

Abstract
Hydroxysafflor yellow A (HSYA), a major constituent in the hydrophilic fraction of the safflower plant, can retard the progress of hepatic fibrosis. However, the anti-inflammatory properties and the underlying mechanisms of HSYA on I/R-induced acute liver injury are unknown. Inhibiting macrophage activation is a potential strategy to treat liver ischemia/reperfusion (I/R) injury. In this study, we investigated the therapeutic effect of HSYA on liver I/R injury and the direct effect of HSYA on macrophage activation following inflammatory conditions. The therapeutic effects of HSYA on I/R injury were tested in vivo using a mouse model of segmental (70%) hepatic ischemia. The mechanisms of HSYA were examined in vitro by evaluating migration and the cytokine expression profile of the macrophage cell line RAW264.7 exposed to acute hypoxia and reoxygenation (H/R). Results showed that mice pretreated with HSYA had reduced serum transaminase levels, attenuated inflammation and necrosis, reduced expression of inflammatory cytokines, and less macrophage recruitment following segmental hepatic ischemia. In vitro HSYA pretreated RAW264.7 macrophages displayed reduced migratory response and produced less inflammatory cytokines. In addition, HSYA pretreatment down-regulated the expression of matrix matalloproteinase-9 and reactive oxygen species, and inhibited NF-κB activation and P38 phosphorylation in RAW264.7 cells. Thus, these data suggest that HSYA can reduce I/R-induced acute liver injury by directly attenuating macrophage activation under inflammatory conditions.
Keywords: Hydroxysafflor yellow A, liver ischemia/reperfusion injury, macrophage, cell migration, inflammatory cytokines, reactive oxygen species
Introduction
Liver ischemia/reperfusion (I/R) injury, an innate immunity-dominated local inflammation, occurs in liver resection, such as surgical interventions, trauma, or transplantation. During these clinical settings, I/R is a mandatory procedure utilized to minimize intraoperative blood loss. However, this procedure also induces liver injury that occasionally leads to postoperative complications and even liver failure [1]. Hepatic I/R injury is also a major cause of morbidity and mortality in patients undergoing liver transplantation. Therefore, minimizing I/R-induced liver injury would greatly improve the safety and efficacy of liver surgeries [2,3].
The pathophysiology of liver I/R injury includes direct cellular damage resulting from the ischemic insult as well as delayed dysfunction and damage resulting from activation of inflammatory pathways. Many studies have shown that the process of I/R-induced liver injury is a cascade of inflammatory events including the generation of reactive oxygen and nitrogen stress in the liver, and the activation of the inflammatory response [4,5]. Previous studies have demonstrated IR injury to be biphasic, with an acute macrophage-dependent phase followed by a later phase characterized by neutrophil recruitment and activation [6,7]. Kupffer’s cells (KCs), the liver resident macrophages, are intimately involved in these early proinflammatory responses.
KCs constitute 80-90% of the tissue macrophages present in the body, are concentrated in the periportal area. As a result, these cells are the first macrophage population to encounter bacteria, bacterial endotoxins, and microbial debris derived from the gastrointestinal tract and transported to the liver via the portal vein. During the sequence of liver I/R injury, KCs are consistently identified as a major contributor of the early post-ischemic oxidant stress and inflammatory reactions [8,9]. For example, upon activation KCs release reactive oxygen species (ROS) and multiple inflammatory cytokines and chemokines. In addition, KCs up-regulate inducible NO synthase in hepatocytes, activate mitogen-activated protein kinases (MAPKs), and accumulate neutrophils, all of which have been identified as contributing events to inflammation-associated liver damage [10,11]. Therefore, agents limiting the activity of macrophages have been proposed as a treatment strategy for improving the clinical outcome of surgical procedures involving liver resections or transplantation [11,12].
Hydroxysafflor Yellow A (HSYA) is a major constituent in the hydrophilic fraction of the safflower plant, and has been widely used in traditional Chinese medicine for the treatment of trauma, and cardiovascular and cerebrovascular diseases [13]. Previous studies suggest that the pharmacological activities of HSYA include suppressing lipid peroxidation, inhibiting thrombosis, attenuating platelet aggregation, and reducing myocardial infarct size in rats [14,15]. Recently, several reports have also reported the anti-inflammatory property of HSYA in brain ischemic [16] and acute lung injury models [17]. Moreover, we previously demonstrated that HSYA attenuates hepatic fibrosis through inhibiting oxidative stress [18] and alleviates oleic acid- mediated acute lung injury through activation of the cAMP-PKA signaling pathway [19]. In this study, we used a mouse model of segmental (70%) hepatic ischemia to investigate the effect of HSYA on I/R-induced liver injury. In addition, the mechanisms of HSYA were examined in vitro by evaluating migration, phagocytosis, and the cytokine expression profile of the macrophage cell line RAW264.7 exposed to periods of acute hypoxia and reoxygenation (H/R) to model in vivo liver IR injury. HSYA pretreatment attenuated hepatic inflammation and I/R-induced injury in mice. Accordingly, HSYA pretreatment significantly inhibited macrophage migration, phagocytosis, cytokine expression, and ROS production. These data support the use of HSYS as an agent to reduce inflammation and oxidative activity during liver surgery and transplantation.
Materials and methods
Reagents and antibodies
HSYA (C27H32O16, m.w. 611.1614 g) [18] was obtained from Shandong Natural Drugs Engineers and Technicians Research Center (Yantai, Shandong Province, China) as a yellow amorphous powder. The purity of HSYA was greater than 99% as determined by HPLC. Prior to use, HSYA was dissolved in double distilled water (pH = 5).
N-acetylcysteine (NAC), p38 MAPkinase inhibitors SB203580 and other common reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM) was obtained from Gibco (Paisley, UK). Fetal bovine serum (FBS) was purchased from Biochrom (Berlin, Germany), and 5-(3-carboxymethoxyphenyl)-2-(4,5-dimethylthiazoly)-3-(4-sulfophenyl) tetrazolium, inner salt (MTS) was purchased from Promega (Madison, USA). 2,7-dichlorofluorescein diacetate (DCFH-DA) was purchased from Invitrogen (Grand Island, NY) and PCR reagents were purchased from Applied Biosystems (Foster City, CA). Mouse interleukin (IL)-6, and tumor necrosis factor (TNF)-α ELISA kits were purchased from ExCell Biology (Shanghai, China). The antibodies against matrix metalloproteinase enzymes (MMP)-9, nuclear factor κB (NF-κB), extracellular regulated protein kinases (ERK), c-Jun N-terminal kinases (JNK), and p38 mitogen-activated protein kinases and respective phosphorylated protein kinases were all purchased from Cell Signaling Technology (Beverly, MA, USA).
