

Abstract
Senescence, a state of cell cycle arrest, has been regarded as an intrinsic barrier to malignance. Although being repressed in most immortal tumors, the genetic program of senescence can be reactivated by critical regulators, including the apoptosis regulator Bcl-2. We showed here that hypoxic condition resulted in an irreversible senescence-like phenotype with increased expression of Bcl-2 in mouse melanoma B16 cells. In CoCl2-simulating hypoxic condition, characteristic morphological alterations and increased activity of senescence-associated β-galactosidase (SA-β-gal) can be detected with high level of Bcl-2, which was confirmed by western blot and co-staining of SA-β-gal and Bcl-2 by immunocytochemistry. Accordingly, Bcl-2 silence by specific siRNA ahead of hypoxia treatment interrupted the senescent development. Moreover Bcl-2 overexpression led to early onset of senescence. We propose that Bcl-2 is required to initiate and maintain the senescent phenotype. In addition, p53 and p16 were not involved in hypoxia-induced senescence according to the expression levels during senescent process. These results suggest that when encountering harmful stress (hypoxia), melanoma cells overexpress Bcl-2 and turn to senescence, a permanent cell-cycle arrest, for prolonged survival.
Keywords: Senescence, melanoma, Bcl-2, hypoxia
Introduction
Senescence was first identified in cells at the end of their replicative lifespan [1]. Subsequent evidence indicates that senescence could be induced prematurely when cells encounter telomere shortening, gene mutations, and stress [2,3]. Senescent cells usually share a combination of functional and morphological changes, including enlarged and flat morphology, activation of senescence-associated beta-galactosidase and enhanced expression of regulatory proteins [4,5]. The p53/p21 and p16/RB pathways are the key regulators of proliferation arrest and irreversibility of the senescent state [6], which engage in most cellular senescence. However, there are exceptions that appear to be independent of these pathways [7,8].
Evidence exists to suggest that many proteins that regulate cell division can also control cell death and vice versa [9]. For instance, under conditions unfavorable for proliferation, certain cell cycle effectors promote apoptosis [10]. As for the Bcl-2 family, a key determinant of cell survival, it is essential for normal development and organ homeostasis [11]. Besides, it has been found to influence the cell cycle. It was reported that Bcl-2 phosphorylation was linked with cell cycle retardation [12,13] and this ability was separate from its role in cell survival [14]. Moreover, there were certain relations between elevated Bcl-2 levels and reduced tumor proliferation rate [15,16]. In addition, neither p53 nor p16 is necessary, because Bcl-2 can promote senescence in cells lacking those genes [17]. Perhaps promoting senescence may provide Bcl-2 with an additional approach to enhance cell survival [14]. However, this exact mechanism remains uncharacterized.
To identify the senescence regulators in melanoma cells, we have used CoCl2 to mimic a mild hypoxic microenvironment, and then analyzed the expression levels of Bcl-2, p16 and p53. We report that Bcl-2 can induce a permanent cell-cycle arrest and may be required to maintain the senescent phenotype. These results suggest that the role of Bcl-2 in suppressing tumor growth may contribute to the ability of promoting senescence.
Materials and methods
Cell culture
B16 cells (Cell Resource Center, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences; School of Basic Medicine, Peking Union Medical College) were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (Invitrogen). Hypoxic condition was established by addition of CoCl2 with final concentrations of 150, 200 and 300 μM for various times (6, 12, 24, 36, 48, 60 and 72 h).
SA-β-gal staining
B16 cells under normoxic or hypoxic conditions as indicated were stained for SA-β-gal according to the staining kit (C0602, Beyotime, Beijing, China).
Western blot analysis
Total proteins of B16 cells at different time points of CoCl2 treatment were collected for Western blot. The primary antibodies were Bcl-2, p53 and p16 (sc-7382; sc-6243; cs-1661, Santa Cruz Biotechnology). The secondary antibody was form Santa Cruz (1:2,000; Santa Cruz, CA, USA). Beta-actin (1:200; Santa Cruz, CA, USA) was detected for protein loading control.
