

Abstract
Tight junction plays a critical role in intestinal defence. The alteration and perturbation of tight junction proteins could induce intestine barrier damage, and lead to the malabsorption of electrolytes and water. Previous studies had showed that colonic infection and inflammation could lead to the alteration of tight junction function, and somatostatin could protect intestinal epithelia. Thus, this study could explore that whether somatostatin could regulate tight junction in colitis mice. Colitis mice with diarrhea were induced by Citrobacter rodentium (CR) and Dextran sulfate sodium (DSS). In CR infected model, cladudin-1 and claudin-3 expression significantly decreased compared with the control mice (P < 0.05); after octreotide treatment, claudin-1 and claudin-3 expression significantly increased compared with untreated CR infected mice (P < 0.05). In DSS colitis model, occludin and claudin-3 expression significantly decreased compared with the control mice (P < 0.05); and octreotide treatment could only significantly upregulate claudin-3 expression compared with untreated DSS colitis mice (P < 0.05). To testify our results in vivo, we repeated the models in caco-2 cells by exposed with enteropathogenic Escherichia coli (E. Coli) and Tumor necrosis factor α (TNF-α). The results in vitro were consistent with in vivo study. The results suggested that somatostatin play a role in intestinal barrier protection by modulating tight junction proteins expression.
Keywords: Tight junction, claudins, occludin, somatostatin, infectious colitis, IBD
Introduction
Tight junctions are located apically in the intercellular junctions, and they contribute to maintaining homeostasis in the intestinal lumen. Multiple proteins that form the tight junction have been identified, including transmembrane proteins of occludin family, claudin family, zona occludens (ZOs), the junction adhesion molecules (JAMs), and the Coxsackievirus and Adenovirus Receptor (CAR) proteins [1]. Abundant studies have documented the crucial position of tight junction in protecting the intestine from toxic substances [2,3]. Decrease in tight junction protein expression could result in the alteration or disruption of the intestinal barrier, which contributes to infection, diarrhea, pyemia, and sepsis. Previous studies have demonstrated that occludin, claudins, ZOs and other tight junction proteins were disrupted in inflammatory bowel diseases (IBD), irritable bowl syndrome (IBS), and infectious diarrhea [4-14]. Thus the integrity of epithelial barrier should be preserved to prevent electrolyte loss and toxin penetration.
Somatostatin, a neuropeptide found in D cells, which are distributed throughout the intestinal tract, functions as a neurotransmitter and a hormone [15]. Studies have shown that somatostatin stimulates Na+ and Cl- absorption, reduces water and electrolyte secretion, and inhibits intestinal mobility [16]. Octreotide, a somatostatin analog, has been used to treat diarrhea for decades [17]. Furthermore, somatostatin had also been shown to stimulate the expression of claudin-4 in human keratinocytes and protect epithelial tight junctions in rats with acute necrotizing pancreatitis. Thus somatostatin likely modulates tight junction proteins [18,19].
In the present study, we explored the effect of somatostatin on tight junction in colitis mice induced by bacterial infection and by intestinal inflammation. Our results suggested that somatostatin could regulate claudin-1 and claudin-3 protein expression in the colon of colitis mice.
Materials and methods
Bacterial strains
E. coli O127:K63 (EPEC) was purchased from China National Research Institute of Food & Fermentation Industries (Beijing, China). Citrobacter rodentium (CR) was purchased from ATCC (Manassas, VA, USA). Both bacteria were grown in LB media at 37°C overnight. CR culture was centrifuged at 1600 g for 5 min, and the bacteria were resuspended in sterile phosphate-buffered saline (PBS) at final concentration of 2.5×108 cfu/ml before use. EPEC culture was centrifuged at 10,000 rpm for 3 min, and the bacteria were suspended in cell culture medium at final concentration of 1×108 cfu/ml before use.
Animals
Six to eight weeks old female C57BL/6 mice were purchased from Animal Experiment Center of West China Hospital (Sichuan University, Chengdu, China). Mice were maintained in specific pathogen-free facility for at least four days before starting experiments. To induce diarrhea via bacterial infection, mice were gavaged with CR (5×107 cfu in 0.2 ml PBS). Thirteen days after gavage, treatment groups were administrated with octreotide at dose of 50 μg/kg body weight three times a day for three days. Sixteen days after gavage, mice were euthanized and colonic tissues were collected for western blot detecting and histological assay. To induce diarrhea via intestinal inflammation, mice were fed with 3% DSS water for seven days. On the eighth day, treatment groups were administrated with octreotide at dose of 50 μg/kg body weight three times a day for three days. On the eleventh day, mice were euthanized and colonic tissues were collected. Diarrheal symptoms were assessed every other day. Diarrheal score was recorded based on fecal shape, color and hardness. Diarrheal score higher than 2.0 is considered to have diarrheal symptom according to previously established standards [20]. The experiments were repeated in three groups with three mice in each group. The experiments were repeated in three groups with three mice in each group. All experiments were approved by the Institutional Animal Care and Use Committee of Sichuan University (Chengdu, China).
