

Abstract
The genetic theory of infectious diseases has proposed that susceptibility to life-threatening infectious diseases in childhood, occurring in the course of primary infection, results mostly from individually rare but collectively diverse single-gene variants. Recent evidence for an ever-expanding spectrum of genes involved in susceptibility to infectious disease indicates the paradigm has important implications for diagnosis and treatment. One such pathology is childhood herpes simplex encephalitis (HSE), which shows a pattern of rare, but diverse disease-disposing genetic variants. The present report shows how proteomics can help to understand susceptibility to childhood HSE and other viral infections, suggests proteomics may have a particularly important role to play, emphasizes that variation over the population is a critical issue for proteomics and notes some new challenges for proteomics and related bioinformatics tools in the context of rare, but diverse genetic defects.
Keywords: Infectious diseases, Herpes simplex encephalitis (HSE), SILAC, herpes simplex virus 1 (HSV-1), TLR3, interferon (IFN), proteomics, genomics, systems biology
Body of the article
Since the 1950s and until recently, it was believed that mutations in a single gene confer vulnerability to multiple infectious diseases. Concomitantly, common infections have been presumed to be associated with the inheritance of mutations in multiple susceptibility genes. In recent work towards a unified genetic theory of disease [1-3], Prof. JL Casanova identified and characterized many new genetic defects that predispose otherwise healthy individuals to a single type of infection [4]. This novel causal relationship has modified the paradigm that dominated the field for several decades. Single-gene inborn errors of immunity in children may confer severe and selective vulnerability to specific infectious illnesses, whereas corresponding infections in adults usually involve more complex gene patterns. Several diseases have been studied including mycobacterial diseases, invasive pneumococcal disease, chronic mucocutaneous candidiasis, severe flu, Kaposi sarcoma and herpes simplex encephalitis (HSE).
Herpes simplex encephalitis
Herpes simplex virus (HSV-1) encephalitis (HSE) is a severe infection of the central nervous system (CNS)[5]. Although HSV-1 is widespread and typically innocuous in human populations, HSE is the most common form of sporadic viral encephalitis in Western countries, where it is estimated to occur in approximately two to four per million individuals per year. Peaks of HSE incidence occur between theages of 6 months to3 years during primary infection with HSV-1. The virus reaches the CNS via a neurotropic route involving the trigeminal and olfactory nerves [6,7]. The mortality rate, which used to be as high as 70%, has declined significantly thanks to treatment with the anti-viral acyclovir [8-10]. In spite of the treatment, up to 60% of patients suffer from long-term neurological sequelae of varying severity [7,11].
Genomic studies, exome sequencing
Exome sequencing, the targeted sequencing of the protein-coding portion of the human genome, has been shown to be a powerful and efficient method for detection of disease variants underlying Mendelian disorders. In the human genome, exons represent about 1% [12]. It is estimated that the protein coding regions of the human genome constitute about 85% of the disease-disposing mutations [13]. Robust sequencing of the complete coding region (exome) has the potential to be clinically relevant in genetic diagnosis as understanding of the functional consequences in sequence variation improves [13]. Currently exome sequencing is discovering inborn errors of immunity in children that confer severe and selective vulnerability to certain infectious diseases [14,15].
Childhood HSE has not been associable with known immunodeficiencies and its pathogenesis remained elusive until identified the first five genetic aetiologies of this condition were identified [16-21]. Autosomal recessive UNC-93B deficiency abolishes Toll-like receptor 3 (TLR3), TLR7, TLR8, and TLR9 signalling [16], whereas autosomal dominant TLR3 deficiency specifically affects TLR3 signalling[21]. Recently an autosomal recessive form of complete TLR3 deficiency has been described as a compound heterozygous for two loss-of-function TLR3 alleles [17]. Moreover an autosomal dominant deficiency in TNF receptor-associated factor 3 (TRAF3) [19], a Toll/IL1R (TIR) domain-containing adaptor inducing IFN-β (TRIF) deficiency [20] and TANK-binding kinase (TBK1)–deficiency [18] have been described.
All of these genetic defects involve the Toll-like receptor 3 (TLR3) signalling pathway and these studies suggested that childhood HSE may result from impaired interferon (IFN)-α/β and IFN-λ production in response to the stimulation of TLR3 by dsRNA intermediates of HSV-1 in the CNS (Fig. 1). However, the study of proteins implicated in the TLR3-IFN pathway for HSE patients revealed that only a small fraction of children with HSE carry mutations in UNC93B1, TLR3, TRAF3, TRIF or TBK1 [16-21]. A larger proportion of patients display an impaired production of IFN type I and III upon TLR3 stimulation of their fibroblasts. Conversely, the study of IFN type I and III production after TLR3 activation in SV40-fibroblasts of HSE patients has shown that 30%, of a total of 89 patients analysed, have IFN type I and III production which is normal. This suggested that in spite of the importance of the TLR3 pathway in HSE immunity, genetic defect(s) responsible for the susceptibility to HSV-1 in the CNS could be due to TLR3-independent pathways, or other TLR3, IFN-dependent pathways that are activated after the initial TLR3 activation.