Cell transfection
For pRNA-U6.1/Neo-shRNA (MSH028734-CU6, Gene Copoeia) transient transfection, B16 cells were seeded in 6-well plates and were transfected with the expression vector of siRNA specific for Bcl-2 at approximately 80% confluence using polyethylenimine (PEI, Polysciences, Inc., Cat#23966). B16 cells were stably transfected with pReceiver-M03-EGFP-Bcl-2 (EXMm30401-M03, Gene Copoeia) by adding G418 (8000 μg/ml) to the medium for 2 or 3 weeks. Clones were picked and expanded for further analysis. In each experiment, untreated controls and mock transfected cells were included. Cells were assayed 24 hours after transfection by GFP detection.
Immunostaining of cultured cells
B16 cells treated with or without CoCl2 were first stained for SA-β-gal and then permeated with 0.5% Triton X-100. The following procedures were same to immunohistochemistry and the primary antibody was Bcl-2.
Reverse transcription PCR
For expression analysis of Bcl-2 in cells transfected with pReceiver-M03-EGFP-Bcl-2 and pRNA-U6.1/Neo-shRNA respectively, total RNA was isolated using Trizol reagent (Invitrogen) according to manufacturer’s instruction. The following primers were used for RT-PCR: GAPDH: 5’-GCCACGGCTGCTTCCAG-3’ (sense) 5’-GGCGTACAGGTCTTTGC-3’ (antisense); Bcl-2: 5’-CATCGCGATGTCGCACGGTA-3’ (sense) 5’-TACGAAAGCGGGGTGGGTTGTG-3’ (antisense); PCR products were separated by 2% agarose gel electrophoresis followed by ethidium bromide staining, quantified by ImageJ (National Institute of Health, USA) and normalized to GAPDH.
Statistical analysis
All data presented were representative of at least three independent experiments. Statistical analyses were performed with the software SPSS version 17 (SPSS Inc.). One-way analysis of variance was used to compare percentages of SA-β-gal -positive cells. Differences were considered significant at P ≤ 0.05.
Results
Hypoxia-induced senescence of melanoma cells
Previous studies indicated that the long term static state of moles, which could transform to malignant melanoma, may be partly attributed to cellular senescence [18]. In addition, for the O2 level is about 1.5-5% in the skin, human melanocytic nevi locate in a mild hypoxic microenvironment [19]. In order to assess whether melanoma cells could also become senescent under hypoxia, CoCl2 was used to simulate hypoxic condition. Cellular senescence was detected at various treatment periods and concentrations with the generally performed marker SA-β-gal. In normoxia, spontaneously senescence occurred in a minority of B16 cells with flat, enlarged cytoplasm and high SA-β-gal activity (Figure 1). Since the hypoxia-mimic agent CoCl2 is harmful for cell metabolism, a large fraction of cells become round, suspended, and died ultimately in a time-and dose-dependent manner. However, a small fraction of cells were still remaining alive and adherent and showed high SA-β-gal activity. Results showed that culturing in 150 μM CoCl2 for 48 h effectively induced senescence, which not only guaranteed the cell viability but also resulted in characteristic senescent phenotype. Moreover, percentages of senescent cells were increasing as the treatment time extended (Figure 1). To investigate further whether hypoxia-induced senescence is transient or irreversible, we examined the SA-β-gal activity of cells culturing in normoxia for 24 h after the CoCl2 removal. Results showed that the morphological alterations and SA-β-gal activity were not recovered to the state of non-treated cells. These results show that CoCl2-simulated hypoxia can induce a permanent senescence in B16 cells.
Figure 1.