Cell cultures
Caco-2 cells were kindly provided by West China School of Pharmacy (Sichuan University, Chengdu, China). Cells were cultured in 75 mm2 flask at 37°C in 95% air -5% CO2. MEM-NEAA medium containing 50 U/ml penicillin, 50 μg/ml streptomycin, and 20% fetal bovine serum was used for cell culture. Cell culture-related products were purchased from Hyclone (Thermal scientific; USA). For the EPEC infection study, 1×108 cfu bacteria (in 10 ml antibiotic-free MEM-NEAA medium) were incubated with Caco-2 cells for 1 hour. Then added 1 μM somatostatin to coincubate for 1 hour. Cells were washed with PBS three times to remove unattached bacteria. For the TNF-α study, Caco-2 cells were incubated with 100 ng/ml TNF-α for 18 hours before adding 1 μM somatostatin. Cells were exposed to somatostatin for 1 hour before harvest.
Protein isolation and western blot
Total protein was prepared from colonic tissues and Caco-2 cells using cell lyses buffer for Western and IP kit (Beyotime; Beijing, China) following manufacturer’s instruction. The following antibody dilutions were used for Western detection: 1:1000 dilution for occludin (Santa cruz; USA); 1:200 dilution for claudin-1 (Invitrogen; USA); 1:2500 for claudin-3 (Invitrogen; USA); detection in tissue; 1:500 dilution for claudin-3 (Invitrogen; USA) detection in cells; and 1:1000 dilution for GAPDH antibody (Good Here; Hangzhou, China). Western blot detection and protein expression quantity were performed using previously described methods [15].
Colonic histology observation and claudin-3 staining in DSS-colitis mice
Colonic tissue was collected from mice and sectioned for hematoxylin-eosin (H&E) stain and claudin-3 immunohistochemical (IHC) assay. Briefly, colonic tissues were fixed in 10% formalin, and embedded in paraffin. For H&E stain, tissues were stained with hematoxylin and eosin; for claudin-3 IHC assay, deparaffinized tissues were treated with 0.3% (v/v) hydrogen peroxide in 60% (v/v) methanol for 30 min to quench endogenous peroxidase. After three washes with TBS, tissue sections were incubated with anti claudin-3 antibody (1:400) overnight. Then tissue sections were incubated with HRP-conjugated anti-rabbit IgG (Bioss; Beijing, China) and detected by DAB (Bioss; Beijing, China). All sections were viewed under microscope.
Somatostatin detection
Colonic tissue was collected from mice for somatostatin radioimmunoassay. Samples were sent to Beijing DORUN International Technology Co. Ltd. (Beijing, China) for the assays.
Statistical analysis
ANOVA-Tukey test was used for comparison among three groups. Student’s t-test was used for comparison between two groups. Data were presented as mean ± SE. P-values < 0.05 was considered significant.
Results
Octreotide treatment improves diarrheal symptoms in CR mice and in DSS colitis mice
To observe the loss of electrolyte and water in colitis mice, we detected the diarrhea scores of two models. In CR-infected mice, diarrhea started on day 7 with diarrheal score of 2.4 ± 0.24 and reached a plateau after day 9 with diarrheal score of 3.0 ± 0.00. After administration of octreotide for three days (day 13 through day 15), octreotide-treated CR-infected mice had a lower diarrheal score than untreated CR-infected mice (2.2 ± 0.2 in octreotide-treated group vs. 3.2 ± 0.2 in untreated group, n = 3, P < 0.05) (Figure 1A).
Figure 1.
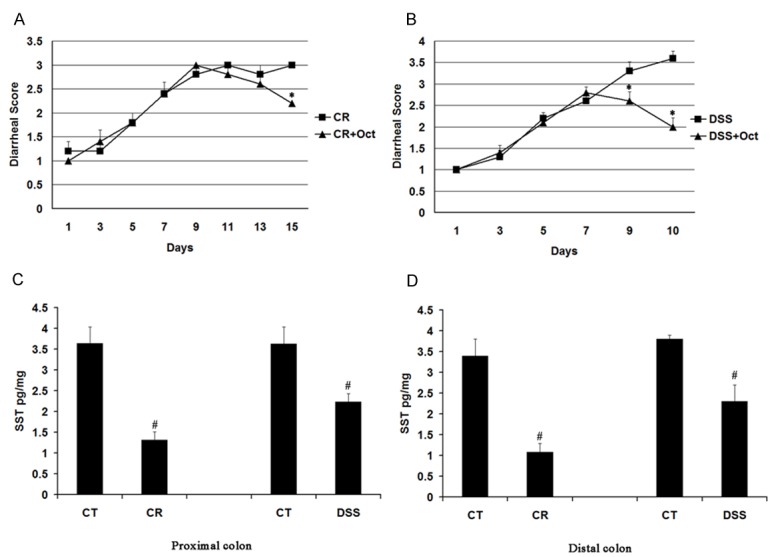
Diarrhea scores and somatostatin assay in CR infected mice and DSS colitis mice. Mice were infected with CR or fed with DSS water before octreotide treatment. Diarrhea scores were recorded and somatostatin was measured. CT: control group; CR: CR infected group; DSS: DSS colitis group; Oct: Octreotide. Data are presented as mean ± SE from three groups of mice (3 mice/group). *p < 0.05 for treatment mice (Oct) vs. CR mice (CR) or DSS mice (DSS); #p < 0.05 for CR mice (CR) and DSS mice vs. control mice (CT). A: Diarrheal score in CR infected mice. B: Diarrheal score in DSS colitis mice. C: Somatostatin levels in the proximal colon. D: Somatostatin level in the distal colon.