Cell culture and H/R procedure
The Murine macrophage cell line RAW264.7 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in DMEM complete medium supplemented with 2 mM L-glutamine, 10% heat-inactivated FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin. The cells were incubated at 37°C in a 5% CO2 humidified incubator.
In vitro H/R injury model was used to mimic I/R injury in vitro [20]. Cell hypoxia was manipulated by placing the cells into a specifically designed hypoxic incubator (Thermo Scientific, HERACELL 150i, Hanau, Germany) purged with a mixture of 95% N2 and 5% CO2. And the hypoxia PO2 was extremely 1%. For exposure to H/R, the cells were changed with serum- and glucose-free deoxygenated DMEM and incubated in the hypoxic incubator for 2 h. After that, the medium was substituted with DMEM containing glucose and the cells were returned to a standard incubator (95% air and 5% CO2, 37°C) with the normoxic PO2, which was 21%. The reoxygenation time was set for 4 h.
Animals and surgery
Male C57BL/6 mice (18~22 g) were obtained from the Academy of Military Medical Sciences (Beijing, China) and housed in specific pathogen free (SPF) conditions. The animals were fed with standard chow and had free access to water. All animal experiments were performed in a humane manner, and also in accordance with the Institutional Animal Care Instructions. This study was conducted under experimental protocols approved by the Ethics Committee for Animals, Binzhou Medical University (approval ID: 2012022).
The mice were divided randomly into three groups: (i) sham group, (ii) hepatic I/R group (vehicle group), and (iii) HSYA-pretreated I/R groups. All animals were anesthetized with chloral hydrate. A model of segmental (70%) hepatic ischemia was performed as described previously [21,22] with minor modifications. Briefly, after a midline laparotomy, all structures in the portal triad (hepatic artery, portal vein and bile duct) to the left and median liver lobes were occluded for 90 min under aseptic conditions. This method of partial hepatic ischemia prevented mesenteric venous congestion by permitting portal decompression through the right and caudate lobes. Reperfusion was initiated by removing the clamp after 2, 6, or 24 hours. In the HSYA-pretreated I/R groups, HSYA (5 mg/kg or 15 mg/kg) was administered with an i.v. injection 1 hour before the operation. An equal volume of normo-saline served as a control. Two, 6, or 24 hours following reperfusion, the mice were anesthetized and blood and liver samples were collected.
Plasma alanine aminotransferase assays
To assess hepatic function and cellular injury following liver ischemia, serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured using the Opera Clinical Chemistry System (Bayer).
Histologic examination
Liver specimens from the ischemic left lobes were fixed in 4% neutral-buffered formalin and embedded in paraffin. Serial sections (5 μm) were mounted on glass slides and stained with hematoxylin-eosin (HE) for histological examination by light microscopy (LeiCA DM 5000B, Leica Microsystems, Wetzlar, Germany). The number of necrotic focus in paraffin-embedded liver samples was counted in a blinded fashion.
Liver specimens from the ischemic left lobes used for immunostaining were embedded in Tissue Tec OCT compound (Miles, Elkhart, IN) and snap frozen in liquid nitrogen. Serial sections (6 μm) were mounted on glass slides and fixed with 4% formaldehyde in phosphate buffer solution (PBS) for 30 min, and then permeabilized with 0.3% Triton X-100 in PBS for 15 min. Nonspecific proteins were further blocked using 2% bovine serum albumin (BSA) for 30 min followed by incubation with diluted F4/80 antibody (1:100) overnight at 4°C. Following the incubation, the slides were washed three times with PBS, and incubated for 1 hour with an Alexa Fluor 594-red anti-rat antibody (1:200 dilution) in PBS with 1% BSA. The slides were then washed three times with PBS, and mounted with aqueous mounting medium containing DAPI. Slide were analyzed and imaged using a Leica confocal microscope in a blind manner by counting F4/80+ staining in 10 high power fields (×400) for each section.
Cytokine ELISA
Serum and the supernatant of cells culture medium concentrations of TNF-α and IL-1β were determined using ELISA kits according to the manufacturer’s instructions. The limit of detection for TNF-α and IL-1β was 5 pg/ml and 5.1 pg/ml, respectively.
Real-time quantitative PCR (RT-qPCR)
The gene expression of inflammatory cytokines in the liver tissue or in RAW264.7 cells was detected by RT-qPCR analysis. Total RNA was isolated from the frozen liver tissue or RAW264.7 cells using the RNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The cDNA was generated from total RNA samples by using of the cDNA Reverse Transcription Kit. The expression of the target genes encoding inflammatory cytokines was determined by comparative quantitative RT-PCR using specific primers. The primers used were as follows: mouse TNF-α sense 5’-GGC AGG TTC TGT CCC TTT CA-3’, antisense 5’-CTG TGC TCA TGG TGT CTT TTC TG-3’; MCP-1 sense 5’-TCT GGG CCT GCTGTT CAC A-3’, antisense 5’-GGA TCA TCT TGC TGG TGA ATG A-3’; IL-1β sense 5’-TCC ATG AGC TTT GTA CAA GGA-3’, antisense 5’-AGC CCA TAC TTT AGG AAG ACA-3’; IL-6 sense 5’-CTC TGG GAAATC GTG GAA ATG-3’, antisense 5’-AAG TGC ATC ATC GTT GTT CATACA-3’; 18S rRNA sense 5’-GTA ACCCGT TGA ACC CCA TT-3’, antisense 5’-CCA TCC AAT CGG TAG TAGCG-3’. 18S rRNA was used as an endogenous control. The comparative cycle threshold (CT) method (ΔΔCT) was used for quantification of gene expression. The average expression level of the Sham + vehicle group data was set to 1.0, and the other data were adjusted relative to this baseline.
