

Abstract
Hepatitis B virus X (HBX) protein plays a crucial role in carcinogenesis, but its mechanism is unclear. The involvement of ataxia telangiectasia mutated (ATM) kinase in the enhanced redox system was investigated by examining the phosphorylation level of ATM in HBX gene-transfected cells and in transgenic mice following redox system manipulation by treatment with hydrogen peroxide (H2O2) or antioxidant. Western blotting and immunostaining showed that phospho-ATM was significantly increased by HBX both in vitro (3.2-fold; p<0.05) and in vivo (4-fold; p<0.05), and this effect was abrogated by antioxidant treatment. The level of PKC-δ in HBX-expressing cells was increased 3.5-fold compared to controls. Nuclear localized NF-E2-related factor 2 (Nrf2) was increased in HBX-expressing cells exposed to H2O2, but remained at lower levels after the treatment with rottlerin, KU55933, or caffeine. The levels of anti-oxidant molecules were increased in HBX expressing cells and in transgenic mice, indicating that HBX stimulates the Nrf2-mediated redox system. The levels of intracellular reactive oxygen species (ROS) were significantly increased in HBX-expressing cells treated with hydrogen peroxide in the presence of ATM inhibitor KU55933 or caffeine. Treatment of HBX-expressing cells with KU55933 or caffeine before the exposure to H2O2 increased the ratio of cell apoptosis to 33 ± 4% (p<0.05) and 22 ± 4% (p<0.05), respectively. Collectively, HBX stimulates the ATM-mediated PKC-δ/Nrf2 pathway, and maintains the enhanced activity of the redox system. Therefore, manipulating ATM kinase activity might be a useful strategy for treating HBX-induced carcinogenesis.
Keywords: Hepatitis B virus X, ataxia telangiectasia mutated, reactive oxygen species
Introduction
Hepatocellular carcinoma (HCC) is one of the most lethal cancers worldwide [1,2]. Many cases of HCC arise in individuals with chronic liver diseases related to hepatitis B virus (HBV), hepatitis C virus (HCV), alcohol abuse, non-alcoholic steatohepatitis (NASH), and hemochromatosis. The etiology of HCC is diverse: HCV-related HCC is prevalent in industrial countries and NASH has now been regarded as a new threat for cancer development in Europe and Unites States. In contrast, HBV infection is more prevalent in Asia, Africa, and the South Pacific Islands and has remains an important risk factor for HCC development in these areas [1-4].
Among the different etiologies of HCC, HBV is a unique causative agent as many studies have implicated this virus as a direct inducer of cancer development. Clinical evidence has shown that HCC arises in some individuals with occult HBV infection without evident chronic liver damage [1,5]. More importantly, a large number of HBV-related HCCs represent the integration of the HBV-encoding HBV X (HBX) gene, which has been identified as a critical mediator of cancer development. HBX-transgenic mice develop HCC without any signs of chronic hepatitis or cirrhosis [6,7], which supports the hypothesis that HBX is the main underlying cause of HBV oncogenesis. Previous studies have reported that HBX constitutively stimulates many oncogenic signaling pathways including Ras/Mitogen-activated Protein Kinase (MAPK) signaling and others involved in DNA damage repair, cell growth, apoptosis, and cell cycle regulation [8-10]. HBX-transduced cells acquire a greater ability for survival and proliferation concomitantly with increased DNA damage [11,12]; therefore, integration of the HBX gene in the host genome might be the ultimate cause of cell transformation.
The mechanism of increased DNA damage in HBX-positive cells is unknown, but HBV induces the generation of reactive oxygen species (ROS) through endoplasmic reticulum stress [13] and many of the HBX-mediated signaling pathways are closely correlated with intracellular ROS levels. Stimulation of HBX by ROS oscillation causes constitutive activation of nuclear factor-kappa B (NF-kappa B), signal transducer and activator of transcription 3 (STAT-3) [14], and Akt/protein kinase B signaling [7]. Intriguingly, HBX induces acquired resistance to oxidative cell death, possibly through enhancement of redox system functions [15,16]. The ROS level is regulated by the balance between oxidant and antioxidant molecules under physiological conditions. The antioxidant/electrophile response element (ARE) pathway mediated by nuclear factor erythroid 2-related factor (Nrf2) is one of the main regulators of redox signaling. Activation of ARE stimulates the production of phase II detoxifying enzymes with antioxidant enzymes that protect cells from oxidative damage. HBV has recently been reported to induce a strong activation of Nrf2/ARE-regulated genes through HBX [15], which suggests that HBX-mediated Nrf2 signaling might be the key to the enhanced cell survival observed in the presence of oxidative stress. At present, the mechanism of HBX-mediated redox signaling is unknown. Investigation of the molecules involved in the protective mechanism preventing oxidative cell damage might be of value for the development of therapies for HBX-induced carcinogenesis.
One critical mediator of antioxidant responses has recently been identified as the ataxia telangiectasia mutated (ATM) kinase. This kinase, originally identified as a DNA damage sensor, has been reported to enhance Nrf2 signaling through protein kinase C (PKC)-δ activation [17]. ATM functions in the defense against oxidative stress induced by oxidized low-density lipoprotein [18], and promotes an antioxidant response by regulating the pentose phosphate pathway [19]. HBX has been implicated in the oxidative DNA damage occurring in host cells; therefore, examining whether ATM plays a role in HBX-mediated redox regulation should provide valuable information. The aim of the present study was to investigate the potential involvement of ATM in HBX-mediated antioxidant signaling, and to examine whether manipulation of ATM kinase activity would be an effective strategy for preventing HBX-mediated carcinogenesis.