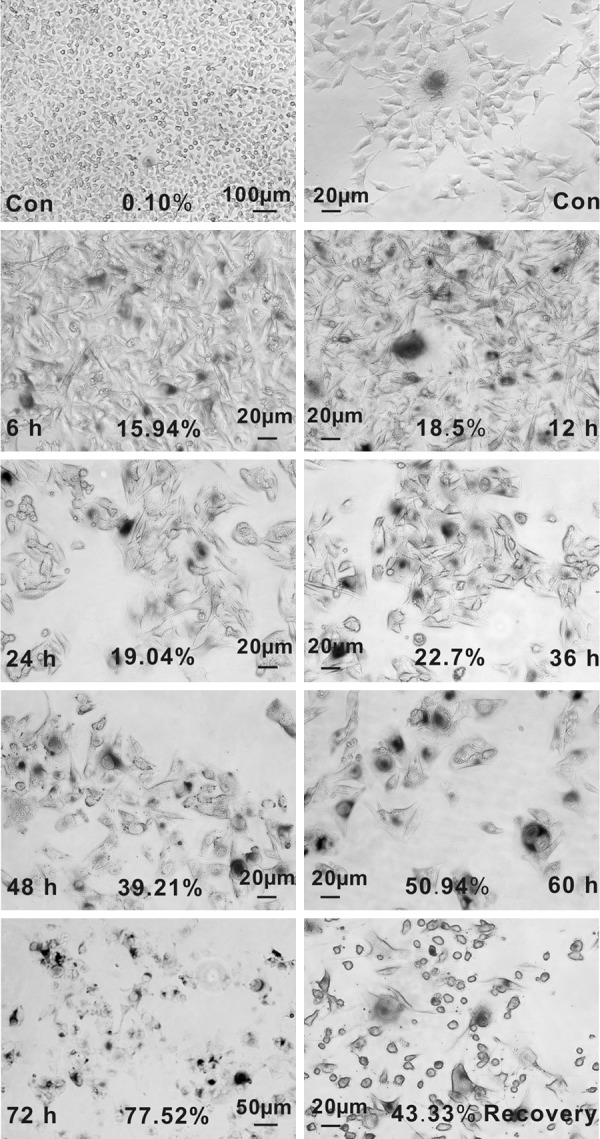
Detection of senescence induced by CoCl2 using SA-β-gal as a marker. SA-β-gal staining of B16 cells cultured in normal medium (Con), treated with 150 μM CoCl2 for different times (6, 12, 24, 36, 48, 60 and 72 h), and those continually cultured in normal medium for 24 h after 24 h treatment of CoCl2 (Recovery). The number of cells positive for SA-β-gal (blue staining around nucleus) activity is reported as percentages of the total cells scored in five fields under a microscope (Nikon ECLIPSE Ti, Japan). (Values are means of three independent experiments. *P < 0.05.).
Expression of cell cycle regulators Bcl-2, p16 and p53 in senescent melanoma cell
Since evidence demonstrated that senescence-inducing signals (DNA-damage response (DDR) and other stresses) usually engage either the p53 or the p16-retinoblastoma protein (pRB) tumor suppressor pathways, we investigated the expression of p53 and p16 at different time points after CoCl2 administration. Western blot results showed that expression of both p53 and p16 did not changed at each time points observed (Figure 2A). CoCl2-induced senescence appears to be independent of these pathways and engage others. Bcl-2 was reported to modulate cell cycle progression, favoring a quiescent state over a proliferative state [20]. Therefore, Bcl-2 expression was tested and data showed that its expression increased gradually from the time CoCl2 added and reached a high level at 24 h and then maintained this state with the occurrence of cellular senescence occurred up to 72 h (determined by detection of SA-β-gal activity synchronously) (Figure 2A). Similarly, statistical results of co-location of SA-β-gal and Bcl-2 expression revealed that 19.2% senescent cells were positive for Bcl-2 at 36 h after CoCl2 treatment, compared with 1.1% at normoxia (Figure 2B). These results reveal that Bcl-2 may be involved in the regulation of hypoxia-induced senescence.
Figure 2.
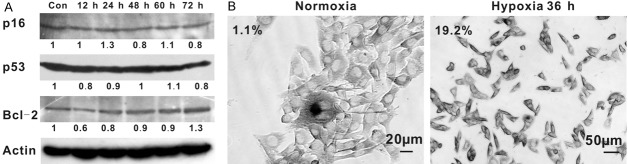
Expression of senescence-associated regulators in hypoxia-induced senescence. A: Shown is the expression of p53, p16 and Bcl-2 at indicated time point after CoCl2 treatment of B16 cells. The relative amounts of the proteins were quantified and normalized to the corresponding β-catenin protein amounts. B: Co-staining of Bcl-2 and SA-β-gal. Percentages of cells positive for both SA-β-gal and Bcl-2 are indicated as percentages of the total cells scored in five fields. In normoxia, only 1.1% cells were double-positive, compared with 19.2% in hypoxia for 36 h.