Figure 1.
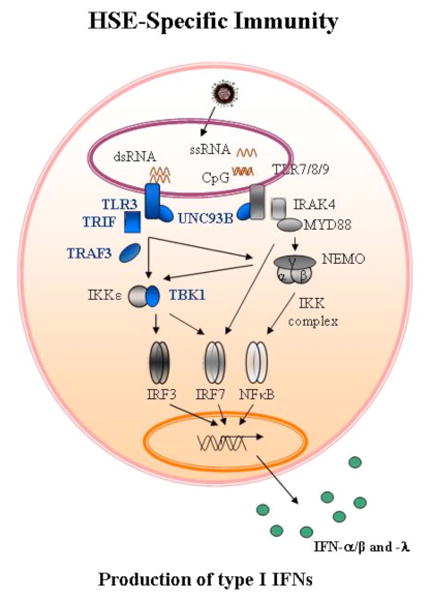
A simplified diagram of the TLR-mediated and interferon (IFN)-mediated immunity in response to viruses. TLR3 is located in the endoplasmic reticulum (ER) and in endosomes, where it recognizes double-stranded RNA produced during the replication of most viruses. Activation of TLR3 induces activation of IRF-3 and NF- κ B via the TRIF adaptor, and the production of IFN-α/β and/or - λ. UNC-93B is required for the trafficking of TLR3, TLR7, TLR8 and TLR9 from the ER to the endosomal compartment. Proteins of the TLR3 pathway for which genetic mutation have been identified and associated with susceptibility to Herpes simplex virus-1 encephalitis (TLR3, TRIF, UNC-93B, TRAF3 and TBK1) are depicted in blue.
Formulating a Proteomics Approach
Conventional attempts to define disease-related genetic defects involving single proteins commonly try to screen large numbers of patient samples to validate single gene defects. This often has the major obstacle that sufficient numbers of patient samples are difficult to obtain. Since TLR3-dependent pathways are clearly involved in HSV-1 susceptibility, it was considered that it might be possible to use a combination of proteomics and systems biology methods to look for other networks and/or proteins[22]. The basic idea behind this strategy is that the increasing amount of available systems biology information has changed the situation. Using only small numbers of healthy controls and patient samples with appropriate functional stimulation, it might now be possible to detect disease-related functional networks by monitoring large numbers of proteins simultaneously, even if the underlying genetic defect in any individual patient cannot be statistically validated by such a study. If successful, definition of such functional networks could already have important diagnostic and therapeutic implications.
In the case of HSE, a large volume of previous biochemical experiments on several hundred patients and healthy individuals provided a strong base of knowledge, well established, highly reproducible sample preparation methods and related experimental tests of cellular response for the proteomics experiments. These previous studies also made it clear that there was a potential problem that is not often considered in present proteomics studies: what constitutes a normal, healthy response? Monitoring the abundance of key proteins (IFN-beta, IFN-lambda, IL6, NFκB and IRF3), cell survival after Vesicular stomatitis virus (VSV) infection, and viral replication [16-21], suggested that several different phenotypes could be distinguished and indicated that there were highly reproducible variations of up to 50 fold in the amounts of IFN type I and III produced by control cells from different healthy individuals in response to dsRNA (Fig 2). In short, for these kinds of proteomics experiments highly reproducible sample preparation for “normal” cell samples that span the range of possible response over the human population seem to be required. Put differently, proteomics experiments should probably only be undertaken in the context of a large base of previous characterization of healthy population variation. With this background in mind, the goal was set out to obtain an initial test of four propositions. (1) Is the variation over population seen by biochemical tests for a few proteins expressed in larger numbers of proteins? (2) Are there proteomics signatures that are characteristic despite population variation? (3) Can significant differences between healthy and patient cells be detected despite population variation? (4) Are the differences of potential genetic, diagnostic or therapeutic interest? Positive results were obtained for all four propositions, but indicated some new challenges for proteomics and bioinformatics (see below).
Figure 2.
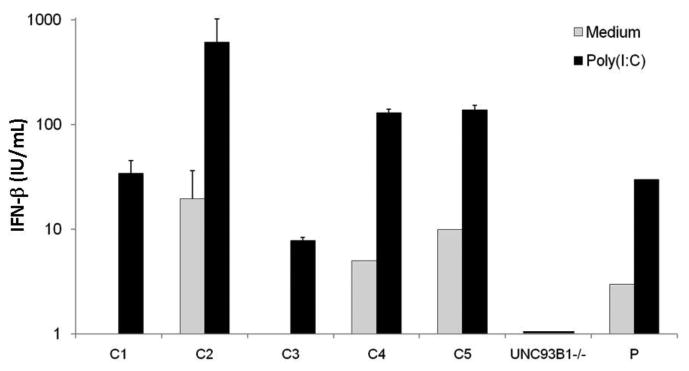
Production of IFN-β by SV40-fibroblasts after poly(I:C) stimulation (25 μg/ml) for 24 hoursas assessed by ELISA. C1-C5 are the positive healthy controls and UNC93B1-/-is the UNC-93B-deficientpatient. Mean values ± SD were calculated from three independent experiments.