In DSS mice, diarrhea started on the fifth day with diarrheal score of 2.2 ± 0.1 and worsened through 10 days of the experiment even after DSS water was replaced with normal water. After administration of octreotide for three days (day 8 through day 10), octreotide-treated DSS mice had a lower diarrheal score than untreated DSS mice (1.7 ± 0.78 in octreotide-treated group vs. 3.6 ± 0.16 in untreated group to, n = 3, P < 0.05) (Figure 1B).
Further study of somatostatin level by radioimmunoassay showed that colonic somatostatin level was decreased in CR-infected mice and DSS mice. In the proximal colon, somatostatin level was reduced from 3.64 ± 0.4 pg/mg in control mice to 1.31 ± 0.2 pg/ml in CR-infected mice and from 3.63 ± 0.4 pg/mg in control mice to 2.23 ± 0.23 pg/mg in DSS mice (n = 3, P < 0.05) (Figure 1C). In the distal colon, somatostatin level was reduced from 3.4 ± 0.4 pg/mg in control mice to 1.08 ± 0.21 pg/mg in CR-infected mice and from 3.80 ± 0.4 pg/mg in control mice to 2.30 ± 0.40 pg/ml in DSS mice (n = 3, P < 0.05) (Figure 1D).
Somatostatin increases claudin-1 and claudin-3 expression in CR-infected mice
Western blot detection showed that CR infection had no effect on occludin expression in the proximal or the distal colon (data not shown). Interestingly, claudin-1 expression was decreased in CR-infected mice in the proximal and the distal colon (0.42 ± 0.02 in CR-infected vs. 0.49 ± 0.02 in control mice in the proximal colon; 0.44 ± 0.02 in CR-infected mice vs. 0.51 ± 0.06 in control mice in the distal colon; n = 3, P < 0.05). After three days of octreotide treatment, claudin-1 protein expression in the colon was restored in treatment group compared with CR-infected mice (0.52 ± 0.02 in octreotide-treated group vs. 0.42 ± 0.02 in untreated group in the proximal colon; 0.58 ± 0.1 in octreotide-treated group vs. 0.44 ± 0.02 in untreated group in the distal colon; n = 3, P < 0.05) (Figure 2A and 2B).
Figure 2.
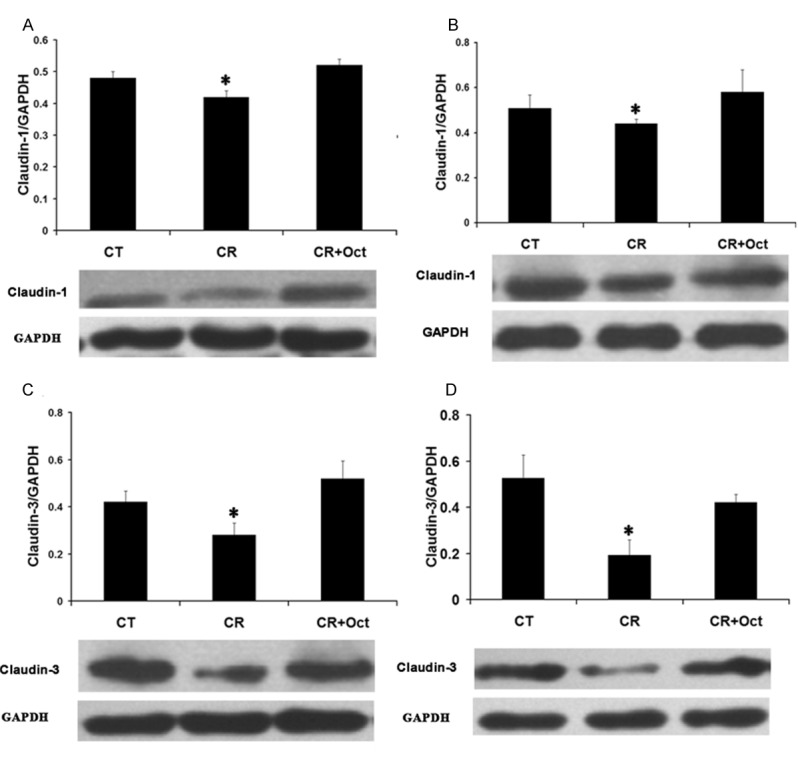
Effect of octreotide on claudin-1 and claudin-3 expression in CR infected mice. Colonic tissues were collected from control mice (CT), CR infected mice (CR) and octreotide treated mice (CR+Oct). Tissue lysate was used to detect claudin-1 and claudin-3 expression by western blot. Data are presented as mean ± SE from 3 groups of mice (3 mice/group). *p < 0.05 for CR mice (CR) vs. control mice (CT) and octreotide treated mice (Oct). A: The ratio of optical density of claudin-1 over GAPDH in the proximal colon. B: The ratio of optical density of claudin-1 over GAPDH in the distal colon. C: The ratio of optical density of claudin-3 over GAPDH in the proximal colon. D: The ratio of optical density of claudin-3 over GAPDH in the distal colon.