Western blot analysis
The cell samples were pretreated with HSYA (5, 50 or 100 μM) for 1 hour and then harvested after 2 h hypoxia and 4 h of reoxygenation. Total protein, nuclear protein or cytoplasmic protein was extracted from each cell sample using a Total Protein Extraction kit or a Nuclear/Cytosolic Fractionation Kit according to the manufacturer’s instructions. The expression of NF-κB/p65 protein was examined in cytoplasmic and nuclear extracts, and the level of the other proteins was examined in total cell lysates. The protein concentrations of the lysates were determined following the method of bicinchoninic acid (BCA) using a Protein Assay Kit. Samples containing 50 or 30 μg of protein were separated using 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After electrophoresis, the proteins in the gel were transferred to a polyvinylidene fluoride (PVDF) membrane for western blot analysis. Primary antibodies against MMP-9 (1:1000), NF-κB/P65 (1:1000), P38 (1:1000), p-P38 (1:1000), ERK (1:1000), p-ERK (1:2000), JNK (1:2000), p-JNK (1:1000), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 1:10,000) were used. The membranes were washed and incubated with goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibodies. Immunoreactive proteins were detected by enhanced chemiluminescence (ECL). The relative density of the protein bands was quantified using Image J software (National Institutes of Health, USA) and signals were normalized to GAPDH expression.
Cell metabolic assays
Cell metabolism was evaluated by measuring MTS activity. RAW264.7 cells (0.5×104 cell per well) were seeded in 96-well plate and incubated overnight with DMEM containing 10% FBS. The following day, the cells were treated with various concentrations of HSYA (5, 50, 100 and 300 μM). After 24, 48, and 72 hours, 20 μl of MTS was added into each well and the plates were incubated for an additional 3 hours at 37°C in a humidified 5% CO2 atmosphere. The absorbance at 490 nm of each well containing cells was measured and compared with the absorbance of wells without cells.
Intracellular ROS measurement
RAW264.7 cells (2×105 per well) were cultured in 6-well plates, with or without HSYAS pretreatment at various concentrations (5, 50, and 100 μM). After 2 h hypoxia and 4 h of reoxygenation 2,7-dichlorofluorescein diacetate (DCFH-DA, Invitrogen) was added to the cells at a concentration of 10uM and incubated at 37°C for 20 min and washed twice with cold PBS. Following the washes, the cells were harvested and the intracellular ROS levels were analyzed by flow cytometry.
Migration assay in vitro
RAW264.7 cells were cultured in complete DMEM medium overnight and then starved with serum-free DMEM medium for 12 hours. The cells were then collected and resuspended in serum-free DMEM medium. Cell migration was assayed using transwell plates (BD Bioscience, Bedford, MA) with a pore size of 8.0 μm. RAW264.7 cells (1×105 cells/well) were added to the top chamber with or without different doses (0, 5, 50, and 100 μM) of HSYA. 5% FBS of DMEM medium was added to the lower chamber. For stimulation, the cells were incubated at 37°C with normoxic or hypoxic (H2h/R4h) for 6 hours. Following the incubation, non-migrating cells were wiped from the upper side of the membrane. Migrated cells were fixed with cold methanol and stained with hematoxylin. For each experiment, the number of cells in five randomly chosen fields of each filter was counted. The results of three independent experiments are presented as the percentage of cell migration relative to the control.
Statistical analysis
The results of multiple observations are presented as the mean ± SD of at least three independent experiments. The data were analyzed with the statistics software SPSS 11.5 and differences between various groups were analyzed using a one-way ANOVA. P value less than 0.05 was considered to be statistically significant.
Results
HSYA attenuates hepatic I/R injury
We previously demonstrated that HSYA attenuates hepatic fibrosis by inhibiting oxidative stress [18]. To determine if HSYA could also prevent hepatocellular damage of the post-ischemic liver, we treated mice with HSYA prior to inducing hepatic ischemia and then measured serum transaminase (ALT and AST) activities. After 90 min of warm ischemia, the clamp was removed and reperfusion was allowed to take place for 2, 6 or 24 hours at which point serum was collected. An increase in transaminase levels was observed in the untreated (vehicle) control group compared with sham-operated animals peaking at 6 hours of reperfusion. However, mice pretreated with HSYA had significantly lower levels of serum transaminase compared to the vehicle control group (Figure 1A and 1B). In addition, this protective effect was concentration-dependent, with > 50% inhibition occurring at a concentration of 15 mg/kg after 24 hours of reperfusion.
Figure 1.
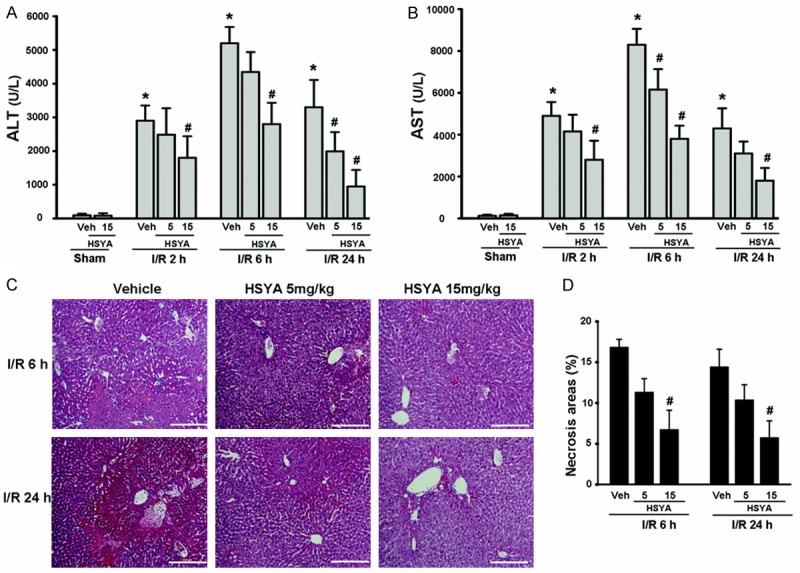
HSYA pretreatment attenuates hepatic I/R injury. Serum transaminase ALT (A) and AST (B) levels in sham-operated mice (n = 5 per group) or mice pretreated with vehicle or HSYA (5 or 15 mg/kg, n = 5-8 per group) prior to 90 min of hepatic ischemia followed by 2, 6, or 24 hours of reperfusion. (C) Representative histopathologic images of ischemic liver lobes at 6 or 24 hours after reperfusion. H&E staining with original magnification ×100, bar equals 200 μm. (D) Quantification of necrotic areas. A minimum of 10 view fields per animal was included. *P < 0.05 vs. vehicle–sham group; #P < 0.05 vs. corresponding control exposed to vehicle-I/R.