A Short Summary of the Biological Findings
The SILAC measurements of differential protein abundance were conducted for six samples: three healthy controls from different individuals that showed weak (C3), medium (C1) and strong (C2) production of IFNs in response to dsRNA that was intended to sample population variation, a healthy control without dsRNA stimulation (C2NS), a patient with an UNC-93B-/- defect that abolishes TLR3 pathway response (UNC) and a patient with an unknown genetic defect (P). As described in the original publication [22], common functional pathways in healthy individuals implicated in transmigration of immune cells, apoptosis and oxidative stress that were abrogated in HSE patients were discovered. Furthermore, a set of new proteins for further investigation of possible disease aetiologies was identified (Fig.3) and evidence was obtained that manipulation of one of these (SOD2) could have therapeutic benefits. The observation of changes in proteins involved in mitochondrial oxidative stress systems (SOD2, PPIF) opened a new, additional perspective apart from nuclear-directedTLR3 pathways that is consistent with other recent studies of response to viral infections [23-25]. For the patient with an unknown gene defect and without the fibroblastic phenotype, a lack of ICAM-1 upregulation, strong upregulation of SOD2, and upregulation of a variety of proteins previously associated with TLR3 pathways, delineated a new cellular phenotype which will help to dissect his genetic aetiology. The details of these results and of their context relative to the biological literature are contained in the original publication, to which the reader is referred – in the following we note some new features of the experimental results that have important consequences for future proteomics studies.
Figure 3.
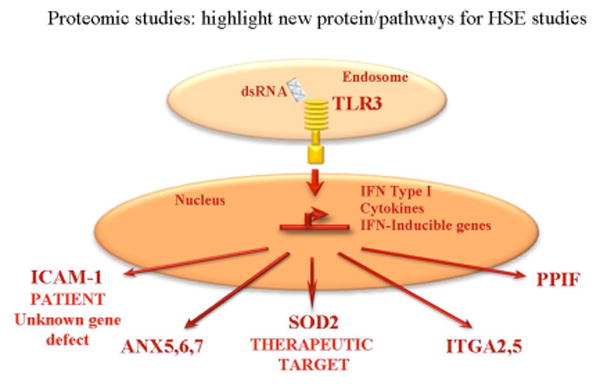
Illustration of the potential biological significance in immunity against HSE for proteins upregulated after TLR3 activation.
New Challenges for Proteomics and Bioinformatics
(1) Population variation is a crucial issue for proteomics
Comparison of the SILAC ratios between the different healthy samples indicated related response with correlation values in the ranges usually accepted as biologically relevant (Fig. 4A). Correlation with the unstimulated sample was in all cases very small. However, in agreement with the large differences in IFN production, the response to dsRNA showed large variation in the overall magnitude of SILAC ratios between different healthy individuals (Fig. 4B). The SILAC ratios revealed that the strong variation in response levels over the population previously detected with small numbers of proteins using western blotting is in fact reflected in the abundance changes for large numbers of proteins. Although the H/L distributions remained approximately Gaussian, for the healthy cells with the strongest response (C2), several hundred proteins showed 2-fold changes in abundance.
Figure 4.
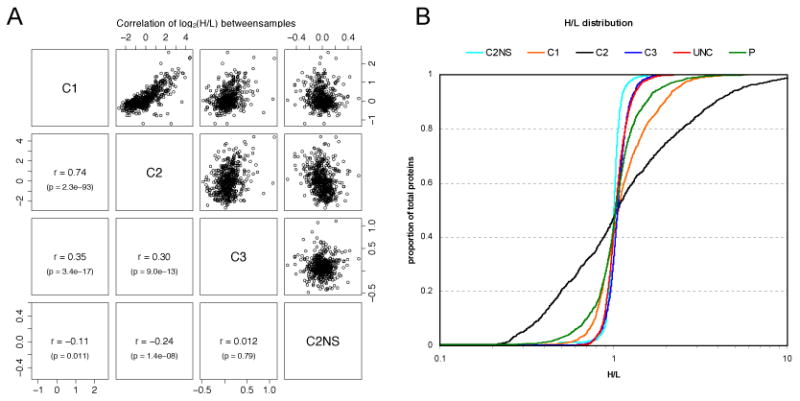
(A) Correlation of log2(H/L) between healthy samples (C1, C2, C3) and the healthy, non-stimulated sample (C2NS). (B) Cumulative proportion of proteins with the indicated H/L ratios for all six samples.