Compared to control mice, claudin-3 expression was also decreased in CR-infected mice (0.28 ± 0.05 in CR-infected mice vs. 0.42 ± 0.05 in control mice in the proximal colon; 0.19 ± 0.06 in CR-infected mice vs. 0.53 ± 0.1 in control mice in the distal colon; n = 3, P < 0.05). Similar to claudin-1, octreotide treatment also increased claudin-3 expression in treatment group compared with CR group (0.52 ± 0.07 in octreotide-treated group vs. 0.28 ± 0.05 in untreated group in the proximal colon; 0.42 ± 0.03 in octreotide-treated group vs. 0.19 ± 0.06 in untreated group in the distal colon; n = 3, P < 0.05) (Figure 2C and 2D).
Somatostatin increases claudin-3 expression in DSS mice
Western blot detection showed that DSS colitis has no effect on claudin-1 expression in mice (data not shown). But occludin expression was significantly reduced in DSS colitis mice (0.37 ± 0.02 in DSS mice vs. 0.67 ± 0.1 in control mice in the proximal colon; 0.54 ± 0.02 in DSS mice vs. 0.78 ± 0.05 in control mice in the distal colon; n = 3, P < 0.05). After three days of octreotide treatment, occludin protein expression in the colon was not restored in treatment group (0.3 ± 0.04 in octreotide-treated group Figure 4. Immunohistochemical assay of claudin-3 in mucosal epithelium in DSS colitis mice. Histological observation of colonic tissues from control mice (CT), DSS mice (DSS) and octrotide treated mice (Oct). Tissues were used for histological assay. A: H&E stain of mice colon tissues. B: Immunohistochemical staining shows Claudin-3 in yellow. While Claudin-3 is decreased in DSS mice colon, treatment with Oct restored Claudin-3 expression. vs. 0.37 ± 0.02 in untreated group in the proximal colon; 0.5 ± 0.03 in octreotide-treated group vs. 0.54 ± 0.02 in untreated group in the distal colon; n = 3, P > 0.05) (Figure 3A and 3B).
Figure 4.
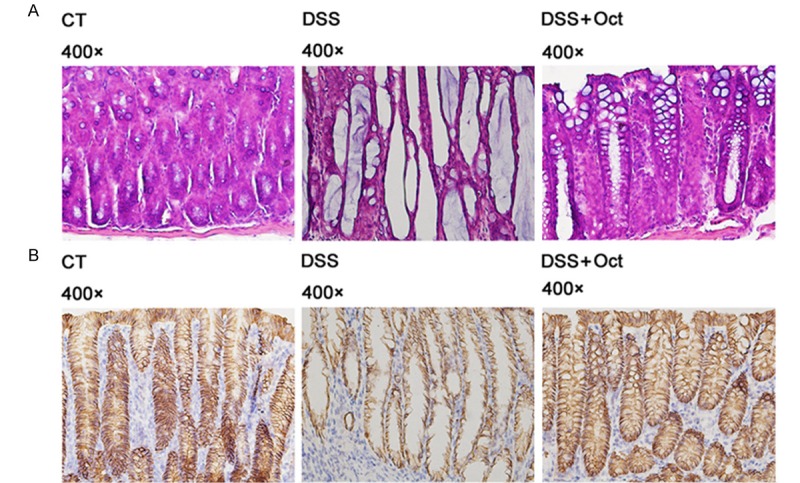
Immunohistochemical assay of claudin-3 in mucosal epithelium in DSS colitis mice. Histological observation of colonic tissues from control mice (CT), DSS mice (DSS) and octrotide treated mice (Oct). Tissues were used for histological assay. A: H&E stain of mice colon tissues. B: Immunohistochemical staining shows Claudin-3 in yellow. While Claudin-3 is decreased in DSS mice colon, treatment with Oct restored Claudin-3 expression.
Figure 3.