Consistent with increased levels of serum transaminase, this method of inducing hepatic I/R also led to increased necrosis in the ischemic lobule with significant inflammatory infiltration in vehicle treated animals. Hepatic I/R damage was significantly improved and inflammation reduced with HSYA pretreatment (Figure 1C and 1D). These data suggest that HSYA can reduce liver damage induced by hepatic ischemia.
Inflammatory cytokines, such as TNF-α and IL-1β, play key roles in the pathophysiology of hepatic I/R injury [1]. To determine if HSYA reduced the levels of these cytokines, we first measured the presence of TNF-α and IL-1β mRNA in liver tissue by Real-time PCR. Mice pretreated with HSYS had significantly reduced expression of TNF-α and IL-1β mRNA in I/R-injured liver tissue (Figure 2A and 2B). We next measured the serum levels of these cytokines following reperfusion. Corresponding to mRNA expression, HSYA pretreatment significantly reduced the levels of systemic TNF-α and IL-1β in the serum of I/R-induced mice compared with vehicle controls after 2 and 6 hours of reperfusion (Figure 2C and 2D). Taken together, these data suggest that HSYA reduces inflammation induced by hepatic I/R injury.
Figure 2.
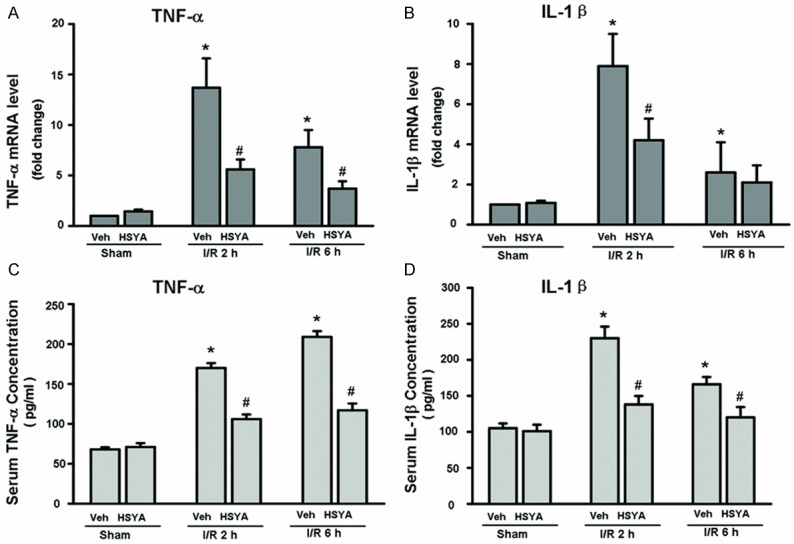
HSYA reduces expression of inflammatory cytokines in liver I/R injury. A, B: Real-time PCR analysis of hepatic proinflammatory cytokine mRNA levels (TNF-α, MCP-1 and IL-1β) at 2 or 6 hours of reperfusion. Pretreatment with HSYA at 15 mg/kg significantly attenuated the I/R-induced hepatic TNF-α and IL-1β expression at time points of reperfusion studied. C, D: The levels of serum TNF-α and IL-1β were measured by ELISA assays. Results are mean ± S.D. of 5-8 mice per group. *P < 0.05 vs. vehicle–sham group; #P < 0.05 vs. corresponding vehicle-I/R mice.
HSYA pretreatment inhibits macrophage recruitment after hepatic I/R injury
Macrophages are activated during the ischemic phase and even more so during reperfusion [23]. Under these circumstances, macrophages play a key role in initiating and propagating cellular damage and death following I/R injury. Therefore, we used immunofluorescence staining to evaluate whether HSYA prevented the recruitment of macrophages following hepatic I/R injury. As shown in Figure 3, HSYA pretreatment significantly decreased the number of F4/80-positive cells in the I/R-injured liver compared to the livers of mice without HSYA administration. These results suggest that HSYA pretreatment reduces the number macrophages present in the liver after hepatic I/R injury.
Figure 3.
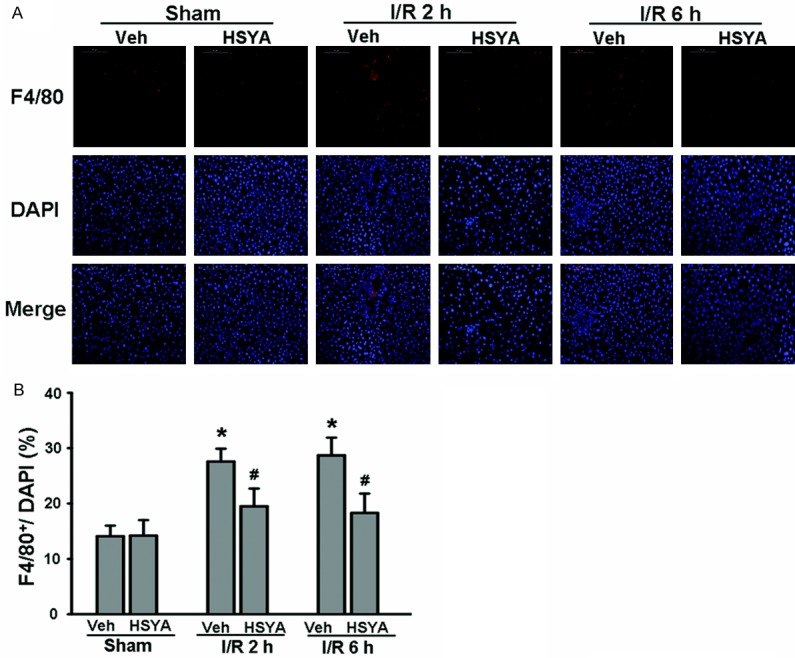
HSYA pretreatment inhibits macrophage recruitment following hepatic I/R injury. A: Immunofluorescence staining using an anti-F4/80 antibody was performed to identify macrophages present in the liver following hepatic I/R. DAPI was used to visualize nuclei (blue). The original magnification was ×200. B: Quantification of the proportion of macrophages in normal and I/R-induced liver with or without HSYA pretreatment (n = 5 per group). *P < 0.05, compared with Sham group. #P < 0.05, compared with corresponding vehicle-I/R group.