(2) Sets of “Most Significant” Proteins are Dependent on Population Variation
Even though similar functional networks were involved, the identity and rank order of proteins with the “most significant” abundance changes differed strongly between different individuals. For example, seven annexins were recorded for all samples. For the healthy samples the general trend for abundance changes was C2 > C1 > C3 in parallel to the changes in IFNs, but the rank order of individual annexins showed C1: ANXA5 > ANXA1 > ANXA7; C2: ANXA7 > ANXA6 > ANXA11; C3: ANXA11 > ANXA7 > ANXA4 (Fig. 5A). This feature complicates the choice of “most significant” proteins for subsequent analysis of functional networks using systems biology tools such as GeneGo. Because the H/L distributions remained approximately Gaussian (Fig. 4B), a Significance B formulation was used[26] to select the most significant changes for each sample type (Fig. 5B). However, for samples such as C1 and C2 the distribution of H/L is not dominated by experimental noise (C2NS), but rather by cellular response (Fig. 4B). Consequently proteins excluded from the C2 most significant set in fact showed substantially stronger abundance changes than proteins accepted for the C1 or C3 data sets (Fig. 5B). As an alternative that was the same for all data sets, a Significance B* factor was calculated relative to the signal intensity/scatter of the unstimulated C2NS data set (Fig. 5C), i.e. relative to real experimental noise. The disadvantage of this was that, because of the large differences in cellular response, the number of proteins accepted for network analysis was heavily dominated by C2, e.g. at Significance B* < 1e-5, the C3/C1/C2 significant data sets included 15/351/842 proteins. Conversely, use of Significance B < 0.05 led to exclusion of large numbers of proteins from the C1 and C2 data sets that had large abundance changes with high reliability relative to real experimental noise. As a compromise, Significance B < 0.05 and H/L cut offs was used to select approximately equal numbers of “most significant” proteins from each sample type [22]. This “equal sampling” was successful in identifying relevant functional networks using GeneGo. However, only a minority of the “most significant” proteins were common to all three healthy individuals, even though other proteins in the union over the healthy samples satisfied the stringent cutoff Significance B* < 1e-5. In the context of small numbers of samples it might be possible to use dosage of the stimulation (amount of dsRNA) to attain similar response levels for different cell samples, but for higher throughput analyses of larger numbers of samples, new computational approaches are needed.
Figure 5.
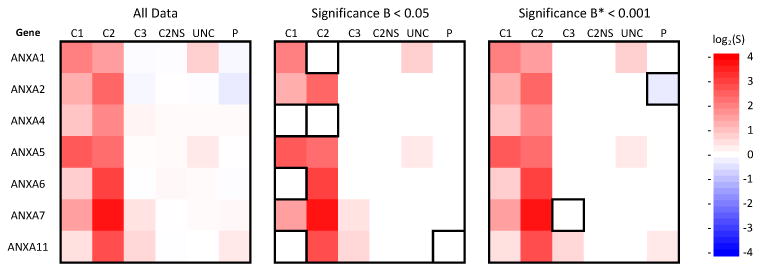
Heat maps showing alternative strategies for selection of “most significant” protein sets for subsequent functional network searches using GeneGo. (A) SILAC ratios recorded for 7 different annexins over the six simple types. The number of ratio counts for individual proteins ranged from 6 to 243 per sample. (B) Proteins retained with a Significance B < 0.05 filter applied to each sample independently. (C) Proteins retained with a Significance B* < 0.001 filter applied across all samples. Boxed regions: proteins deleted that had |log2(S)| equal to or greater than “significant” proteins retained in other samples.
Such population variation has been seen in other recent proteomics studies. For example, measurements for 90 genetically different strains of yeast showed that most variation in protein abundance was due to variability in translation and/or protein stability rather than in transcript levels [27]. Similarly, a recent study of four patients with acute myeloid leukemia, five patients with acute lymphoid leukemia and 8 healthy controls compared the basal abundances of 639 different proteins using alignment-based quantitation of LC-MS/MS data sets [28] and found population variation similar to that shown in Fig. 5.
(3) Current systems biology tools need adaptation to analysis of population variation
The ultimate goal of a population-wide, network-based analysis of function would be to identify common networks across the population and to specify for different individuals the extent to which a common stimulation engages the different networks. Such networks will not be easy to define since they are likely to be highly intertwined (buffered networks in the terminology of complex adaptive systems theory [29]) and the “output” of any sub-network may be diverse and may include: changes in protein abundance, post-translational state and subcellular spatial distribution [30,31] (from proteomics), changes in abundance of metabolites, co-factors, etc. (metabolomics) and genetic changes (epigenetics, micro-RNAs, etc.). The conceptual model of similar networks turned on to different degrees in different individuals that are reflected in protein abundance changes (Fig. 6A) is a testable model. Across the space of icell samples from different individuals, all proteins k that belong to network j have a vector of measured H/L ratios of the form: in which ajk represents an amplitude for “unit engagement” of the network jfor each protein k and nij represents the amplitude to which the network is engaged in each individual i. That is, in a multidimensional space with H/L ratios for different cell samples as the orthogonal axes, there is an axis described by the vector (n1j,n2j, …,nij) that is the same for all proteins k in network j (Fig. 6B).
