

Abstract
In this paper, an unsupervised artificial neural network was implemented to identify the patters of specific signatures. The network was based on the differential expression of miRNAs (under or over expression) found in healthy or cancerous gastric tissues. Among the tissues analyzes, the neural network evaluated 514 miRNAs of gastric tissue that exhibited significant differential expression. The result suggested a specific expression signature nine miRNAs (hsa-mir-21, hsa-mir-29a, hsa-mir-29c, hsa-mir-148a, hsa-mir-141, hsa-let-7b, hsa-mir-31, hsa-mir-451, and hsa-mir-192), all with significant values (p-value < 0.01 and fold change > 5) that clustered the samples into two groups: healthy tissue and gastric cancer tissue. The results obtained “in silico” must be validated in a molecular biology laboratory; if confirmed, this method may be used in the future as a risk marker for gastric cancer development.
Keywords: miRNA, Gastric Cancer, Artificial Neural Network, Bioinformatics, Risk Biomarker
Background
Gastric cancer (GC) is a complex and heterogeneous disease that results from multiple epigenetic and genetic steps. This type of cancer involves a gain of function of oncogenes and a loss of function of tumor suppressor genes [1]. In some genetic disorders, several alterations are selected to provide proliferative advantages to the carcinogenic cells.
Gastric cancer occurs when malignant cells are found in the stomach tissue, although they can spread to adjacent tissue. Each year, approximately one million people worldwide die due to this disease, and the survival rate five years after diagnosis is 9–10%. It is believed that preventive measures (identification of risk markers) along with early diagnosis can be crucial to reduce the death rate of this neoplasia.
The microRNAs (miRNAs) are small non-coding RNAs with a length ranging from 17 to 25 nucleotides. They were conserved throughout evolution and are capable of regulating gene expression at the post-transcriptional level by either degrading or repressing the translation of messenger RNA (mRNA) markers [2]. miRNAs have been implicated in most major cellular functions, such as proliferation, differentiation, apoptosis, stress response, and transcriptional regulation [3].
The recognition of miRNAs that are differentially expressed between tumor tissues and healthy tissues may help to identify miRNAs that are involved in human cancers and to further establish the pathogenic role of miRNAs in cancers [3]. miRNAs modify gene expression by epigenetic mechanisms and affect the mRNAs responsible for maintaining balance, such as those corresponding to oncogenes and tumor suppressor genes [4].
Since the first studies on the involvement of miRNAs in carcinomas, changes in their expression have been described in several types of tumors, including gastric cancer. Furthermore, the differential expression of these molecules suggests that they can be used to observe genetic profiles of gastric cancer [5]. Therefore, the anomalous miRNA expression in tumors is important in the mechanism of carcinogenesis. Consequently, the tumor development, the type of tissues affected, and their staging directly influence the risk markers, diagnosis, and therapeutics.
An artificial neural network (ANN) is a computational technique inspired by the neuronal structure of intelligent organisms. ANNs are able to acquire knowledge through experience, usually by comparing the input data stimuli with the corresponding output pattern; that is, applying an iterative training over the available data until the patterns are learned. The self-organizing maps (SOM) is a specific class of ANN, that is able to identify the existing similarity patterns in the data without the need of an output variable to be used as reference for comparison. In the SOM, the neurons are connected in a grid topology (map), in which, during the training, clusters are formed by grouping samples with common characteristics.
In this paper, an ANN was created by unsupervised learning with self-organizing maps algorithm (SOM) to identify the expression profile of miRNAs, given the type of differential expression (under or over expression) observed in healthy subjects regarding GC.
Methodology
The methodology was divided in three parts: pre-processing of the data, processing of the data and the analysis of the generated clusters.
In the pre-processing, the archives were obtained from the next generation sequencing (NGS) SOLiD platform (Life Technologies, CA, US) as described by Ribeiro-dos-Santos [6]. A total of ten tissue samples, or barcodes, were sequenced: Healthy Tissue Cardia [6], Healthy Tissue Antrum [7], and eight GC samples (Tumor Tissue and Adjacent Tissue) extracted from four patients at the Hospital Universitário João de Barros Barreto, Belém, Pará - Brazil. The classification of each sample follows the Tumor-Node-Metastasis (TNM) pattern for GC, as shown in Table 1 (see supplementary material). After sequencing, the generated archives were processed on quality filter software so that the low quality reads (reads that were randomly sequenced) could be removed from the data. A second filter was then applied to remove the last 10 nucleotides of the read sequence, which had 35 nucleotides. Finally, all 514 miRNAs had a length of 25 nucleotides (mature miRNA).