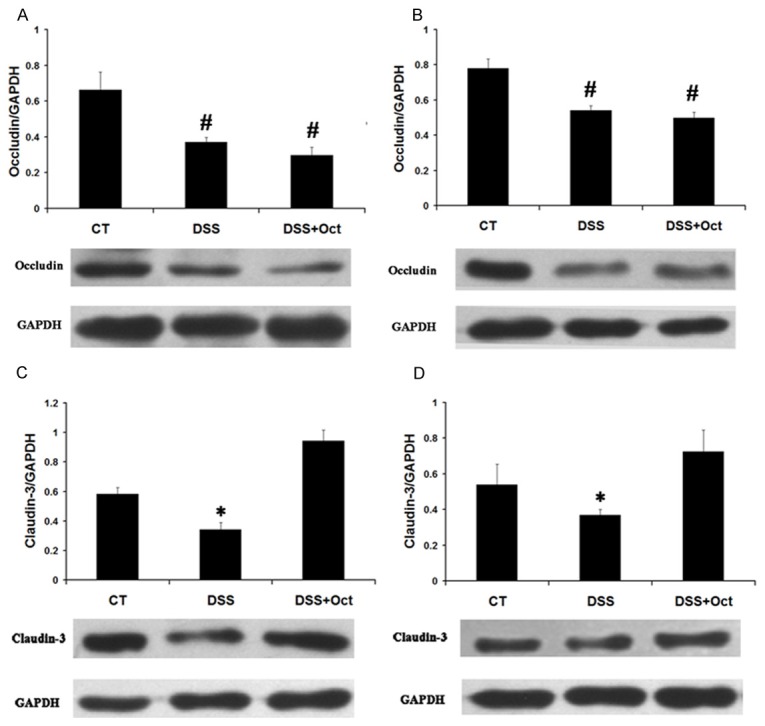
Effect of octreotide on occludin and claudin-3 expression in DSS colitis mice. Colonic tissues were collected from control mice (CT), DSS mice (DSS) and octrotide treated mice (DSS+Oct). Tissue lysate was used to detect occludin and claudin-3 expression by western blot. Tissue lysate was used to detect occludin and claudin-3 expression by western blot. Data are presented as mean ± SE from 3 groups of mice (3 mice/group). #p < 0.05 for DSS mice (DSS) and octreotide mice (Oct) vs. control mice; *p < 0.05 for DSS mice (DSS) vs. control mice (CT) and octreotide treated mice (Oct). A: The ratio of optical density of occludin over GAPDH in the proximal colon. B: The ratio of optical density of occludin over GAPDH in the distal colon. C: The ratio of optical density of claudin-3 over GAPDH in the proximal colon. D: The ratio of optical density of claudin-3 over GAPDH in the proximal colon.
DSS colitis also affected claudin-3 expression in mice. The expression of claudin-3 was decreased in DSS mice compared with control mice (0.34 ± 0.04 in DSS mice vs. 0.58 ±0.04 in control mice in the proximal colon; 0.36 ± 0.03 in DSS mice vs. 0.53 ± 0.1 in control mice in the distal colon; n = 3, P < 0.05). After three days of octreotide treatment, claudin-3 protein expression was increased from 0.34 ± 0.04 in untreated group to 0.94 ± 0.07 in octreotide-treated group in the proximal colon (n = 3, P < 0.05) and from 0.36 ± 0.03 in untreated group to 0.73 ± 0.1 in octreotide-treated group in the distal colon (n = 3, P < 0.05) (Figure 3C and 3D).
To verify the results of western blot and detect Claudin-3 location, we performed histological assay on colonic tissues. As claudin-3 protein expression changed in both two models and up-regulated by octreotide in same pattern, we selectively analyzed the claudin-3 proteins in the colon of DSS colitis mice. H&E stain showed that DSS disrupted the epithelial layer structure in the colon, and octreotide treatment partially restored the epithelial layer structure (Figure 4A). Claudin-3 IHC stain indicated that Claudin-3 locates between epithelial cells and reduced in DSS mice, treatment with octreotide restored Claudin-3 expression (Figure 4B).
Somatostatin increases claudin-1 and claudin-3 in Caco-2 cells
Caco-2 cells were exposed to 1×107 cfu/ml EPEC for 1 hour or 100 ng/ml TNF-α for 18 hours before they were treated with 1 μM somatostatin for 1 hour. Total protein was extracted for Western blot detection. Western blot showed that the expression of claudin-3 was decreased in both EPEC-infected and TNF-α-treated cells (0.73 ± 0.05 in control cells, 0.4 ± 0.06 in EPEC-infected cells, and 0.48 ± 0.03 in TNF-α treated cells, n = 3, P < 0.05). After somatostatin treatment, claudin-3 protein expression increased in EPEC-infected cells (0.4 ± 0.06 in untreated cells vs. 0.9 ± 0.2 in treated cells, n = 3, P < 0.05). In cells exposed to TNF-α, somatostatin similarly increased claudin-3 protein expression (0.48 ± 0.03 in untreated cells vs. 0.69 ± 0.05 in treated cells, n = 3, P < 0.05) (Figure 5A & 5B).
Figure 5.