Effect of Bcl-2 expression on cellular senescence development
To confirm whether loss of Bcl-2 directly interrupts senescence and whether overexpression of Bcl-2 promotes premature senescence in hypoxic condition, we initially transfected B16 cells with a Bcl-2 expression vector and shRNA targeting Bcl-2. Control cells were transfected with corresponding control vectors. G418 was used for selection of stable transfectants for 2 weeks. Transfection efficacy was assessed by Western blotting and GFP expression (Figure 3A). Stable transfectants (Bcl-2-overexpressing) and transient transfectants (Bcl-2-silencing) B16 cells were cultured with CoCl2 and were detected for SA-β-gal at different time points. Overexpression of Bcl-2 in B16 cells resulted in distinctive morphological alterations and high SA-β-gal activity under hypoxia at 36 h (39.26% positive for SA-β-gal) earlier than parental and control cells that senesce at 48 h. While, Bcl-2-silencing cells assumed an apoptosis-like morphology and typical senescence did not efficiently establish (Figure 3B). These results suggest that Bcl-2 was required for the onset of senescent process.
Figure 3.

Role of Bcl-2 in the development of senescence. A: Transfection efficiency was detected by Western blot and PCR. Bcl-2 expression and mRNA levels of B16 cells transfected with pReceiver-Bcl-2 was higher than control and, cells transfected with pshRNA-Bcl-2 lower than control. The relative amounts of the proteins and mRNA were quantified and normalized to the corresponding β-catenin protein and GAPDH amounts. B: Upper panel: B16 cells after pReceiver-Bcl-2 transfection were flat with enlarged cytoplasm and the percentage of SA-β-gal positive cells is 39.26% in hypoxia for 36 h. Lower panel: B16 cells after pshRNA-Bcl-2 transfection were round and suspended, and the percentage of SA-β-gal positive cells is 6.56% in hypoxia for 36 h. To avoid transfect efficiency, senescent cells were counted by SA-β-gal staining vs. GFP-expressing.
Discussion
Cellular senescence is a response to exogenous and endogenous stress by adopting a state of permanent cell-cycle arrest, which would inhibit damaged, stressed or oncogene-expressing cells to proliferate. Understanding the causes and consequences of cellular senescence helps to understand how cells react to stress, and how this cellular response can affect various processes such as cancer, wound healing and ageing.
It has been demonstrated that several different stimuli can trigger senescence in normal and neoplastic cells. Hypoxic condition (CoCl2) induced the expression of several distinctive features of cellular senescence in our study. We hypothesized that B16 cells initially respond to such a stress by entering into a static state, but still metabolically active. Similarly, the O2 level in the skin is about 1.5% to 5% [21], in such hypoxic microenvironment, human melanocytic nevi undergoes irreversible senescence. The mechanism of hypoxia-induced senescence may result from reactive oxygen species during hypoxia, which lead to DNA damage and then senescence [22,23].
The mechanism of senescence is generally elucidated by two tumor suppressor pathways: p53 and pRB/p16INK4a [24]. Depending on various research backgrounds, no unified recognition of contribution of p16 in senescence development was obtained [25-27]. As for p53, which functions to eliminate damaged cells by apoptosis or senescence, is involved in replicative and Ras-induced senescence [2]. Moreover imbalance of survival and apoptotic factors in Bcl-2 family is related with status of p53 activity [28,29]. Thus, although the p53 pathway, p16-pRB pathway, or both are involved in most cellular senescence, there are examples of senescence in which other pathways engage [7,8].
In this study, we determined that Bcl-2 upregulation is necessary for initiation of hypoxia-induced senescence. Based on our findings that expression level of Bcl-2 increased at the beginning and maintained a relatively long time, it is likely that Bcl-2 is required for initiation of senescence. In addition, hypoxia-induced senescence occurred earlier with overexpression of Bcl-2, while siRNA silencing of Bcl-2 resulted in cell death. Bcl-2 not only functions as a survival agonist by suppressing apoptosis but also inhibits cell division cycle progression and exhibits antioxidant properties [17,30,31]. In this study we focused on Bcl-2 which may be implicated in the onset of senescence according to previous studies involving Bcl-2 effect on cell survival. As an antiapoptotic molecule, Bcl-2 enhances tumorigenesis by prolonging survival, while the anti-proliferative activity of Bcl-2 has been shown to inhibit tumor progression. In breast cancers, high Bcl-2 expression has been associated with low proliferative potential and better prognosis [32]. Thus, dual-effects of Bcl-2 on oncogenesis explain the long latency in tumor formation resulting from Bcl-2 overexpression [33]. In addition, it has been demonstrated that the mechanism of anti-apoptosis and cell cycle delay functions can be genetically separated [34].