Materials and methods
Reagents
For in vitro experiment, KU55933 (a specific ATM inhibitor) (Calbiochem, Gibbstown, NJ, USA) and rottlerin (a PKC-δ inhibitor) (Calbiochem) were dissolved in dimethyl sulfoxide and used at 10 μM and 5 μM, respectively. Caffeine (a prototype of an ATM inhibitor) (Latoxan, Rosans, France) was diluted with culture medium and used at 5 mM. N-acetyl-L-cysteine (NAC) (LKT Laboratories, St. Paul, MN, USA) was freshly prepared by dissolving in water at pH 7.4 and used at 5 mM. For western blotting, polyclonal antibodies against phospho-ATM (Ser1981), phospho-Chk2 (Thr68), PKC-δ and heme oxygenase-1 (HO-1) were obtained from Cell Signaling Technology (Beverly, MA, USA), and anti-NAD(P)H:quinone oxidoreductase 1 (Nqo1) antibody was from OriGene (Rockville, MD, USA). Antibodies specific for FLAG M2 and β-actin were from Sigma Chemical Co. (St. Louis, MO, USA).
Cell culture
Human hepatoma cell lines HepG2 cells (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s (DME) medium containing 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a humidified atmosphere containing 5% CO2. In all experiments, the chemical inhibitors were added to the culture medium 1 h before the addition of hydrogen peroxide (H2O2) at 100 μM.
HBX-expressing plasmid
HBX expression plasmid pCAG-FLAG-HBX (pHBX) was derived from a plasmid containing the CAG (cytomegalovirus enhancer-chicken β-actin hybrid) promoter. The NotI-BglII fragment containing the FLAG-tagged HBX encoding sequence was obtained from pNK-FLAG-HBx (kind gift from Dr. Murakami, Kanazawa University, Japan), and was inserted into the EcoRI site downstream of the CAG promoter in pCAG-IRES-Puro (kind gift from Dr. Jun-ichi Miyazaki of Osaka University Graduate School of Medicine). After the verification of DNA sequence, HepG2 cells were transfected with mock plasmid pCAG-IRES-Puro or pCAG-FLAG-HBx using FuGENE6 (Roche Diagnostics, Indianapolis, IN, USA) with MA lipofection enhancer (IBA, St. Louis, MO, USA). Stable transfected cells were established by transfection with the linearized pCAG-IRES-Puro or pCAG-FLAG-HBx, and selected using 2 μg/ml puromycin (Cayla, Toulouse, France).
Annexin V assay
Cells were maintained in serum-free media overnight, and exposed to H2O2 (100 μM) for 24 h. The percentage of apoptotic cells was evaluated using an Annexin V FITC Kit (Invitrogen, Karlsruhe, Germany), according to the manufacturer’s instructions. Fluorescence was visualized with a fluorescence microscope (BZ-9000; Keyence, Osaka, Japan). To assess the annexin V-positive apoptotic cells, 300 cells were counted in a given microscopic field. The results were obtained from three independent experiments.
Evaluation of intracellular ROS production
Cells were cultured in pyruvate-free DMEM containing 1% charcoal-stripped FBS overnight, and treated with H2O2 (100 μM) for 15 min followed by an incubation with a fluorescent probe 5-(and-6)-chloromethyl-2’,7’-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Invitrogen, Carlsbad, CA, USA). Intracellular ROS generation was visualized using a fluorescence microscope (BZ-9000; Keyence). Quantification of fluorescence intensity changes reflecting ROS levels was performed by a luminometer (Fluoroskan Ascent Microplate Fluorometer, Labsystems, Helsinki, Finland) using an excitation wavelength of 485 nm and an emission wavelength of 530 nm.
Immunofluorescence
Cells were cultured on coverslips with or without the treatment with H2O2 (20 μM) for 20 min. Cells were fixed in 4% paraformaldehyde, reacted with an anti-phospho-ATM (Ser1981) antibody (Abgent, San Diego, CA, USA) or anti-Nrf2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight, and incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Molecular Probes, Carlsbad, CA, USA). Fluorescence images were visualized with a fluorescence microscope (BZ-9000; Keyence, Osaka, Japan) and 300 cells were counted in a given microscopic field. The results were obtained from three independent experiments.
Animal model
Male HBX transgenic mouse model at the age of 6 months [6] and age-matched control wild mice of same strain were maintained in a pathogen-free facility at the Niigata University. Liver tissue samples were obtained from the animals and processed for western blotting and immunohistochemical analysis. NAC (Sigma) dissolved in phosphate buffer saline (PBS) (pH 7.4) was administered to mice intraperitoneally at 100 mg/kg of body weight for 5 days as previously described [20]. For control, the same number of mice were administered PBS (5 wild mice, 5 NAC-treated wild mice, 5 HBX mice and 5 NAC-treated HBX mice). All the experimental protocols were approved by the Institutional Animal Care and Use Committee of Niigata University and performed in accordance with the National Institute of Health Guidelines.
Western blotting
Cultured cells were lysed in a modified radioimmunoprecipitation assay buffer supplemented with complete protease inhibitor. Tissue samples were homogenized with the same lysis buffer on ice using Tissue Ruptor (Qiagen, Tokyo, Japan). Aliquots of protein samples (20 μg) were electrophoresed on 5-20% SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes. The membranes were probed with appropriate primary antibodies and with horseradish peroxidase-conjugated secondary antibodies, and reacted with an enhanced ECL western blotting detection system GE Healthcare, (Uppsala, Sweden). Protein band intensity was normalized by the β-actin band, and was quantified using image analysis software (Image-J, ver. 1.44; NIH, Bethesda, MD).
Immunohistochemistry
Tissue sections were reacted with antibodies against phospho-ATM (Ser1981) (Abgent, San Diego, CA, USA; 1:100) or phospho-Chk2 (Thr68) (Abnova, Taipei, Taiwan) overnight at 4°C using a Vector Elite ABC kit (Vector Laboratories, Burlingame, CA, USA). The immunohistochemical staining was evaluated by two independent observers and evaluated the labeling indices by counting 200 cells at three different microscopic fields.