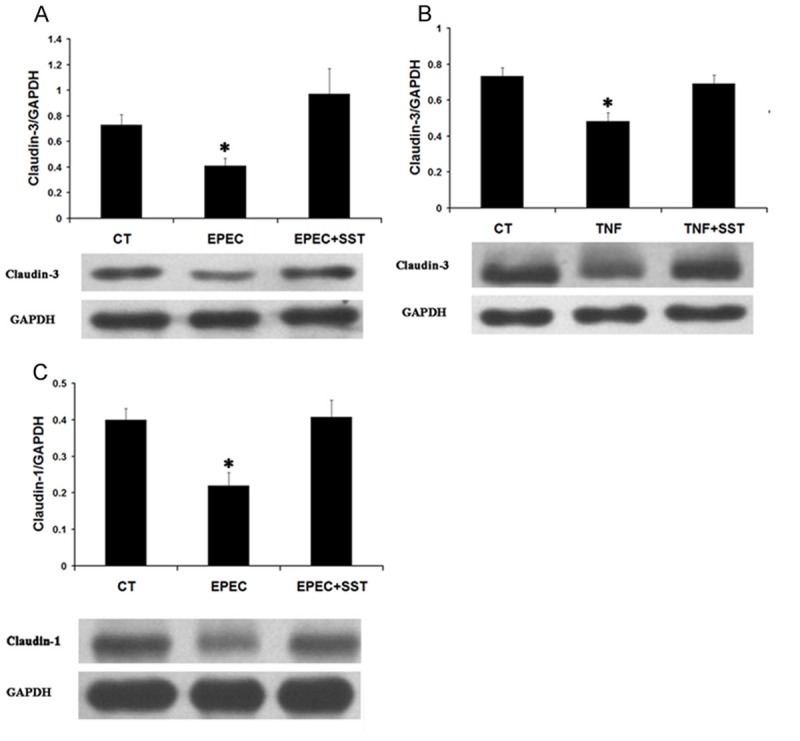
Effect of somatostatin on claudin-1 and claudin-3 expression in Caco-2 cells. Caco-2 cells were preincubated with 1×107 cfu/ml EPEC for 1 hour or 100 ng/ml TNF-α for 18 hours before adding 1 μM somatostatin. Cell lysate was used to detect claudin-1 and claudin-3 expression by western blot. Data are presented as Mean ± SE from 3 separate experiments. *p < 0.05 for EPEC or TNF-α treated cells vs. control cells and somatostatin treated cells. A: The ratio of optical density of claudin-3 over GAPDH in EPEC infected cells. B: The ratio of optical density of claudin-3 over GAPDH in TNF-α treated cells. C: The ratio of optical density of claudin-1 over GAPDH in EPEC infected cells.
Interestingly, claudin-1 expression in Caco-2 cells was only affected by EPEC treatment and not by TNF-α treatment. In EPEC-treated Caco-2 cells, claudin-1 expression was reduced from 0.4 ± 0.03 in control cells to 0.22 ± 0.03 in EPEC-infected cells (n = 3, P < 0.05). After somatostatin treatment, claudin-1 protein Figure 5. Effect of somatostatin on claudin-1 and claudin-3 expression in Caco-2 cells. Caco-2 cells were preincubated with 1×107 cfu/ml EPEC for 1 hour or 100 ng/ml TNF-α for 18 hours before adding 1 μM somatostatin. Cell lysate was used to detect claudin-1 and claudin-3 expression by western blot. Data are presented as Mean ± SE from 3 separate experiments. *p < 0.05 for EPEC or TNF-α treated cells vs. control cells and somatostatin treated cells. A: The ratio of optical density of claudin-3 over GAPDH in EPEC infected cells. B: The ratio of optical density of claudin-3 over GAPDH in TNF-α treated cells. C: The ratio of optical density of claudin-1 over GAPDH in EPEC infected cells. expression in EPEC-infected cells increased (0.22 ± 0.03 in untreated cells vs. 0.41 ± 0.04 in treated cells, n = 3, P < 0.05) (Figure 5C).
Discussion
The intestinal epithelia dysfunction is an important contributing factor to intestinal dysfunction in IBD and bacterial infection. This dysfunction often involves the tight junction, which is an important protective barrier in the intestine. Tight junction structure may be disrupted by bacterial pathogens [21], and they has been shown to be impaired in IBD patients [4]. We have previously shown that somatostatin stimulates NHE8 expression in intestine [15], and others showed that somatostatin up-regulates tight junction claudin-4 expression in human keratinocytes [18]. Therefore, somatostatin may modulate tight junction function in pathologic status.
Reduction of somatostatin levels has been reported in IBD patients [22]. To explore if the same pattern exists in mice suffering diarrhea, we used CR infection and DSS colitis models. To establish CR infection mouse model, mice were gavaged with CR bacteria to induce diarrhea. Other groups have used gavage to introduce A/E pathogen infection in mice [23,24]. To establish colonic inflammation, mice were fed with 3% DSS water for seven days; this is a widely accepted method to induce acute colitis in rodents [25]. Our results showed that mice in both models developed diarrhea and the level of somatostatin in the colon was significantly reduced in CR infected mice and in DSS colitis mice. These observations suggested that somatostatin level was decreased in diarrheal conditions.
Tight junction protein alteration has been reported in IBD patients and mice [4,26,27]. In the current study, we observed tight junction changes in CR-infected mice and in DSS mice. In CR-infected mice, expression of claudin-1 and claudin-3 was significantly reduced by ~15% and ~50% respectively. In DSS mice, expression of occludin and claudin-3 were both significantly reduced by ~36%. Interestingly, occludin expression was not altered in CR-infected mice while claudin-1 expression was not altered in DSS mice. These observations suggested that the expressions of tight junction proteins are not uniformly affected by diarrhea states. Because somatostatin level was decreased in both diarrhea mouse models, we treated these mice with octreotide, an analog of somatostatin. Octreotide treatment not only improved diarrhea symptoms but also stimulated tight junction protein expression in these mice. In CR-infected mice, octreotide treatment restored the expression of claudin-1 and claudin-3 to control level. In DSS mice, octreotide treatment also restored the expression of claudin-3. Interestingly, octreotide treatment did not restore the expression of occludin in CR-infected mice or DSS mice, which suggests that the role of somatostatin on tight junction is protein-specific. In spite of not affecting occludin, results from CR-infected mice and DSS mice on expression of claudin proteins suggest that somatostatin could stimulate colonic tight junction protein expression in colitis mice.