Studies in vivo and in vitro have demonstrated that the cycle delay by Bcl-2 is S phase from quiescence [35], and CDC2-mediated phosphorylation of Bcl-2 maybe involved in the negative regulations during mitosis [36]. Besides, it has been reported that upon certain oxidative stress, overexpression of Bcl-2 triggered senescence induction by an increased expression of cell cycle inhibitors, JNK or oncogenic Ras [37-40]. H. Brady reported that p27KIP1 acts as a senescence effector downstream of Bcl-2 [41]. Is Bcl-2 routinely engaged in the modulation of various forms of cellular senescence? Thus, it is indispensable to explore the mechanism of interaction between Bcl-2 and other well known senescence regulators.
Disclosure of conflict of interest
None.
References
- 1.Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 2.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 4.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 7.Olsen CL, Gardie B, Yaswen P, Stampfer MR. Raf-1-induced growth arrest in human mammary epithelial cells is p16-independent and is overcome in immortal cells during conversion. Oncogene. 2002;21:6328–6339. doi: 10.1038/sj.onc.1205780. [DOI] [PubMed] [Google Scholar]
- 8.Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst C, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAF(E600)-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 9.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 10.Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- 11.Kerr LE, Birse-Archbold JL, Short DM, McGregor AL, Heron I, Macdonald DC, Thompson J, Carlson GJ, Kelly JS, McCulloch J, Sharkey J. Nucleophosmin is a novel Bax chaperone that regulates apoptotic cell death. Oncogene. 2007;26:2554–2562. doi: 10.1038/sj.onc.1210044. [DOI] [PubMed] [Google Scholar]
- 12.Deng X, Gao F, May WS Jr. Bcl2 retards G1/S cell cycle transition by regulating intracellular ROS. Blood. 2003;102:3179–3185. doi: 10.1182/blood-2003-04-1027. [DOI] [PubMed] [Google Scholar]
- 13.Deng X, Gao F, Flagg T, May WS Jr. Mono- and multisite phosphorylation enhances Bcl2’s antiapoptotic function and inhibition of cell cycle entry functions. Proc Natl Acad Sci U S A. 2004;101:153–158. doi: 10.1073/pnas.2533920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vairo G, Innes KM, Adams JM. Bcl-2 has a cell cycle inhibitory function separable from its enhancement of cell survival. Oncogene. 1996;13:1511–1519. [PubMed] [Google Scholar]
- 15.Campani D, Esposito I, Boggi U, Cecchetti D, Menicagli M, De Negri F, Colizzi L, Del Chiaro M, Mosca F, Fornaciari G, Bevilacqua G. Bcl-2 expression in pancreas development and pancreatic cancer progression. J Pathol. 2001;194:444–450. doi: 10.1002/path.925. [DOI] [PubMed] [Google Scholar]
- 16.Biden KG, Simms LA, Cummings M, Buttenshaw R, Schoch E, Searle J, Gobe G, Jass JR, Meltzer SJ, Leggett BA, Young J. Expression of Bcl-2 protein is decreased in colorectal adenocarcinomas with microsatellite instability. Oncogene. 1999;18:1245–1249. doi: 10.1038/sj.onc.1202413. [DOI] [PubMed] [Google Scholar]
- 17.O’Reilly LA, Huang DC, Strasser A. The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 1996;15:6979–6990. [PMC free article] [PubMed] [Google Scholar]
- 18.Gray-Schopfer V. Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br J Cancer. 2006;95:496–505. doi: 10.1038/sj.bjc.6603283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Hara JA, Goda F, Dunn JF, Swartz HM. Potential for EPR oximetry to guide treatment planning for tumors. Adv Exp Med Biol. 1997;411:233–242. doi: 10.1007/978-1-4615-5865-1_28. [DOI] [PubMed] [Google Scholar]
- 20.Quinn LM, Richardson H. Bcl-2 in cell cycle regulation. Cell Cycle. 2004;3:7–9. [PubMed] [Google Scholar]