HSYA pretreatment inhibits the activity of RAW264.7 macrophages
The reduced number of liver infiltrating macrophages observed in HSYA pretreated mice could either be the result of increased cell death or reduced migration. To further explore the mechanisms of HSYA, we used the macrophage cell line RAW264.7. First, we tested the toxicity of HSYA on RAW264.7 cells using a MST viability assay. HSYA did not exert a significant toxic effect on RAW264.7 macrophages at the concentrations 5, 50, 100, and 300 μM examined after 4 days of treatment (Figure 4). To address the effects of HSYA on macrophage recruitment, we evaluated migration in transwell plates. The results indicated that pretreatment with HSYA significantly prevented the migration of RAW264.7 macrophages in response to H/R activation (Figure 5A and 5B). Taken together, these data suggest that HSYA does not directly kill macrophages, but rather alters the migration of these cells to the liver.
Figure 4.

HSYA does not induce RAW264.7 cell death. RAW264.7 macrophages were pretreated with different concentrations of HSYA for the indicated times, and then metabolic activity was assessed via MTS assay. Data are presented as the mean ± S.D. of three independent experiments. *P < 0.05, compared with control (vehicle group).
Figure 5.

HSYA pretreatment inhibits the migration of RAW264.7 macrophages,and decreases the production of intracellular ROS and MMP-9 protein expression. A and B: HSYA inhibited the migration of RAW264.7 macrophages. RAW264.7 cells were pretreated with different concentrations of HSYA for 1 hour in the upper chamber of transwell plates and the cells were treated with normal oxy or H/R for 6 hours. A: Representative images of RAW264.7 cells that migrated in response to H/R stimulation. Original magnification, ×100. B: Quantification of RAW264.7 cells migrations in each group. HSYA pretreatment decreased the migration of RAW264.7 cells in a dose-dependent manner in comparison with H/R group. *p < 0.05 vs. control group, #p < 0.05 vs. H/R group (H/R challenge alone). C: RAW264.7 cells were pretreated with different concentrations of HSYA or 5 mM NAC for 1 hour, followed by H2h/R4h for 6 hours. Representative Western blot data from three independent experiments is shown. MMP-9 levels were calculated with reference to an H/R-stimulated culture. The data are presented as mean ± S.D. for three different experiments performed in triplicate. *p < 0.05, compared with control group (vehicle group), #p < 0.05, compared with H/R-alone group. D: RAW264.7 cells were incubated in NAC (5 mM) or various concentrations of HSYA, and then exposed to 2 h hypoxia and 4 h of reoxygenation. NAC reduces ROS and served as a positive control. Intracellular ROS production was detected by DCF fluorescence using flow cytometry.
Given that HSYA pretreatment reduced the presence of inflammatory cytokines in mice following hepatic I/R damage, we also evaluated the effect of HSYA on early stage TNF-α and IL-1β secretion via ELISA. Compared with untreated cells, HSYA pretreatment significantly decreased the expression of TNF-α, IL-1β, MCP-1 in RAW264.7 macrophages in a dose-dependent manner (Figure 6). These data suggest that the reduced liver damage observed in HSYA pretreated mice results from the ability of HSYA to prevent macrophage activation, specifically migration and the production of inflammatory cytokines.
Figure 6.

HSYA decreases the production of inflammatory molecules in RAW264.7 macrophages. A: HSYA down-regulated the expression of inflammatory molecules in RAW264.7 macrophages upon H/R challenge. RAW264.7 cells were pretreatment with various concentrations of HSYA, and then exposed to H/R for 2 h hypoxia and 4 h of reoxygenation. The mRNA levels of TNF-α, MCP-1, IL-1β and IL-6 were measured by real-time PCR. B: TNF-α and IL-1β concentration in the supernatant of culture medium TNF-α and IL-1β were measured by ELISA assays. The cells were pre-incubated with p38 MAPK inhibitor SB203580 (10 μM) for 2 h followed by stimulation with H/R for 6 h. The supernatants of culture medium were collected for analyzing TNF-α and IL-1β release. Data are presented as the mean ± S.D. of three independent experiments. *P < 0.05, compared with H/R challenge alone.
HSYA down-regulates the expression of ROS and MMP-9 in RAW264.7 macrophages
To further establish the anti-inflammatory activity of HSYA, we examined effects of HSYA on H/R-induced ROS and MMP-9 protein production by RAW264.7 macrophages. First, RAW264.7 macrophages were exposed to H/R and increasing concentrations of HSYA. After six hours the cells were harvested and ROS expression was evaluated by flow cytometry. As shown in Figure 5D, HSYA significantly reduced expression of ROS in a dose dependent manner with the highest dose (100 μM) inhibiting ROS levels in manner similar to the antioxidant NAC.
Activation of MMP-9 is important for migration of macrophages across the extracellular matrix [24]. To determine if HSYA pretreatment decreased the secretion of this protein, we examined the presence of MMP-9 by Western blot. H/R activated RAW264.7 macrophages pretreated with HSYA expressed reduced levels of MMP-9 protein in a dose dependent manner (Figure 5C). Together these data suggest that HSYA reduces the production of ROS and inhibits migration by reducing the expression of MMP-9.
HSYA inhibits LPS-induced NF-κB activation and P38 phosphorylation in RAW264.7 macrophages
To further characterize the mechanism underlying the effects of HSYA the activation and translocation of NF-κB were examined. Nuclei were isolated, and the NF-κB in the nuclear fraction was quantified by Western blot for the p65 subunit. As shown in Figure 7A and 7B, significant levels of NF-κB p65 accumulated in the nucleus of RAW264.7 macrophages after H/R stimulation (3.82 ± 0.45 folds of control group, p < 0.05); however HSYA inhibited the H/R-induced nuclear translocation of p65 (Figure 7A and 7B).
Figure 7.