Statistical analysis
Data were analyzed using SPSS software (Statistical Product and Service Solutions 11.5 for Windows; SPSS Inc., Chicago, IL), and are presented as the means ± SD of individual triplicate experiments. For data comparisons between the groups, Student’s t-test and two-way analysis of variance (ANOVA) with the Bonferroni’s correction was used. Differences were considered significant for values of p<0.05.
Results
ATM is activated by HBX
Western blotting of cultured cells showed that the phosphorylation levels were increased for ATM in transiently transfected HBX cells in parallel with the level of gene transduction. Phospho-ATM was also increased 3.2-fold in the stably transfected cells (p<0.05) (Figure 1A). The immunostaining results confirmed the western blotting findings, as the percentage of phospho-ATM-positive cells showed a significant increase (60 ± 7%) compared to the control cells (5 ± 3%, p<0.05) (Figure 1B). Western blotting also showed that the levels of phospho-ATM were increased 4-fold in the liver of HBX-transgenic mice compared to wild type mice (Figure 1C). Immunohistochemical staining showed that phospho-ATM was expressed in non-parenchymal cells in the liver of both wild type and transgenic mice (Figure 1D, arrows). The transgenic mice showed expression of phospho-ATM in 47 ± 6% of the hepatocytes, compared to 5 ± 3% in the wild type (p<0.05). Expression was predominantly nuclear (Figure 1D).
Figure 1.
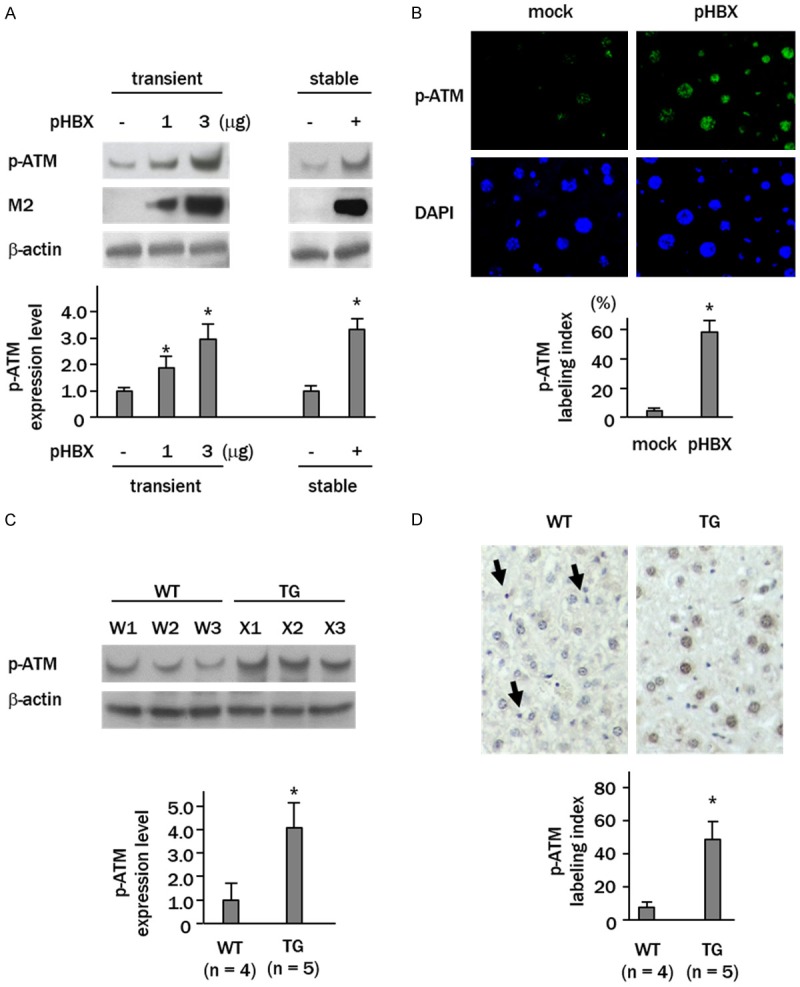
ATM is activated by HBX both in vitro and in vivo. (A) Western blot analyses showing up-regulation of phospho-ATM (p-ATM) in HepG2 cells transfected stably with or transiently with increasing amounts of HBX-expressing plasmid (pHBX). M2, anti-FLAG M2 antibody; mock, cells transfected empty vector; pHBX, cells transfected pHBX. (B) Immunofluorescent staining of p-ATM (green) in HepG2 cells stably transfected with pHBX. Nuclei were stained with DAPI (blue). (C) Western blot analysis showing increased p-ATM expression in livers from wild (WT) and HBX gene transgenic (TG) mice. W1-3, wild mice; X1-3, transgenic mice. (D) Immunohistochemical staining of p-ATM in livers. Arrows indicate p-ATM-positive non-parenchymal cells (magnification x 40). Columns in (A and C) show relative band intensities expressed as the fold changes of control cells or wild mice. Columns in (B and D) represent p-ATM labeling indices. Data are expressed as mean ± SD of three independent experiments (A and B, *p<0.05 vs. control plasmid transfected cells; C and D, *p<0.05 vs. wild mice).
Phospho-ATM is induced through ROS generation
Sensitization to H2O2 increased the levels of phospho-ATM 4.8- and 7.9-fold in the control and HBX-transfected cells, respectively. Increased levels of phospho-Chk2, a down-streaming signaling molecule of ATM, were also detected in H2O2 treated cells (Figure 2A). In contrast, treatment of HBX-transfected cells with the antioxidant NAC significantly decreased phosphorylation levels of both ATM and Chk2 (p<0.05, p<0.05, respectively, vs. HBX-transfected cells) (Figure 2B). Immunohistochemical analysis showed considerable expression of phospho-ATM and Chk2 in hepatocytes of the HBX transgenic mice, both mainly in the nuclei. Administration of NAC to these animals caused significant decreases in the numbers of phospho-ATM and phospho-Chk2-positive hepatocytes (phospho-ATM; 48 ± 4% vs. 31 ± 4%, p<0.05) (Figure 2C).