Figure 6.
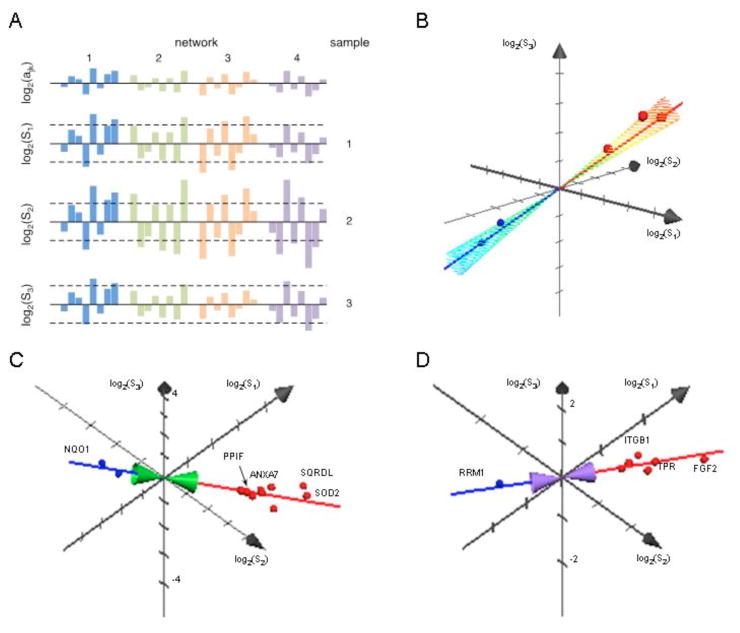
(A) Model of abundance changes for four networks with intrinsic abundance changes ajk for different proteins for unit turn-on of the network. For three cell samples from healthy individuals each network is turned on to different degrees. These results in changes in the set of “most significant” proteins selected with Significance B filters (dashed lines) and their rank order for each cell sample. (B) Relationships in the 3D space of SILAC ratios [log2(S1), log2(S2), log2(S3)] for proteins from a single network. The red/blue spheres and axis indicate increased/decreased abundance. The relative amplitude to which the network is turned on in the different cell samples is given by the axis log2(S1):log2(S2):log2(S3) = 1:1:1 for equal activation in all cell samples. (C) Putative network for proteins involved in redox responses following stimulation of the healthy samples with dsRNA. log2(S1):log2(S2):log2(S3) = 0.42:0.91:0.13. (D) Putative network for proteins involved in nuclear processes following stimulation of the healthy samples with dsRNA. log2(S1):log2(S2):log2(S3) = 0.69:0.73:0.26.
The model implies the need to search for correlation amongst functionally related proteins (systems biology functional correlations) in functional data (H/L ratios) for high dimensional spaces (many individual samples) – a feature that seems not to be available in current publicly accessible systems biology tools. There are strong indications of such relations in the present data (Fig. 6C, D), but new, more sophisticated analysis and statistical validation is required.
Final conclusions
Even for a disease like HSE of apparent aetiological diversity, and in the face of cellular diversity amongst individuals, proteomics provided an overview of cellular responses that may be crucial to HSE susceptibility. At present the systems biology knowledge encoded in data bases such as GeneGo ascribed the groups of proteins for which upregulation was detected to several different related, but fragmented pathways. Much greater depth than in these initial proteomics experiments is certainly possible. Future studies of the pathways should contribute to a better understanding of HSV-1 infection in the CNS, should contribute to finding new genetic aetiologies for HSE patients and could provide crucial input to diagnostic and therapeutic developments.
Although a relatively small number of patient and healthy samples were used, the results also provide a promising “proof of principle” for the strategy of comparing large numbers of proteins in individual patient cell samples with a database of “possible” responses by healthy cells. Importantly, in this strategy: (1) greater reliability requires a larger number and greater depth for easy to obtain healthy cell samples to ensure that the background against which individual patient samples are compared is adequate, as opposed to the large numbers of hard to obtain patient samples required in more conventional approaches; and, (2) the “output” being measured is closer to function than features such as genetic defects, which may aid in subsequent diagnostic and therapeutic efforts. In this sense the appreciable differences in response for different healthy individuals may represent an advantage if this means the “range of possible responses” can be established with moderate numbers of healthy samples. Furthermore, cellular response to dsRNA is a common feature of viral infection and approaches of the type outlined here can be expected to apply to a wide range of viruses.
It may also be noted that the proteomics results seem not to be predictable with current gene network tools. For example, only 9 of described set of 152 most significant proteins by Perez et all [22] showed overlap with the 601 genes predicted (p < 0.05) to be most closely related to the TLR3 pathway with the Human Gene Connectome approach [32]. Amongst many possibilities, this might arise because systems biology data bases are still too sparse, because aggregation of data from many cell types over many functional contexts obscures relationships for specific contexts, because other information such as post-translational state and subcellular spatial distribution of proteins as well as epigenetic modifications are insufficiently represented, or because proteomics is also detecting responses more distant from the TLR3 pathway. Similar differences in classificatory features have been observed between transcriptomic and proteomic results in the case of human leukemia cells [28]. In short, information from proteomics seems to be highly complementary to other approaches.