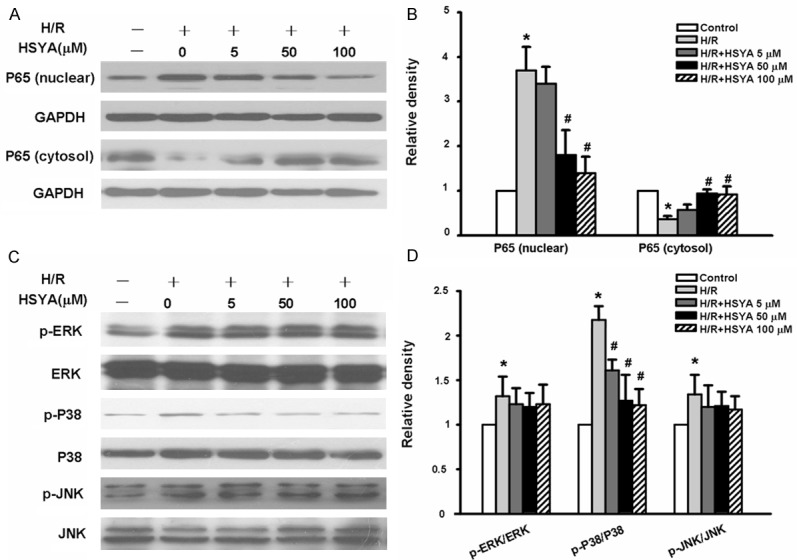
HSYA inhibits H/R-induced NF-κB/p65 nuclear translocation and p38 MAPK activation in RAW264.7 cells. RAW264.7 cells were pretreated with different concentrations of HSYA for 1 hour and then stimulated with 2 h hypoxia and 4 h of reoxygenation. A representative blot (A) and quantitative data (B) from three independent experiments examining the expression of NF-κB/P65 in the nucleus and cytosol are shown. A representative blot (C) and quantitative data (D) on the activation of ERK, p38, and JNK from three independent experiments are shown. The data were presented as mean ± S.D. for experiments performed in triplicate. *p < 0.05 vs. control group. #p < 0.05 vs. H/R group (H/R challenge alone).
The three MAP kinases, ERK1/2, p38 MAPK, and JNK, are important upstream factors leading to the activation of NF-κB [25]. We further addressed the question of whether HSYA could counteract the effect of H/R at this level. As shown in Figure 7C and 7D, the activity of MAPK/ERK1/2, MAPK/JNK and MAPK/P38 were also increased following H/R activation (MAPK/ERK1/2 1.28±0.06 fold versus control group, p < 0.05; MAPK/JNK 1.29±0.07 folds versus control group, p < 0.05; MAPK/P38 2.19±0.04 folds versus control group, p < 0.01) but HSYA only suppressed H/R induced phosphorylation of p38 MAPK but not that of ERK1/2 and JNK. The total amounts of ERK1/2, p38 MAPK, and JNK were not changed by either H/R or HSYA. In order to confirm the causal link of MAP kinase inhibition by HSYA with its suppression of H/R-induced TNF-α and IL-1β formation, the effects of SB 203580, p38 MAPK inhibitors, on H/R were further examined. Similar inhibition of the inflammatory effects of H/R by these inhibitors was identified (Figure 6B). The results thus indicate that inhibition of MAP kinase is an upstream mechanism responsible for the anti-inflammatory effects of HSYA on H/R induced macrophage production the proinflammatory cytokine.
Discussion
The flower of the safflower plant (Carthamus tinctorius) has been regarded as one of the most important traditional Chinese medicine for invigorating blood circulation reducing stasis. Therefore, this plant is used extensively to treat ischemic diseases including, myocardial ischemia, cerebral ischemia, coronary heart disease, and cerebral thrombosis [16,26,27]. Hydroxysafflor yellow A (HSYA) is the major constituent in the hydrophilic fraction of safflower, and exhibits antioxidant, platelet aggregation inhibition, anti-apoptosis, and anti-inflammatory pharmacological activities [14,17,28]. In addition, we previously reported that HSYA attenuates hepatic fibrosis induced by oxidative stress through de-activating PPARgamma [18]. Moreover, HSYA can protect against oleic acid-induced acute lung injury by regulating the PKA pathway [19]. However, the anti-inflammatory functions of HSYA remain unclear. In this study, we demonstrate that preoperative administration of HSYA significantly attenuated hepatic I/R injury. Moreover, the beneficial effects of HSYA on I/R-induced liver injury were, in part, due to attenuation of macrophage recruitment, and production of pro-inflammatory cytokines.
In response to inflammatory signals, macrophages migrate and accumulate at the sites of inflammation. The multifaceted signaling cascade initiated by the inflammatory stimuli leads to macrophage activation and production of a vast array of pro-inflammatory cytokines and chemokines [29]. Liver resident macrophages, named KCs, contribute to the systemic response, local inflammation, clearance of pathogen-derived soluble molecules and toxins from the circulation, and engulfment of apoptotic bodies. These cells are also professional phagocytes and have been widely implicated in various liver injuries [30]. During hepatic I/R injury, KCs are consistently identified as a major contributor of the early post-ischemic oxidant stress and inflammatory reactions, and inactivation/depletion of KCs results in attenuation of injury [31,32]. In this study, we demonstrate that administration of HSYA prior to inducing segmental (70%) hepatic ischemia inhibited recruitment of macrophages after hepatic I/R injury. Accordingly, HSYA pretreatment attenuated I/R-induced necrosis and expression of pro-inflammatory cytokines. Recent studies have revealed that a majority of the proinflammatory cytokines present during I/R injury are produced by KCs, and these cytokines promote hepatic neutrophil infiltration that further contributes to hepatocellular injury [33]. Consistent with these reports, our results suggest that HSYA administration attenuates hepatic I/R injury through suppressing macrophage recruitment and activation.
To understand the underlying mechanism of potential pharmacological activities of HSYA in I/R-induced liver injury, we tested the effects of HSYA on the murine macrophage cell line RAW264.7 in vitro cellular H/R model. We found that HSYA inhibited H/R-induced activation of murine RAW264.7 macrophages. Specifically, HSYA pretreatment decreased the phagocytic activity, reduced migration, and down-regulated the expression of inflammatory cytokines by RAW264.7 cells. Previous studies have also demonstrated that activated KCs generate ROS, which are signals promoting the release of proinflammatory mediators [34]. In addition, blocking hepatic ROS production prevents hepatic I/R injury, suggesting that ROS are major contributors to injury in I/R [35,36]. Similarly, we observed that treatment with HSYA significantly reduced the ROS production in RAW264.7 macrophages upon H/R challenge.