Figure 2.
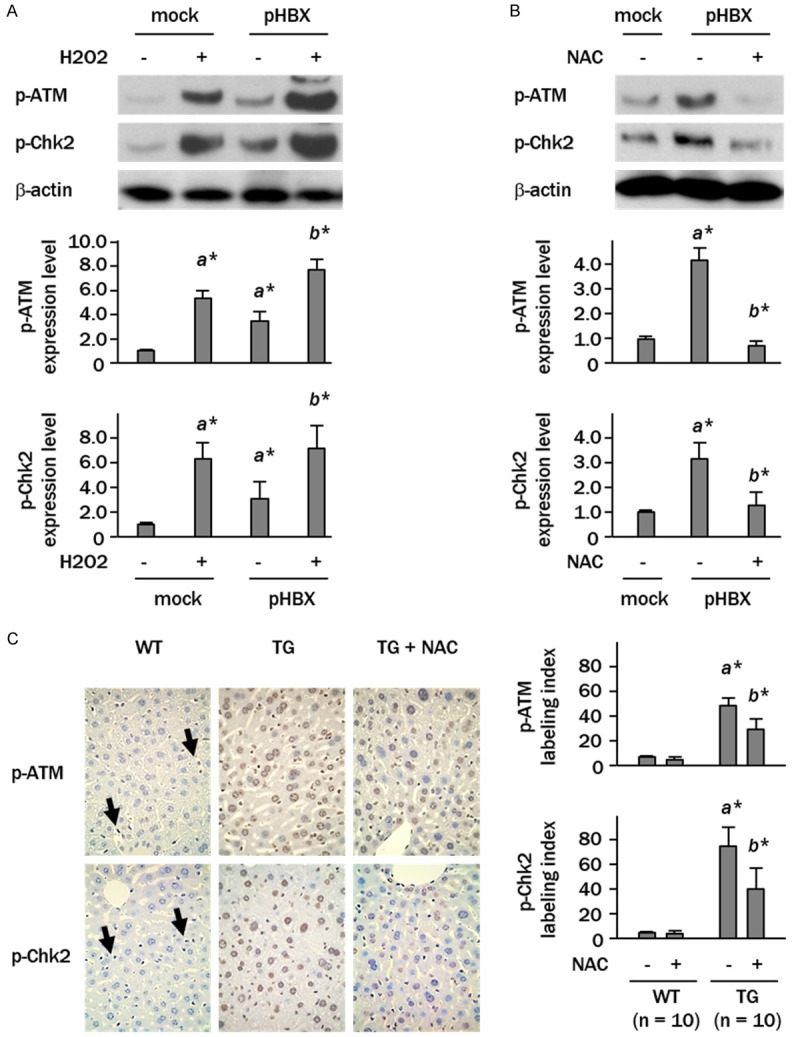
Phosphorylation of ATM is oxidant-dependent. (A and B) Cells were treated with H2O2 (100 μM) for 30 min or with antioxidant NAC (5 mM) for 2 h. Levels of phospho-ATM (p-ATM) and phospho-Chk2 (p-Chk2) were determined by western blotting. Columns represent relative band intensities normalized against β-actin expressed as fold changes relative to control plasmid-transfected cells (a*, p<0.05 vs. mock plasmid transfected cells; b*, p<0.05 vs. pHBX-transfected cells). (C) Immunohistochemical staining of p-ATM and p-Chk2 in livers. Arrows indicate p-ATM- or p-Chk2-positive non-parenchymal cells (magnification x 40). Columns show labeling indices in hepatocytes. Data are expressed as mean ± SD of three independent experiments (a*, p<0.05 vs. wild mice; b*, p<0.05 vs. HBX transgenic mice).
ATM regulates PKC-δ/Nrf2 signaling pathway in HBX-transduced cells
The regulation of PKC-δ/Nrf2 signaling by HBX induced ATM activity was then examined by western blotting. The level of PKC-δ in HBX-expressing cells was increased 3.5-fold compared to the controls (Figure 3A). Treatment of HBX-expressing cells with KU55933 or caffeine decreased the levels of phospho-Chk2 to one third of the level seen in non-treated cells, indicating that these reagents effectively suppressed the ATM kinase activity at the supplied doses. The KU55933- or caffeine-treated HBX-expressing cells showed decreased expression of PKC-δ compared to non-treated controls (0.3- and 0.8-fold, respectively) (Figure 3A). Immunostaining revealed Nrf2 expression in the nuclei of 10 ± 4% of the HBX-expressing cells, suggesting that Nrf2 is constitutively activated in some percentage of HBX-positive cells. Increased expression of nuclear localized Nrf2 was detected immediately after the H2O2 treatment, but remained at 1.5-, 0.9-, and 1.2-fold of the control levels in cells treated with rottlerin, KU55933, and caffeine, respectively (Figure 3B).
Figure 3.