Key issues.
Herpes simplex virus (HSV-1) encephalitis (HSE) is a devastating infection of the central nervous system, the most common form of sporadic viral encephalitis in Western countries and typically produces neurological sequelae in patients, especially young children.
In keeping with the genetic theory of infectious diseases, mutations in five diverse genes have been implicated in susceptibility to HSE, but only a low percentage of patients exhibit these mutations. They show that the toll-like receptor 3 (TLR3) signalling pathway is crucial for primary infection.
Different phenotypes have been identified amongst patients, e.g. about 30% of HSE patients have normal Interferon type I and III production after TLR3 stimulation, implying that pathways beyond the TLR3-IFN pathway must be involved.
Use of a new strategy in which large numbers of proteins in patient samples are compared with population variation for healthy individuals and with systems biology functional information helped identify additional pathways and proteins that are implicated in susceptibility to HSE.
Initial evidence has been obtained that new pathways and proteins identified with this strategy can contribute to therapeutic treatments.
The results indicate that normal variation over the population in healthy individuals is a crucial issue for proteomics and these new measures of significance and new forms of data analysis are needed to deal with population variation.
The results were obtained in the context of a unified theory of genetic predisposition to infectious disease, but similar paradigms should be expected for complex genetic diseases and population variation.
Efforts to approach these complex problems from the perspective of the genetic theory of infectious disease may have a crucial advantage in that “infection” can provide a defined, controllable tool to initiate cellular responses in ways that can be easily exploited by proteomics.
Response of cells to dsRNA is a characteristic of many viral infections and the approach described here should be widely applicable to viral infection.
Integration of proteomics information with genetic, metabolomics and medical information is important to develop “functional-network-based” conceptual models of normal function, disease, diagnostics and therapy.
Acknowledgments
We thank the members of the Laboratory of Human Genetics of Infectious Diseases. We thank the patients and their families for their participation in this study, which was supported by the Groupement d'Intérêt Scientifique Maladies Rares, the Action Concertée Incitative de Microbiologie, the March of Dimes, the Agence Nationale pour la Recherche, the Eppley Foundation, the National Institute of Allergy and Infectious Diseases grant number R01AI088364, the Thrasher Research Fund, the Jeffrey Modell Foundation, Talecris Biotherapeutics, the St. Giles Foundation, the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH) grant number 8UL1TR000043, and the Rockefeller University. R.P.D. is supported by the “Ramon y Cajal” programme (MINECO, Spain). J.-L.C. was an international investigator of the Howard Hughes Medical Institute since 2013. The work was supported by a Wellcome Trust Grant to J. G-Z.
Footnotes
Expert commentary: “vision of the author”: In depth analysis of changes in abundance for networks of proteins after Herpes simplex virus-1 (HSV-1) infection is possible with current proteomic methods. Correlation with systems biology information provides an overview of the cellular responses that contribute to susceptibility to diseases such as herpes simplex encephalitis (HSE) that are based on rare, but diverse genetic defects. The studies contribute to better understanding of HSV-1 infection in the central nervous system, can contribute to finding new genetic aetiologies for HSE patients and could provide crucial input to diagnostic and therapeutic developments. The approach should be more generally applicable to viral infections. Furthermore, the new strategy that is used takes variation over the population into account and should be applicable both to susceptibility to infectious diseases as well as to genetic diseases that are based on rare, but diverse genetic defects.