In addition to ROS, MMP-9 secreted by macrophages is a critical mediator of monocyte and leukocyte migration in hepatic I/R injury [24]. Pretreatment of RAW264.7 cells with HSYA strongly inhibited the expression of MMP-9 and inhibited cell migration in RAW264.7 macrophages. These results confirm previous reports demonstrating that ROS and MMP-9 play an important role in regulating migratory response to macrophages [24,36], and suggest that HYSA prevention hepatic injury by down-regulating the expression of these proteins.
Several intersecting signaling pathways have been implicated in the development of I/R injury. The mammalian transcription factor NF-κB is a pleiotropic regulator of various genes involved in inflammatory processes [37]. During activation of the inflammatory cascade, the p65 subgroup of NF-κB translocates to the nucleus [38]. In this study, we demonstrate that HSYA inhibited NF-κB/p65 nuclear translocation after H/R stimulation. In addition to NF-kB, mitogen-activated protein kinases (MAPKs), which are serine/threonine-specific kinases responding extracellular stimuli, are important for regulating various cellular activities. These kinases are classified into three distinct groups, including ERK1/2, c-Jun N-terminal kinase (JNK) and p38 MAP kinase [39]. MAPKs modulate the transcription of many genes involved in the inflammatory process. Among them, p38 MAPK plays an important role in transferring inflammatory signals and cytokine synthesis in H/R-induced acute liver injury. What we further showed was that HSYA significantly inhibited the phosphorylation, or activation, of p38 MAPK by H/R with the total protein levels of MAPK remaining unchanged, indicating that HSYA blocked the activation but not biosynthesis of MAPKs. The inhibition on p38MAPK activation by HSYA could be a causal factor for its suppression of HSYA-induced the proinflammatory cytokine formation especially TNF-α and IL-1β. As shown in this paper, specific inhibitors of p38 MAPK also suppressed TNF-α and IL-1β formation by H/R. Put together, our results indicate that the anti-inflammatory effects of HSYA are initiated by suppression of p38 MAPK, resulting in inhibition of NF-κB activation and the proinflammatory cytokine formation.
Although multiple mechanisms are likely involved, we have demonstrated that the protective effect of HSYA on I/R-induced liver injury is, in part, mediated by the direct modulation of the macrophage activities. The major mechanisms of these beneficial effects may include the direct suppression of macrophage migration and production of ROS and inflammatory cytokines. Thus, this study supports the use of HSYA pretreatment as a potential therapeutic for attenuating I/R-induced liver injury during liver surgery.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81073123) and the Development planning Program of Shandong (Grant No. 2011YD18001).
Disclosure of conflict of interest
All authors have no conflict of interest.
References
- 1.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 2.Bahde R, Spiegel HU. Hepatic ischaemia-reperfusion injury from bench to bedside. Br J Surg. 2010;97:1461–1475. doi: 10.1002/bjs.7176. [DOI] [PubMed] [Google Scholar]
- 3.Belghiti J, Hiramatsu K, Benoist S, Massault P, Sauvanet A, Farges O. Seven hundred forty-seven hepatectomies in the 1990s: an update to evaluate the actual risk of liver resection. J Am Coll Surg. 2000;191:38–46. doi: 10.1016/s1072-7515(00)00261-1. [DOI] [PubMed] [Google Scholar]
- 4.Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10:79–89. doi: 10.1038/nrgastro.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klune JR, Tsung A. Molecular biology of liver ischemia/reperfusion injury: established mechanisms and recent advancements. Surg Clin North Am. 2010;90:665–677. doi: 10.1016/j.suc.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant. 2011;11:1563–1569. doi: 10.1111/j.1600-6143.2011.03579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abu-Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver ischemia/reperfusion injury: processes in inflammatory networks--a review. Liver Transpl. 2010;16:1016–1032. doi: 10.1002/lt.22117. [DOI] [PubMed] [Google Scholar]
- 8.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26:1175–1186. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 9.Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Pina E, Geller DA. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res. 2008;147:153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhogal RH, Sutaria R, Afford SC. Hepatic liver ischemia/reperfusion injury: processes in inflammatory networks--a review. Liver Transpl. 2011;17:95. doi: 10.1002/lt.22205. author reply 96. [DOI] [PubMed] [Google Scholar]
- 11.Tomiyama K, Ikeda A, Ueki S, Nakao A, Stolz DB, Koike Y, Afrazi A, Gandhi C, Tokita D, Geller DA, Murase N. Inhibition of Kupffer cell-mediated early proinflammatory response with carbon monoxide in transplant-induced hepatic ischemia/reperfusion injury in rats. Hepatology. 2008;48:1608–1620. doi: 10.1002/hep.22482. [DOI] [PubMed] [Google Scholar]
- 12.Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32:169–173. doi: 10.1053/jhep.2000.9323. [DOI] [PubMed] [Google Scholar]
- 13.Feng ZM, He J, Jiang JS, Chen Z, Yang YN, Zhang PC. NMR solution structure study of the representative component hydroxysafflor yellow A and other quinochalcone C-glycosides from Carthamus tinctorius. J Nat Prod. 2013;76:270–274. doi: 10.1021/np300814k. [DOI] [PubMed] [Google Scholar]
- 14.He H, Yang X, Shi M, Zeng X, Yang J, Wu L, Li L. Protective effects of hydroxysafflor yellow A on acute and chronic congestive cardiac failure mediated by reducing ET-1, NOS and oxidative stress in rats. J Pharm Pharmacol. 2008;60:115–123. doi: 10.1211/jpp.60.1.0015. [DOI] [PubMed] [Google Scholar]
