Mesallamy HO, Hamdy NM, Rizk HH, El-Zayadi AR. Apelin serum level in Egyptian patients with chronic hepatitis C. Mediators Inflamm. 2011;2011:703031. doi: 10.1155/2011/703031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yasuzaki H, Yoshida S, Hashimoto T, Shibata W, Inamori M, Toya Y, Tamura K, Maeda S, Umemura S. Involvement of the apelin receptor APJ in Fas-induced liver injury. Liver Int. 2013;33:118–26. doi: 10.1111/liv.12006. [DOI] [PubMed] [Google Scholar]
- 23.Chen W, Oue T, Ueno T, Uehara S, Usui N, Fukuzawa M. Apelin is a marker of the progression of liver fibrosis and portal hypertension in patients with biliary atresia. Pediatr Surg Int. 2013;29:79–85. doi: 10.1007/s00383-012-3210-7. [DOI] [PubMed] [Google Scholar]
- 24.O’Carroll AM, Don AL, Lolait SJ. APJ receptor mRNA expression in the rat hypothalamic paraventricular nucleus: regulation by stress and glucocorticoids. J Neuroendocrinol. 2003;15:1095–101. doi: 10.1046/j.1365-2826.2003.01102.x. [DOI] [PubMed] [Google Scholar]
- 25.Wang G, Kundu R, Han S, Qi X, Englander EW, Quertermous T, Greeley GH Jr. Ontogeny of apelin and its receptor in the rodent gastrointestinal tract. Regul Pept. 2009;158:32–39. doi: 10.1016/j.regpep.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vodyanik MA, Yu J, Zhang X, Tian S, Stewart R, Thomson JA, Slukvin II. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell. 2010;7:718–29. doi: 10.1016/j.stem.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bissell DM, Roulot D, George J. Transforming growth factor beta and the liver. Hepatology. 2001;34:859–67. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- 28.Bissell DM, Wang SS, Jarnagin WR, Roll FJ. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest. 1995;96:447–455. doi: 10.1172/JCI118055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melgar-Lesmes P, Pauta M, Reichenbach V, Casals G, Ros J, Bataller R, Morales-Ruiz M, Jiménez W. Hypoxia and proinflammatory factors upregulate apelin receptor expression in human stellate cells and hepatocytes. Gut. 2011;60:1404–11. doi: 10.1136/gut.2010.234690. [DOI] [PubMed] [Google Scholar]
- 30.Jeong WI, Do SH, Yun HS, Song BJ, Kim SJ, Kwak WJ, Yoo SE, Park HY, Jeong KS. Hypoxia potentiates transforming growth factor-beta expression of hepatocyte during the cirrhotic condition in rat liver. Liver Int. 2004;24:658–68. doi: 10.1111/j.1478-3231.2004.0961.x. [DOI] [PubMed] [Google Scholar]
- 31.Stover DG, Bierie B, Moses HL. A delicate balance: TGF-beta and the tumour microenvironment. J Cell Biochem. 2007;101:851–861. doi: 10.1002/jcb.21149. [DOI] [PubMed] [Google Scholar]
- 32.Hyytiäinen M, Taipale J, Heldin CH, Keski-Oja J. Recombinant latent transforming growth factor-binding protein 2 assembles to fibroblast extracellular matrix and is susceptible to proteolytic processing and release. J Biol Chem. 1998;273:20669–20676. doi: 10.1074/jbc.273.32.20669. [DOI] [PubMed] [Google Scholar]
- 33.Breitkopf K, Lahme B, Tag CG, Gressner AM. Expression and matrix deposition of latent transforming growth factor-beta binding proteins in normal and fibrotic rat liver and transdifferentiating hepatic stellate cells in culture. Hepatology. 2001;33:387–396. doi: 10.1053/jhep.2001.21996. [DOI] [PubMed] [Google Scholar]
- 34.Yang L, Roh YS, Song J, Zhang B, Liu C, Loomba R, Seki E. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology. 2014;59:483–95. doi: 10.1002/hep.26698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. 2001;276:14466–73. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- 36.Kwak HJ, Park MJ, Cho H, Park CM, Moon SI, Lee HC, Park IC, Kim MS, Rhee CH, Hong SI. Transforming growth factor-beta1 induces tissue inhibitor of metalloproteinase-1 expression via activation of extracellular signal-regulated kinase and Sp1 in human fibrosarcoma cells. Mol Cancer Res. 2006;4:209–220. doi: 10.1158/1541-7786.MCR-05-0140. [DOI] [PubMed] [Google Scholar]
- 37.Paik SY, Park YN, Kim H, Park C. Expression of transforming growth factor-beta1 and transforming growth factor-beta receptors in hepatocellularcarcinoma and dysplastic nodules. Mod Pathol. 2003;16:86–96. doi: 10.1097/01.MP.0000047308.03300.9C. [DOI] [PubMed] [Google Scholar]
- 38.Adinolfi LE, Restivo L, Marrone A. The predictive value of steatosis in hepatitis C virus infection. Review. Expert Rev Gastroenterol Hepatol. 2013;7:205–13. doi: 10.1586/egh.13.7. [DOI] [PubMed] [Google Scholar]
- 39.Li G, Qin L, Ye Q, Dong Q, Ren N, Jia H. Organ microenvironment affects growth and metastasis of hepatocellular carcinoma via the TGF-β/Smad pathway in mice. Exp Ther Med. 2013;5:133–137. doi: 10.3892/etm.2012.752. [DOI] [PMC free article] [PubMed] [Google Scholar]