Previous study has shown that somatostatin is the main inhibitory peptide in inflammation [28]. To determine whether the stimulation of tight junction protein expression is a mechanism of somatostatin’s anti-inflammatory function, we studied the effect of somatostatin in cultured intestinal epithelial cells. Because treating cells with EPEC and TNF-α are established models of infection and inflammation, respectively [29,30], we studied the effect of somatostatin on Caco-2 cells exposed to EPEC and on Caco-2 cells treated with TNF-α. Our cell culture results mirrored in vivo study results. In Caco-2 cells, EPEC infection reduced claudin-1 and claudin-3 expression by ~45%, and somatostatin treatment completely restored their expression. Similarity, claudin-3 expression was decreased by ~35% after TNF-α treatment, and somatostatin treatment could restore the expression level. These results suggested that the anti-inflammatory effect of somatostatin acts directly on epithelial cells through alteration of tight junction proteins.
In summary, our study showed that mice colitis caused by bacterial infection or intestinal inflammation results in reduced somatostatin level and altered epithelial tight junction protein expression. Administration of somatostatin improved diarrheal symptom in mice and restored tight junction protein expression in the intestinal epithelial cells. This is the first study to show somatostatin directly regulates tight junction function in the intestinal epithelial cells.
Acknowledgements
This investigation was funded by Natural Science Fund of China grant 81070294.
Disclosure of conflicts of interest
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- 1.González-Mariscal L, Betanzos A, Nava P, Jaramillo BE. Tight junction proteins. Prog Biophys Mol Biol. 2003;81:1–44. doi: 10.1016/s0079-6107(02)00037-8. [DOI] [PubMed] [Google Scholar]
- 2.Guttman JA, Li Y, Wickham ME, Deng W, Vogl AW, Finlay BB. Attaching and effacing pathogen-induced tight junction disruption in vivo. Cell Microbiol. 2006;8:634–45. doi: 10.1111/j.1462-5822.2005.00656.x. [DOI] [PubMed] [Google Scholar]
- 3.Guttman JA, Finlay BB. Tight junctions as targets of infectious agents. Biochim Biophys Acta. 2009;1788:832–841. doi: 10.1016/j.bbamem.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 4.Zeissig S, Bürgel N, Günzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vetrano S, Rescigno M, Cera MR, Correale C, Rumio C, Doni A, Fantini M, Sturm A, Borroni E, Repici A, Locati M, Malesci A, Dejana E, Danese S. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology. 2008;135:173–184. doi: 10.1053/j.gastro.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Piche T, Barbara G, Aubert P, Bruley des Varannes S, Dainese R, Nano JL, Cremon C, Stanghellini V, De Giorgio R, Galmiche JP, Neunlist M. Impaired intestinal barrier integrity in the colon of patients with irritable bowel syndrome: involvement of soluble mediators. Gut. 2009;58:196–201. doi: 10.1136/gut.2007.140806. [DOI] [PubMed] [Google Scholar]
- 7.Mazzon E, Crisafulli C, Galuppo M, Cuzzocrea S. Role of peroxisome proliferator-activated receptoralpha in ileum tight junction alteration in mouse model of restraint stress. Am J Physiol Gastrointest Liver Physiol. 2009;297:G488–505. doi: 10.1152/ajpgi.00023.2009. [DOI] [PubMed] [Google Scholar]
- 8.McClane BA, Chakrabarti G. New insights into the cytotoxic mechanisms of Clostridium perfringens enterotoxin. Anaerobe. 2004;10:107–114. doi: 10.1016/j.anaerobe.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Muza-Moons MM, Schneeberger EE, Hecht GA. Enteropathogenic Escherichia coli infection leads to appearance of aberrant tight junctions strands in the lateral membrane of intestinal epithelial cell. Cell Microbiol. 2004;6:783–93. doi: 10.1111/j.1462-5822.2004.00404.x. [DOI] [PubMed] [Google Scholar]
- 10.Gassler N, Rohr C, Schneider A, Kartenbeck J, Bach A, Obermüller N, Otto HF, Autschbach F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001;281:G216–228. doi: 10.1152/ajpgi.2001.281.1.G216. [DOI] [PubMed] [Google Scholar]
- 11.Marin ML, Greenstein AJ, Geller SA, Gordon RE, Aufses AH Jr. A freeze fracture study of Crohn’s disease of the terminal ileum: changes in epithelial tight junction organization. Am J Gastroenterol. 1983;78:537–547. [PubMed] [Google Scholar]