- 15.Tian Y, Yang ZF, Li Y, Qiao Y, Yang J, Jia YY, Wen AD. Pharmacokinetic comparisons of hydroxysafflower yellow A in normal and blood stasis syndrome rats. J Ethnopharmacol. 2010;129:1–4. doi: 10.1016/j.jep.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 16.Ye SY, Gao WY. Hydroxysafflor yellow A protects neuron against hypoxia injury and suppresses inflammatory responses following focal ischemia reperfusion in rats. Arch Pharm Res. 2008;31:1010–1015. doi: 10.1007/s12272-001-1261-y. [DOI] [PubMed] [Google Scholar]
- 17.Sun CY, Pei CQ, Zang BX, Wang L, Jin M. The ability of hydroxysafflor yellow a to attenuate lipopolysaccharide-induced pulmonary inflammatory injury in mice. Phytother Res. 2010;24:1788–1795. doi: 10.1002/ptr.3166. [DOI] [PubMed] [Google Scholar]
- 18.Wang CY, Liu Q, Huang QX, Liu JT, He YH, Lu JJ, Bai XY. Activation of PPARgamma is required for hydroxysafflor yellow A of Carthamus tinctorius to attenuate hepatic fibrosis induced by oxidative stress. Phytomedicine. 2013;20:592–599. doi: 10.1016/j.phymed.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Wang C, Huang Q, Wang C, Zhu X, Duan Y, Yuan S, Bai X. Hydroxysafflor yellow A suppresses oleic acid-induced acute lung injury via protein kinase A. Toxicol Appl Pharmacol. 2013;272:895–904. doi: 10.1016/j.taap.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 20.Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol. 2002;282:C227–241. doi: 10.1152/ajpcell.00112.2001. [DOI] [PubMed] [Google Scholar]
- 21.Jiang SJ, Li W, An W. Adenoviral gene transfer of hepatic stimulator substance confers resistance against hepatic ischemia-reperfusion injury by improving mitochondrial function. Hum Gene Ther. 2013;24:443–456. doi: 10.1089/hum.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abe Y, Hines IN, Zibari G, Pavlick K, Gray L, Kitagawa Y, Grisham MB. Mouse model of liver ischemia and reperfusion injury: method for studying reactive oxygen and nitrogen metabolites in vivo. Free Radic Biol Med. 2009;46:1–7. doi: 10.1016/j.freeradbiomed.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanschen M, Zahler S, Krombach F, Khandoga A. Reciprocal activation between CD4+ T cells and Kupffer cells during hepatic ischemia-reperfusion. Transplantation. 2008;86:710–718. doi: 10.1097/TP.0b013e3181821aa7. [DOI] [PubMed] [Google Scholar]
- 24.Hamada T, Fondevila C, Busuttil RW, Coito AJ. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology. 2008;47:186–198. doi: 10.1002/hep.21922. [DOI] [PubMed] [Google Scholar]
- 25.Nakano H, Shindo M, Sakon S, Nishinaka S, Mihara M, Yagita H, Okumura K. Differential regulation of IkappaB kinase alpha and beta by two upstream kinases, NF-kappaB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc Natl Acad Sci U S A. 1998;95:3537–3542. doi: 10.1073/pnas.95.7.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu YN, Zhou ZM, Chen P. Evidence that hydroxysafflor yellow A protects the heart against ischaemia-reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Clin Exp Pharmacol Physiol. 2008;35:211–216. doi: 10.1111/j.1440-1681.2007.04814.x. [DOI] [PubMed] [Google Scholar]
- 27.Nie PH, Zhang L, Zhang WH, Rong WF, Zhi JM. The effects of hydroxysafflor yellow A on blood pressure and cardiac function. J Ethnopharmacol. 2012;139:746–750. doi: 10.1016/j.jep.2011.11.054. [DOI] [PubMed] [Google Scholar]
- 28.Liu SX, Zhang Y, Wang YF, Li XC, Xiang MX, Bian C, Chen P. Upregulation of heme oxygenase-1 expression by hydroxysafflor yellow A conferring protection from anoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes. Int J Cardiol. 2012;160:95–101. doi: 10.1016/j.ijcard.2011.03.033. [DOI] [PubMed] [Google Scholar]
- 29.Ivashkiv LB. Inflammatory signaling in macrophages: transitions from acute to tolerant and alternative activation states. Eur J Immunol. 2011;41:2477–2481. doi: 10.1002/eji.201141783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, Gores GJ. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38:1188–1198. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]
- 31.Hsu CM, Wang JS, Liu CH, Chen LW. Kupffer cells protect liver from ischemia-reperfusion injury by an inducible nitric oxide synthase-dependent mechanism. Shock. 2002;17:280–285. doi: 10.1097/00024382-200204000-00007. [DOI] [PubMed] [Google Scholar]
- 32.Nace GW, Huang H, Klune JR, Eid RE, Rosborough BR, Korff S, Li S, Shapiro RA, Stolz DB, Sodhi CP, Hackam DJ, Geller DA, Billiar TR, Tsung A. Cellular-specific role of toll-like receptor 4 in hepatic ischemia-reperfusion injury in mice. Hepatology. 2013;58:374–387. doi: 10.1002/hep.26346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perry BC, Soltys D, Toledo AH, Toledo-Pereyra LH. Tumor necrosis factor-alpha in liver ischemia/reperfusion injury. J Invest Surg. 2011;24:178–188. doi: 10.3109/08941939.2011.568594. [DOI] [PubMed] [Google Scholar]
- 34.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 35.Lehmann TG, Wheeler MD, Schwabe RF, Connor HD, Schoonhoven R, Bunzendahl H, Brenner DA, Jude Samulski R, Zhong Z, Thurman RG. Gene delivery of Cu/Zn-superoxide dismutase improves graft function after transplantation of fatty livers in the rat. Hepatology. 2000;32:1255–1264. doi: 10.1053/jhep.2000.19814. [DOI] [PubMed] [Google Scholar]
- 36.Soumyarani VS, Jayakumari N. Oxidatively modified high density lipoprotein promotes inflammatory response in human monocytes-macrophages by enhanced production of ROS, TNF-alpha, MMP-9, and MMP-2. Mol Cell Biochem. 2012;366:277–285. doi: 10.1007/s11010-012-1306-y. [DOI] [PubMed] [Google Scholar]
- 37.Schwabe RF, Brenner DA. Nuclear factor-kappaB in the liver: friend or foe? Gastroenterology. 2007;132:2601–2604. doi: 10.1053/j.gastro.2007.04.058. [DOI] [PubMed] [Google Scholar]
- 38.Llacuna L, Mari M, Lluis JM, Garcia-Ruiz C, Fernandez-Checa JC, Morales A. Reactive oxygen species mediate liver injury through parenchymal nuclear factor-kappaB inactivation in prolonged ischemia/reperfusion. Am J Pathol. 2009;174:1776–1785. doi: 10.2353/ajpath.2009.080857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Narang H, Krishna M. Mitogen-activated protein kinases: specificity of response to dose of ionizing radiation in liver. J Radiat Res. 2004;45:213–220. doi: 10.1269/jrr.45.213. [DOI] [PubMed] [Google Scholar]