Five year view: Diseases of diverse aetiology are difficult to study, but the present results suggest that functional networks and proteins with correlation to HSE can be identified with proteomics. In addition proteomics analysis even identified differences in individual patients that could eventually have consequences for choices of therapeutic regime. Although these results were obtained in the context of a unified theory of genetic predisposition to infectious disease, attentive readers may have noted that there are substantial similarities to complex genetic diseases. For example, in complex genetic diseases such as Parkinson's disease, where patient cohorts of substantial size can be recruited, large scale genome-wide-association-strategies have been crucial to defining many genes that can contribute to disease susceptibility. About 20 genes that can contribute to Parkinson's disease have been identified [33-38]. However, these only account for 50-60% of inheritable susceptibility and some of the most commonly occurring “gene defects” are known to be neither necessary nor sufficient for the disease[38]. Consequently, current genome-wide-association-strategies, and the even more powerful nucleotide sequencing technologies, are already being seen as being incomplete for development of medical diagnostics or therapies for complex diseases. For this reason, efforts are rapidly moving on to integrate the genetic information with proteomics, metabolomics and clinical information to develop “functional-network-based” conceptual models of normal function, disease, diagnostics and therapy[38]. Proteomics has a vital role to play in these developments and that the work on HSE indicates indicate some of the challenges that will be faced. “Normal” networks are not a clearly defined concept against which to detect aberrations. “Normal” variation over the population may be exactly why infections like HSE and diseases like Parkinson's disease are complex and multi-genomic without any specific“gene defect” being necessary or sufficient, i.e. a gene is only defective in the context of the genome/proteome of an individual. In this situation, higher level, functional-network-based approaches that analyze the dynamics of large networks [39] seem essential. This is a forte of proteomics, but the proteomics studies will have to have a well characterized background of samples representative of variation of function over the population to be relevant. We will also need new tools for evaluation of significance and new computational and bioinformatics tools of kinds hinted at above to interpret the results. Efforts to approach these complex problems from the perspective of the genetic theory of infectious disease may have a crucial advantage in that “infection” can provide a defined, controllable tool to initiate cellular responses in ways that can be easily exploited by proteomics.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Casanova JL, Abel L. Human genetics of infectious diseases: a unified theory. EMBO J. 2007;26(4):915–922. doi: 10.1038/sj.emboj.7601558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2*.Casanova JL, Abel L. The Genetic Theory of Infectious Diseases: A Brief History and Selected Illustrations. Annu Rev Genomics Hum Genet. 2013;14:215–243. doi: 10.1146/annurev-genom-091212-153448. Very good review on history and current developement in genetics of infectious disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alcais A, Abel L, Casanova JL. Human genetics of infectious diseases: between proof of principle and paradigm. J Clin Invest. 2009;119(9):2506–2514. doi: 10.1172/JCI38111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casanova JL, Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317(5838):617–619. doi: 10.1126/science.1142963. [DOI] [PubMed] [Google Scholar]
- 5.Whitley RJ. Herpes simplex encephalitis: adolescents and adults. Antiviral Res. 2006;71(2-3):141–148. doi: 10.1016/j.antiviral.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 6.De Tiege X, Rozenberg F, Heron B. The spectrum of herpes simplex encephalitis in children. Eur J Paediatr Neurol. 2008;12(2):72–81. doi: 10.1016/j.ejpn.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Abel L, Plancoulaine S, Jouanguy E, et al. Age-Dependent Mendelian Predisposition to Herpes Simplex Virus Type 1 Encephalitis in Childhood. J Pediatr. 2010;157(4):623–629. doi: 10.1016/j.jpeds.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 8.Bustamante J, Picard C, Fieschi C, et al. A novel X-linked recessive form of Mendelian susceptibility to mycobaterial disease. J Med Genet. 2007;44(2):e65. doi: 10.1136/jmg.2006.043406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitley RJ, Alford CA, Hirsch MS, et al. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med. 1986;314(3):144–149. doi: 10.1056/NEJM198601163140303. [DOI] [PubMed] [Google Scholar]
- 10.Whitley RJ, Lakeman F. Herpes simplex virus infections of the central nervous system: therapeutic and diagnostic considerations. Clin Infect Dis. 1995;20(2):414–420. doi: 10.1093/clinids/20.2.414. [DOI] [PubMed] [Google Scholar]
- 11.Kneen R, Michael BD, Menson E, et al. Management of suspected viral encephalitis in children - Association of British Neurologists and British Paediatric Allergy, Immunology and Infection Group national guidelines. J Infect. 2012;64(5):449–477. doi: 10.1016/j.jinf.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byun M, Ma CS, Akcay A, et al. Inherited human OX40 deficiency underlying classic Kaposi sarcoma of childhood. J Exp Med. 2013;210(9):1743–1759. doi: 10.1084/jem.20130592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong XF, Bousfiha A, Rouissi A, et al. A novel homozygous p.R1105X mutation of the AP4E1 gene in twins with hereditary spastic paraplegia and mycobacterial disease. PLoS One. 2013;8(3):e58286. doi: 10.1371/journal.pone.0058286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16**.Casrouge A, Zhang SY, Eidenschenk C, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314(5797):308–312. doi: 10.1126/science.1128346. Describes autosomal-recessive UNC-93B deficiency in abolishing Toll-like receptors 3, TLR7, TLR8 and TLR9 signalling. [DOI] [PubMed] [Google Scholar]