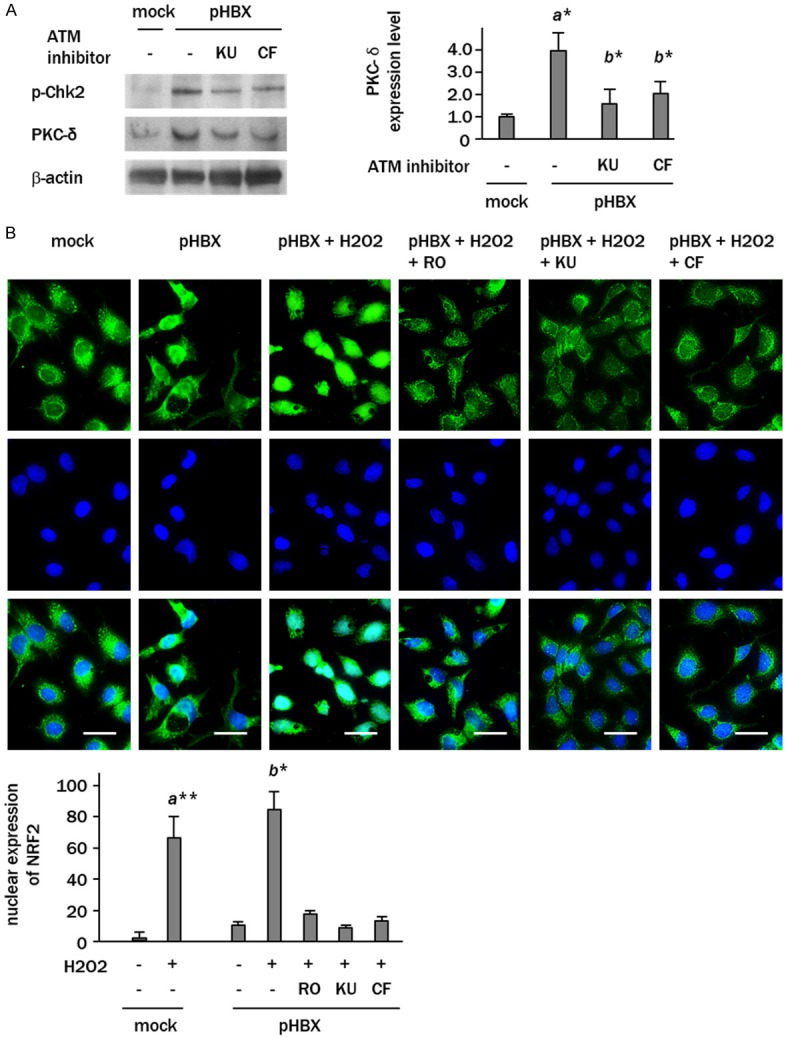
ATM regulates PKC-δ/Nrf2 signaling in HBX-expressing cells. A: Western blot analyses showing the increased level of PKC-δ expression in pHBX-transfected cells, which was attenuated by KU55933 and caffeine. B: Immunofluorescence staining of Nrf2 (green) in HBX-expressing cells treated with H2O2 with or without the treatment with rottlerin (RO), KU55933 (KU) and caffeine (CF). Scale bars represent 20 μm. Upper panel, Nrf2; middle panel, DAPI; bottom panel, merge images. Columns represent the data expressed as mean ± SD of triplicate experiments (a*, p<0.05, vs. mock-transfected cells; b*, p<0.05 vs. HBX-expressing cells).
HBX increases antioxidant molecules
Western blots showed 2.8- and 6.2-fold increases in the levels of HO-1 and Nqo1, respectively, in HBX-expressing cells compared with controls (Figure 4A). Treatment with KU55933 effectively decreased the expression of these antioxidant molecules. Similar increases in the levels of antioxidant molecules were also observed in the livers of mice expressing the HBX transgene (a 3.2-fold increase in HO-1 expression and a 8.0-fold increase in Nqo1 expression, compared to wild type mice) (Figure 4B).
Figure 4.
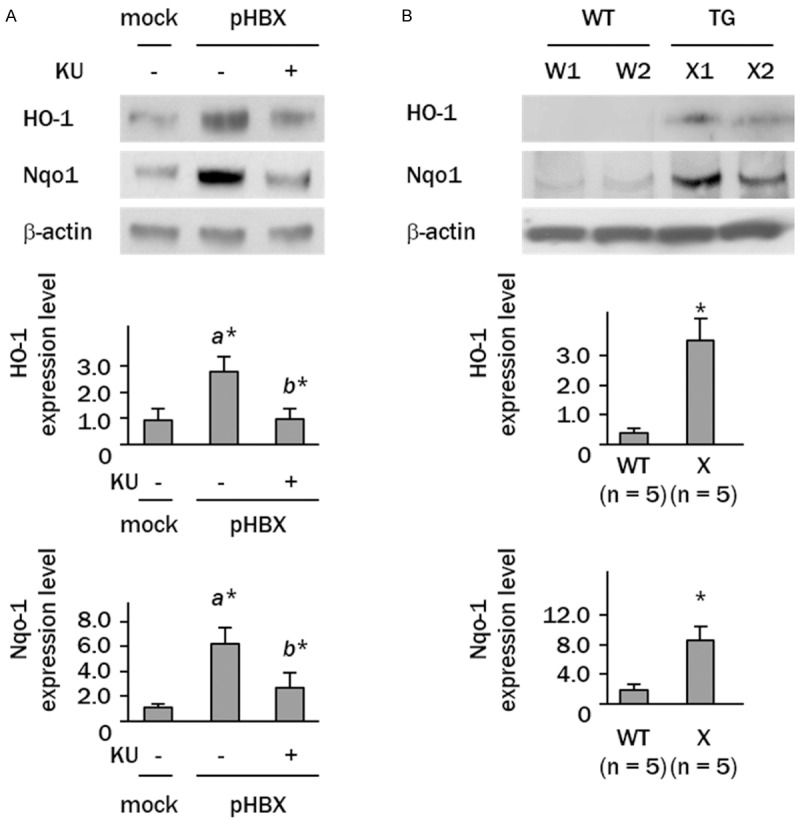
Antioxidant molecules are induced by HBX. A: Western blot analyses showing up-regulation of HO-1 and Nqo1 in HBX-expressing cells (a*, p<0.05, vs. mock-transfected cells; b*, p<0.05 vs. HBX-expressing cells). B: Western blot analysis showing increased expression of HO-1 and Nqo1 in livers from wild (WT) and HBX gene transgenic (TG) mice. W1 and W2, wild mice; X1 and X2, transgenic mice (*p<0.05).