- 21.O’Hara JA, Goda F, Dunn JF, Swartz HM. Potential for EPR oximetry to guide treatment planning for tumors. Adv Exp Med Biol. 1997;411:233–242. doi: 10.1007/978-1-4615-5865-1_28. [DOI] [PubMed] [Google Scholar]
- 22.Clanton TL. Hypoxia-induced reactive oxygen species formation in skeletal muscle. J Appl Physiol. 2007;102:2379–2388. doi: 10.1152/japplphysiol.01298.2006. [DOI] [PubMed] [Google Scholar]
- 23.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130:1715–1725. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haferkamp S, Scurr LL, Becker TM, Frausto M, Kefford RF, Rizos H. Oncogene-induced senescence does not require the p16(INK4a) or p14ARF melanoma tumor suppressors. J Invest Dermatol. 2009;129:1983–1991. doi: 10.1038/jid.2009.5. [DOI] [PubMed] [Google Scholar]
- 27.Romagosa C, Simonetti S, Lopez-Vicente L, Mazo A, Lleonart ME, Castellvi J, Ramon y Cajal S. p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene. 2011;30:2087–2097. doi: 10.1038/onc.2010.614. [DOI] [PubMed] [Google Scholar]
- 28.Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 29.Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- 30.Mazel S, Burtrum D, Petrie HT. Regulation of cell division cycle progression by bcl-2 expression: a potential mechanism for inhibition of programmed cell death. J Exp Med. 1996;183:2219–2226. doi: 10.1084/jem.183.5.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 32.de La Coste A, Mignon A, Fabre M, Gilbert E, Porteu A, Van Dyke T, Kahn A, Perret C. Paradoxical inhibition of c-myc-induced carcinogenesis by Bcl-2 in transgenic mice. Cancer Res. 1999;59:5017–5022. [PubMed] [Google Scholar]
- 33.Vail ME, Chaisson ML, Thompson J, Fausto N. Bcl-2 expression delays hepatocyte cell cycle progression during liver regeneration. Oncogene. 2002;21:1548–1555. doi: 10.1038/sj.onc.1205212. [DOI] [PubMed] [Google Scholar]
- 34.Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, Andrews D, Knudson CM, Yang E. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 2003;22:5459–5470. doi: 10.1093/emboj/cdg533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greider C, Chattopadhyay A, Parkhurst C, Yang E. BCL-x(L) and BCL2 delay Myc-induced cell cycle entry through elevation of p27 and inhibition of G1 cyclin-dependent kinases. Oncogene. 2002;21:7765–7775. doi: 10.1038/sj.onc.1205928. [DOI] [PubMed] [Google Scholar]
- 36.Furukawa Y, Iwase S, Kikuchi J, Terui Y, Nakamura M, Yamada H, Kano Y, Matsuda M. Phosphorylation of Bcl-2 protein by CDC2 kinase during G2/M phases and its role in cell cycle regulation. J Biol Chem. 2000;275:21661–21667. doi: 10.1074/jbc.M906893199. [DOI] [PubMed] [Google Scholar]
- 37.Vairo G. Bcl-2 retards cell cycle entry through p27Kip1, pRb relative p130, and altered E2F regulation. Mol Cell Biol. 2000;20:4745–53. doi: 10.1128/mcb.20.13.4745-4753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tombor B, Rundell K, Oltvai ZN. Bcl-2 promotes premature senescence induced by oncogenic Ras. Biochem Biophys Res Commun. 2003;303:800–807. doi: 10.1016/s0006-291x(03)00402-9. [DOI] [PubMed] [Google Scholar]
- 39.Greider C, Chattopadhyay A, Parkhurst C, Yang E. Bcl-xL and Bcl-2delay Myc-induced cell cycle entry through elevation of p27 and inhibition of G1 cyclin-dependent kinases. Oncogene. 2002;21:7765–7775. doi: 10.1038/sj.onc.1205928. [DOI] [PubMed] [Google Scholar]
- 40.Lee JJ, Lee JH, Ko YG, Hong SI, Lee JS. Prevention of premature senescence requires JNK regulation of Bcl-2 and reactive oxygen species. Oncogene. 2010;29:561–575. doi: 10.1038/onc.2009.355. [DOI] [PubMed] [Google Scholar]
- 41.Crescenzi E, Palumbo G, Brady HJ. Bcl-2 activates a programme of premature senescence in human carcinoma cells. Biochem J. 2003;375:263–274. doi: 10.1042/BJ20030868. [DOI] [PMC free article] [PubMed] [Google Scholar]