- 12.Olson TS, Reuter BK, Scott KG, Morris MA, Wang XM, Hancock LN, Burcin TL, Cohn SM, Ernst PB, Cominelli F, Meddings JB, Ley K, Pizarro TT. The primary defect in experimental ileitis originates from a nonhematopoietic source. J Exp Med. 2006;203:541–552. doi: 10.1084/jem.20050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poritz LS, Garver KI, Green C, Fitzpatrick L, Ruggiero F, Koltun WA. Loss of the tight junction protein ZO-1 in dextran sulfate sodium induced colitis. J Surg Res. 2007;140:12–19. doi: 10.1016/j.jss.2006.07.050. [DOI] [PubMed] [Google Scholar]
- 14.Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159:2001–2009. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang C, Xu H, Chen H, Li J, Zhang B, Tang C, Ghishan FK. Somatostatin stimulates intestinal NHE8 expression via p38 MAPK pathway. Am J Physiol Cell Physiol. 2011;300:C375–382. doi: 10.1152/ajpcell.00421.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dharmsathaphorn K, Gorelick FS, Sherwin RS, Cataland S, Dobbins JW. Somatostatin Decreases Diarrhea in Patients with the Short-Bowel Syndrome. J Clin Gastroenterol. 1982;4:521–524. doi: 10.1097/00004836-198212000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Lewin MJ. The somatostatin receptor in the GI tract. Annu Rev Physiol. 1992;54:455–468. doi: 10.1146/annurev.ph.54.030192.002323. [DOI] [PubMed] [Google Scholar]
- 18.Vockel M, Breitenbach U, Kreienkamp HJ, Brandner JM. Somatostatin regulates tight junction function and composition in human keratinocytes. Exp Dermatol. 2010;19:888–894. doi: 10.1111/j.1600-0625.2010.01101.x. [DOI] [PubMed] [Google Scholar]
- 19.Deng Q, Li ZL, Lu LR. Somatostatin ameliorates gut mucosal barrier damages in acute necrotizing pancreatitis. Chin J Gen Surg. 2000;15:752–754. [Google Scholar]
- 20.Boshuizen JA, Reimerink JH, Korteland-van Male AM, van Ham VJ, Koopmans MP, Büller HA, Dekker J, Einerhand AW. Changes in Small Intestinal Homeostasis, Morphology, and Gene Expression during Rotavirus Infection of Infant Mice. J Virol. 2003;77:13005–13016. doi: 10.1128/JVI.77.24.13005-13016.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guttman JA, Samji FN, Li Y, Deng W, Lin A, Finlay BB. Aquaporins contribute to diarrhoea caused by attaching and effacing bacterial pathogens. Cell Microbiol. 2007;9:131–141. doi: 10.1111/j.1462-5822.2006.00773.x. [DOI] [PubMed] [Google Scholar]
- 22.Koch TR, Morris VA, Go VLW. Somatostatin in the Idiopathic Inflammatory bowel diseases. Dis Colon Rectum. 1988;31:198–203. doi: 10.1007/BF02552546. [DOI] [PubMed] [Google Scholar]
- 23.Deng W, Vallance BA, Li Y, Puente JL, Finlay BB. Citrobacter rodentium translocated intimin receptor (Tir) is an essential virulence factor needed for actin condensation, intestinal colonization and colonic hyperplasia in mice. Mol Microbiol. 2003;48:95–115. doi: 10.1046/j.1365-2958.2003.03429.x. [DOI] [PubMed] [Google Scholar]
- 24.Borenshtein D, McBee ME, Schauer DB. Utility of the Citrobacter rodentium infection model in laboratory mice. Curr Opin Gastroenterol. 2008;24:32–37. doi: 10.1097/MOG.0b013e3282f2b0fb. [DOI] [PubMed] [Google Scholar]
- 25.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 26.Poritz LS, Harris LR, Kelly AA, Koltun WA. Increase in the Tight Junction Protein Claudin-1 in Intestinal Inflammation. Dig Dis Sci. 2011;56:2802–2809. doi: 10.1007/s10620-011-1688-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil Transmigration in Inflammatory Bowel Disease Is Associated with Differential Expression of Epithelial Intercellular Junction Proteins. Am J Pathol. 2001;159:2001–2009. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Bergeijk JD, Wilson JH. Somatostatin in inflammatory bowel disease. Mediators Inflamm. 1997;6:303–309. doi: 10.1080/09629359791424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.A Borthakur A, Gill RK, Hodges K, Ramaswamy K, Hecht G, Dudeja PK. Enteropathogenic Escherichia coli inhibits butyrate uptake in Caco-2 cells by altering the apical membrane MCT1 level. Am J Physiol Gastrointest Liver Physiol. 2006;290:G30–35. doi: 10.1152/ajpgi.00302.2005. [DOI] [PubMed] [Google Scholar]
- 30.Mashukova A, Wald FA, Salas PJ. Tumor Necrosis Factor Alpha and Inflammation Disrupt the Polarity Complex in Intestinal Epithelial Cells by a Posttranslational Mechanism. Mol Cell Biol. 2011;32:756–765. doi: 10.1128/MCB.00811-10. [DOI] [PMC free article] [PubMed] [Google Scholar]