ATM acts as an antioxidant in HBX-expressing cells
The H2DCFDA fluorescence staining revealed no apparent differences in the levels of ROS generation between control and HBX-transfected cells. In contrast, ROS generation was increased in HBX-transfected cells treated with the ATM inhibitors KU55933 and caffeine (Figure 5A). Fluorescence measurements in HBX-expressing cells indicated 1.6 and 1.4-fold increases in relative absorbance after the treatment with KU55933 or caffeine, respectively (p<0.05, p<0.05, vs. non-treated HBX-expressing cells) (Figure 5B).
Figure 5.
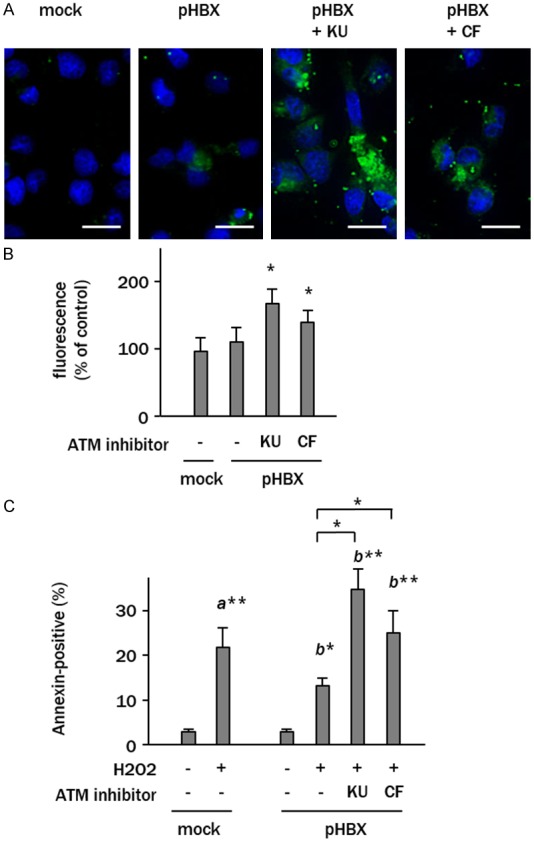
ATM acts as a redox regulator in HBX-expressing cells. A: Representative images of CM-H2DCFDA fluorescence (green) in cells analyzed in B. Nuclei were stained with DAPI (blue). Scale bars represent 20 μm. B: Relative fluorescence intensities reflecting the ROS production. Data representing the fold changes of mock plasmid-transfected cells are expressed as mean ± SD of triplicate experiments (*p<0.05, vs. untreated pHBX-transfected cells). C: The percentages of annexin V-positive apoptotic cells treated with H2O2 (100 μM) in serum-free culture media. The data represent the means ± SD of triplicate experiments (a*, p<0.05 vs. control plasmid transfected cells; b*, p<0.05 vs. H2O2-treated pHBX-transfected cells). KU, KU55933; CF, caffeine.
The involvement of the redox role of ATM in cell survival was examined in cells exposed to H2O2. Control cells exposed to H2O2 showed cell apoptosis rates of 22 ± 4% as assessed by Annexin V assay (p<0.01) (Figure 5C). The HBX-expressing cells showed lower rates of apoptosis (13 ± 3%, p<0.05) in response to H2O2 treatment. Pretreatment of HBX-expressing cells with KU55933 or caffeine before H2O2 exposure increased the rate of cell apoptosis to 33 ± 4% and 22 ± 4%, respectively (p<0.05, p<0.05 vs. control plasmid transfected cells and H2O2-treated HBX-expressing cells) (Figure 5C).
Discussion
This study showed constitutive activation of serine/threonine kinase ATM by HBX both in vitro and in vivo. ATM is a widely recognized DNA damage sensor that responds to stimuli such as ionized radiation. It cooperates with Rad3-related (ATR) and is recruited to damaged DNA sites, where it is activated by phosphorylation to stimulate its down-stream signaling molecules [11]. No clear relationship between HBX and ATM has yet been reported, but previous studies indicate that HBX is involved in the function of several DNA damage response factors. For example, HBX binds and inactivates the tumor suppressor p53, rendering cells susceptible to increased DNA damage [21-23]. HBX interacts with DNA damage-binding (DDR) proteins, which might explain the increased DNA damage seen in HBV-infected cells [24]. HBX also inhibits the intra-S-phase checkpoint through the ATR-Chk1 pathway [25]. However, drawing a simple picture of the role of HBX in DNA damage is difficult because the functional role of DNA damage response proteins is diverse and differs according to the type of DNA damage [11]. Nevertheless, the available evidence, together with the data provided in the present study, strongly suggests that HBX directly regulates elements of the DNA damage response.
ATM appears to have an additional role in ROS regulation, independent of its originally proposed role as a DNA damage sensor [26,27]. In the present study, H2O2 treatment increased the phosphorylation of ATM in both HBX-negative and positive cells, and this increase was strongly suppressed by NAC. Levels of phospho-ATM were also decreased by NAC in the livers of transgenic HBX mice, suggesting that the phosphorylation status of ATM might reflect HBX-induced oxidative stress rather than DNA damage. The H2DCFDA assay indicated no appreciable differences in the endogenous intracellular ROS levels in HBX-expressing cells and control cells, but the ROS levels were significantly elevated in the presence of ATM inhibitors. The HBX-expressing cells showed a relatively better cell survival against oxidative stress, which was abrogated by ATM inhibitors. These lines of evidence support the idea that ATM is a critical regulator of ROS generation in HBX-expressing cells, and that ATM activity might be tightly controlled to maintain homeostasis during oxidative stress through adaptations of the redox system.