- 17.Guo Y, Audry M, Ciancanelli M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. 2011;208(10):2083–2098. doi: 10.1084/jem.20101568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herman M, Ciancanelli M, Ou YH, et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med. 2012;209(9):1567–1582. doi: 10.1084/jem.20111316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19*.de Diego Perez R, Sancho-Shimizu V, Lorenzo L, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33(3):400–411. doi: 10.1016/j.immuni.2010.08.014. This paper demonstrates the implication of autosomal dominant deficiency in TNF receptor associated factor 3 on TLR3 response in HSE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20*.Sancho-Shimizu V, de Diego Perez R, Lorenzo L, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest. 2011;121(12):4889–4902. doi: 10.1172/JCI59259. This paper demonstrates TRIF deficiency as a protein downstream TLR3 pathway implicated in HSE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21**.Zhang SY, Jouanguy E, Ugolini S, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317(5844):1522–1527. doi: 10.1126/science.1139522. Describes for the first time TLR3deficiency in patients with herpes simplex encephalitis showing that TLR3 is specific for immunity against HSV-1. [DOI] [PubMed] [Google Scholar]
- 22*.de Diego Perez R, Mulvey C, Crawford M, et al. The proteome of Toll-like receptor 3-stimulated human immortalized fibroblasts: Implications for susceptibility to herpes simplex virus encephalitis. J Allergy Clin Immunol. 2013;131(4):1157–1166. doi: 10.1016/j.jaci.2013.01.008. Presents large scale proteomics analysis of pathways that may contribute to HSE susceptibility. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hill JM, Lukiw WJ, Gebhardt BM, et al. Gene expression analyzed by microarrays in HSV-1 latent mouse trigeminal ganglion following heat stress. Virus Genes. 2001;23(3):273–280. doi: 10.1023/a:1012517221937. [DOI] [PubMed] [Google Scholar]
- 24.Rakkola R, Matikainen S, Nyman TA. Proteome analysis of human macrophages reveals the upregulation of manganese-containing superoxide dismutase after toll-like receptor activation. Proteomics. 2007;7(3):378–384. doi: 10.1002/pmic.200600582. [DOI] [PubMed] [Google Scholar]
- 25.Saha RN, Pahan K. Differential regulation of Mn-superoxide dismutase in neurons and astroglia by HIV-1 gp120: Implications for HIV-associated dementia. Free Radic Biol Med. 2007;42(12):1866–1878. doi: 10.1016/j.freeradbiomed.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26**.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. Paper describes MaxQuant, an untegrated suite of algorithms developed for quantitative mass spectrometry based proteomics. [DOI] [PubMed] [Google Scholar]
- 27**.Foss EJ, Radulovic D, Shaffer SA, Goodlett DR, Kruglyak L, Bedalov A. Genetic variation shapes protein networks mainly through non-transcriptional mechanisms. PLoS Biol. 2011;9(9):e1001144. doi: 10.1371/journal.pbio.1001144. Very interesting article showing that transcriptomics and proteomics monitor different features of genetic variation across populations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Foss EJ, Radulovic D, Stirewalt DL, et al. Proteomic classification of acute leukemias by alignment-based quantitation of LC-MS/MS data sets. J Proteome Res. 2012;11(10):5005–5010. doi: 10.1021/pr300567r. Interesting example of population variation as seen by proteomics measurements of basal protein abundances in human leukemia cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitacre JM, Bender A. Networked buffering: a basic mechanism for distributed robustness in complex adaptive systems. Theor Biol Med Model. 2010;7:20. doi: 10.1186/1742-4682-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulvey CM, Tudzarova S, Crawford M, Williams GH, Stoeber K, Godovac-Zimmermann J. Subcellular proteomics reveals a role for nucleo-cytoplasmic trafficking at the DNA replication origin activation checkpoint. J Proteome Res. 2013;12(3):1436–1453. doi: 10.1021/pr3010919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henke RM, Dastidar RG, Shah A, et al. Hypoxia elicits broad and systematic changes in protein subcellular localization. Am J Physiol Cell Physiol. 2011;301(4):C913–928. doi: 10.1152/ajpcell.00481.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itan Y, Zhang SY, Vogt G, et al. The human gene connectome as a map of short cuts for morbid allele discovery. Proc Natl Acad Sci U S A. 2013;110(14):5558–5563. doi: 10.1073/pnas.1218167110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butcher NJ, Kiehl TR, Hazrati LN, et al. Association Between Early-Onset Parkinson Disease and 22q11.2 Deletion Syndrome: Identification of a Novel Genetic Form of Parkinson Disease and Its Clinical Implications. JAMA Neurol. 2013 doi: 10.1001/jamaneurol.2013.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flinn LJ, Keatinge M, Bretaud S, et al. TigarB causes mitochondrial dysfunction and neuronal loss in PINK1 deficiency. Ann Neurol. 2013 doi: 10.1002/ana.23999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manzanillo PS, Ayres JS, Watson RO, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013 doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puschmann A. Monogenic Parkinson's disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord. 2013;19(4):407–415. doi: 10.1016/j.parkreldis.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 37.Tsika E, Moore DJ. Contribution of GTPase activity to LRRK2-associated Parkinson disease. Small GTPases. 2013;4(3) doi: 10.4161/sgtp.25130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38**.Scholz SW, Mhyre T, Ressom H, Shah S, Federoff HJ. Genomics and bioinformatics of Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2(7):a009449. doi: 10.1101/cshperspect.a009449. Excellent article on application of genomics and bioinformatics in understanding the nature of complex diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39*.Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol. 2012;8:565. doi: 10.1038/msb.2011.99. Short review of the need for differential dynamic measurements of cellular networks. [DOI] [PMC free article] [PubMed] [Google Scholar]