Among several regulatory pathway known to control ROS production, the PKC-δ-mediated Nrf2 pathway is a master regulator of redox signaling. PKC-δ is a unique PKC subtype that contributes to ROS homeostasis by operating through intermediate pathways such as p38MAPK. PKC-δ plays a crucial role in the phosphorylation of the oxidative stress sensor Nrf2 at serine40, which causes the dissociation of Nrf2 from an adaptor protein Keap1 (INrf2) [28,29]. The nuclear localized Nrf2 binds with an antioxidant response element (ARE), leading to the induction of antioxidant defensive molecules such as HO-1 and Nqo1 and resulting ultimately in protection of the cells from oxidative stress [30]. In our study, total amount of PKC-δ was significantly increased in HBX-expressing cells. ATM inhibitors attenuated the induction of PKC-δ, indicating that HBX regulates PKC-δ through ATM activity. Although there have been only a few studies of the correlation of ATM and PKC-δ, previous study may support our obtained data, showing that PKC-δ expression is decreased in ATM gene deleted osteoblasts [17]. Because PKC-δ plays diverse roles depending on the site of phosphorylation [31-33], investigating the phosphorylation sites of PKC-δ may help to understand the regulatory mechanism of HBX/ATM/PKC-δ signaling pathway.
Our immunostaining showed that Nrf2 is localized in the nuclei of a small number of HBX-expressing cells, suggesting that Nrf2 might be constitutively activated by HBX-induced oxidative stress in some sets of these cells. Exposure to H2O2 resulted in a significant increase in the nuclear sequestration of Nrf2 in HBX-expressing cells, which was abrogated either by a PKC-δ inhibitor or by ATM inhibitors. We therefore surmise that constitutively activated ATM might play a part in the activation of the PKC-δ/Nrf2 pathway in HBX-expressing cells. The observation that defense molecules such as HO-1 and Nqo1 were considerably increased in HBX-positive cells, both in vitro and in vivo, supports this idea.
In summary, we investigated the molecular mechanism of HBX-mediated redox system and found that ATM might be a critical mediator that balances the increased oxidative stress and activation of the redox system. HBX might activate ATM through ROS generation, while activated ATM might play an adaptive defense role in HBX-induced ROS generation through the PKC-δ/Nrf2 pathway. Our findings suggest that manipulating ATM kinase activity might be effective for controlling the HBX-mediated enhancement of redox activity that promotes cell survival in the presence of oxidative DNA damage. Confirmation of the potential for ATM to regulate HBX-induced carcinogenesis would require further studies on the protective effect of ATM inhibitors in HBX transgenic mice and/or HBV-positive individuals. In this regard, recent study has shown that a blockade of ATM results in a decrease in immature tumor vessels via increased ROS generation, suggesting that ATM-mediated oxidative defense might be an important target for treating neovascular diseases [34]. Further understanding the molecular mechanism of HBV-related hepatocarcinogenesis will require more extensive investigation of the potential of ATM as a redox mediator.
Acknowledgements
We thank Dr. S. Murakami for providing pNK-FLAG-HBx. This work was partly supported by a Grant-in-Aid for Scientific Research (no. 24590962) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Disclosure of conflict of interest
None.
References
- 1.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 3.Schutte K, Bornschein J, Malfertheiner P. Hepatocellular carcinoma--epidemiological trends and risk factors. Dig Dis. 2009;27:80–92. doi: 10.1159/000218339. [DOI] [PubMed] [Google Scholar]
- 4.Gao J, Xie L, Yang WS, Zhang W, Gao S, Wang J, Xiang YB. Risk factors of hepatocellular carcinoma--current status and perspectives. Asian Pac J Cancer Prev. 2012;13:743–752. doi: 10.7314/apjcp.2012.13.3.743. [DOI] [PubMed] [Google Scholar]
- 5.Brechot C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: old and new paradigms. Gastroenterology. 2004;127:S56–61. doi: 10.1053/j.gastro.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 6.Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317–320. doi: 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- 7.Ha HL, Yu DY. HBx-induced reactive oxygen species activates hepatocellular carcinogenesis via dysregulation of PTEN/Akt pathway. World J Gastroenterol. 2010;16:4932–4937. doi: 10.3748/wjg.v16.i39.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koike K, Shirakata Y, Yaginuma K, Arii M, Takada S, Nakamura I, Hayashi Y, Kawada M, Kobayashi M. Oncogenic potential of hepatitis B virus. Mol Biol Med. 1989;6:151–160. [PubMed] [Google Scholar]
- 9.Benn J, Schneider RJ. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci U S A. 1994;91:10350–10354. doi: 10.1073/pnas.91.22.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Motavaf M, Safari S, Saffari Jourshari M, Alavian SM. Hepatitis B virus-induced hepatocellular carcinoma: the role of the virus x protein. Acta Virol. 2013;57:389–396. doi: 10.4149/av_2013_04_389. [DOI] [PubMed] [Google Scholar]
- 11.Matsuda Y, Ichida T. Impact of hepatitis B virus X protein on the DNA damage response during hepatocarcinogenesis. Med Mol Morphol. 2009;42:138–142. doi: 10.1007/s00795-009-0457-8. [DOI] [PubMed] [Google Scholar]
- 12.De Mitri MS, Cassini R, Bernardi M. Hepatitis B virus-related hepatocarcinogenesis: molecular oncogenic potential of clear or occult infections. Eur J Cancer. 2010;46:2178–2186. doi: 10.1016/j.ejca.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 13.Hung JH, Su IJ, Lei HY, Wang HC, Lin WC, Chang WT, Huang W, Chang WC, Chang YS, Chen CC, Lai MD. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J Biol Chem. 2004;279:46384–46392. doi: 10.1074/jbc.M403568200. [DOI] [PubMed] [Google Scholar]
- 14.Waris G, Huh KW, Siddiqui A. Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-kappa B via oxidative stress. Mol Cell Biol. 2001;21:7721–7730. doi: 10.1128/MCB.21.22.7721-7730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaedler S, Krause J, Himmelsbach K, Carvajal-Yepes M, Lieder F, Klingel K, Nassal M, Weiss TS, Werner S, Hildt E. Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. J Biol Chem. 2010;285:41074–41086. doi: 10.1074/jbc.M110.145862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srisuttee R, Koh SS, Park EH, Cho IR, Min HJ, Jhun BH, Yu DY, Park S, Park do Y, Lee MO, Castrillon DH, Johnston RN, Chung YH. Up-regulation of Foxo4 mediated by hepatitis B virus X protein confers resistance to oxidative stress-induced cell death. Int J Mol Med. 2011;28:255–260. doi: 10.3892/ijmm.2011.699. [DOI] [PubMed] [Google Scholar]
- 17.Li B, Wang X, Rasheed N, Hu Y, Boast S, Ishii T, Nakayama K, Nakayama KI, Goff SP. Distinct roles of c-Abl and Atm in oxidative stress response are mediated by protein kinase C δ. Genes Dev. 2004;18:1824–1837. doi: 10.1101/gad.1223504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Semlitsch M, Shackelford RE, Zirkl S, Sattler W, Malle E. ATM protects against oxidative stress induced by oxidized low-density lipoprotein. DNA Repair (Amst) 2011;10:848–860. doi: 10.1016/j.dnarep.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011;30:546–555. doi: 10.1038/emboj.2010.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yalcin S, Zhang X, Luciano JP, Mungamuri SK, Marinkovic D, Vercherat C, Sarkar A, Grisotto M, Taneja R, Ghaffari S. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J Biol Chem. 2008;283:25692–25705. doi: 10.1074/jbc.M800517200. [DOI] [PubMed] [Google Scholar]
- 21.Takada S, Kaneniwa N, Tsuchida N, Koike K. Cytoplasmic retention of the p53 tumor suppressor gene product is observed in the hepatitis B virus X gene-transfected cells. Oncogene. 1997;15:1895–1901. doi: 10.1038/sj.onc.1201369. [DOI] [PubMed] [Google Scholar]
- 22.Prost S, Ford JM, Taylor C, Doig J, Harrison DJ. Hepatitis B x protein inhibits p53-dependent DNA repair in primary mouse hepatocytes. J Biol Chem. 1998;273:33327–33332. doi: 10.1074/jbc.273.50.33327. [DOI] [PubMed] [Google Scholar]
- 23.Ogden SK, Lee KC, Barton MC. Hepatitis B viral transactivator HBx alleviates p53-mediated repression of alpha-fetoprotein gene expression. J Biol Chem. 2000;275:27806–27814. doi: 10.1074/jbc.M004449200. [DOI] [PubMed] [Google Scholar]
- 24.Becker SA, Lee TH, Butel JS, Slagle BL. Hepatitis B virus X protein interferes with cellular DNA repair. J Virol. 1998;72:266–272. doi: 10.1128/jvi.72.1.266-272.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu XY, Qian JJ, Lin Y, Zheng MH. Hepatitis B virus X protein disrupts DNA interstrand crosslinking agent mitomycin C induced ATR dependent intra-S-phase checkpoint. Eur J Cancer. 2008;44:1596–1602. doi: 10.1016/j.ejca.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 26.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujimaki S, Matsuda Y, Wakai T, Sanpei A, Kubota M, Takamura M, Yamagiwa S, Yano M, Ohkoshi S, Aoyagi Y. Blockade of ataxia telangiectasia mutated sensitizes hepatoma cell lines to sorafenib by interfering with Akt signaling. Cancer Lett. 2012;319:98–108. doi: 10.1016/j.canlet.2011.12.043. [DOI] [PubMed] [Google Scholar]
- 28.Niture SK, Jain AK, Jaiswal AK. Antioxidant-induced modification of INrf2 cysteine 151 and PKC-δ-mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. J Cell Sci. 2009;122:4452–4464. doi: 10.1242/jcs.058537. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Lee SE, Yang H, Jeong SI, Jin YH, Park CS, Park YS. Induction of heme oxygenase-1 inhibits cell death in crotonaldehyde-stimulated HepG2 cells via the PKC-δ-p38-Nrf2 pathway. PLoS One. 2012;7:e41676. doi: 10.1371/journal.pone.0041676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Hung AC, Ng PY, Nakayama K, Hu Y, Li B, Porter AG, Dhakshinamoorthy S. PKCδ mediates Nrf2-dependent protection of neuronal cells from NO-induced apoptosis. Biochem Biophys Res Commun. 2009;386:750–756. doi: 10.1016/j.bbrc.2009.06.129. [DOI] [PubMed] [Google Scholar]
- 31.Konishi H, Yamauchi E, Taniguchi H, Yamamoto T, Matsuzaki H, Takemura Y, Ohmae K, Kikkawa U, Nishizuka Y. Phosphorylation sites of protein kinase C δ in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc Natl Acad Sci U S A. 2001;98:6587–6592. doi: 10.1073/pnas.111158798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kikkawa U, Matsuzaki H, Yamamoto T. Protein kinase C δ (PKC δ): activation mechanisms and functions. J Biochem. 2002;132:831–839. doi: 10.1093/oxfordjournals.jbchem.a003294. [DOI] [PubMed] [Google Scholar]
- 33.Hall KJ, Jones ML, Poole AW. Coincident regulation of PKCδ in human platelets by phosphorylation of Tyr311 and Tyr565 and phospholipase C signalling. Biochem J. 2007;406:501–509. doi: 10.1042/BJ20070244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okuno Y, Nakamura-Ishizu A, Otsu K, Suda T, Kubota Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat Med. 2012;18:1208–1216. doi: 10.1038/nm.2846. [DOI] [PubMed] [Google Scholar]