In data processing, the relative quantification of miRNA profiling was performed, and the ANN was developed. The data from pre-processing were normalized to a value of 3,000 reads. After normalizing the data, the miRNAs that had an initial expression level greater than 10 miRNA per tissue were selected. During the differential expression analysis, only the tissues that exhibited significant differences (p-value < 0.01 and fold change > 5) were considered. From the total of 514 miRNAs, only 76 miRNAs were of interest for the ANN processing. Table 2 (see supplementary material) shows the 76 miRNAs used in the ANN.
The network was arranged as follows: a 76 x 10 input matrix, representing the 76 miRNAs (from pre-processing) over 10 samples, represented on a rectangular topology. The matrix׳s values consist of the number of reads observed for each of the 76 miRNAs found in each sample, as shown in Table 2. The initial weights were randomly determined so that no bias occurred at the time of allocation of the weights for each input. During the training process, the Euclidean distance function Equation 1 (see supplementary material for equation and explanation) was used to differentiate the cluster (neuron grid) for assigning each data sample. The weight update for iteratively improve the cluster distinction is given by Equation 2 (see supplementary material for equation and explanation).
The third and last part consists of the analysis of the generated clusters. These clusters formed at the end of running the network represent Healthy Gastric Tissue and Gastric Cancer. Several simulations were performed with the SOM type of ANN, and training took 2,000 iterations. In the following simulations, the number of iterations was changed to 10,000.
Results
The result reduced the input data that had two dimensions (quantity of miRNA and input weights) to a single dimension corresponding to the type of tissue (Healthy Gastric Tissue or Gastric Cancer) with no loss of information.
Of the ANNs generated, two of them (network 1 and 2) showed more significant results according to the study's goal, and the second network had the best result. The first network, Network 1, grouped the samples in two clusters: Healthy Gastric Tissue and Gastric Cancer, where samples 1, 2, 3, 5, 7, 8, and 10 where clustered as Healthy and samples 4, 6, and 9 were clustered as GC. By comparing the results with the samples organization as shown in (Table 1) was observed in sample 7 (extracted from a Diffused Tumor Tiseu at stage T4) was classified incorrectly by the network.
The second network, identified as Network 2, was generated by increasing the filter on the miRNAs used as input. For this network, only the miRNAs that showed specificity or differential expression in the different types of tissues (Healthy Gastric Tissue and GC Tissue) were selected [6, 7]. The findings showed the molecular signature of nine miRNAs, as follows: hsa-miR-21, hsa-miR-29a, hsa-miR-29c, hsa-miR-148a, hsa-miR-141, hsa-let-7b, hsa-miR-31, hsa-miR-451, and hsa-miR-192. The findings from this network indicated a clustering (grouping) of samples 1, 2, 3, 5, 8, and 10, representing the healthy group, and samples 4, 6, 7, and 9, representing GC. Figure 1 corroborates the data listed in (Table 1).
Figure 1.
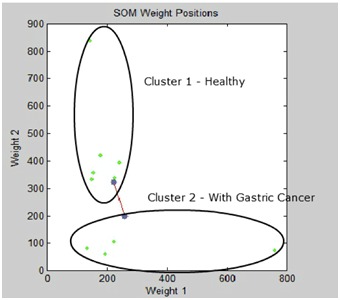
Results clustering of miRNAs. Iindicated a clustering (grouping) of samples 1, 2, 3, 5, 8, and 10, representing the healthy group, and samples 4, 6, 7, and 9, representing GC.
Discussion
The differential expression of the candidate miRNAs has been shown to be an excellent tool for understanding the role of miRNAs in cancer pathogenesis. Many papers report expression patterns of these molecules in both tumor tissues and healthy tissues in prostate, lung, ovarian, colon, brain, and liver cancers [8, 9, 10, 11, 12], which clearly indicates the aberrant expression of miRNAs in cancer. These results support the hypothesis that miRNAs play important roles in all cancers [5]. The present work demonstrated that with a SOM ANN, it was possible to reproduce the expression patterns (or signatures) in different types of tissue samples – Healthy and GC. Similar results were reported in several papers that used artificial neural networks to classify different types of cancer using different input data, namely pathology imaging, computed tomography (CT), magnetic resonance, gene selection, and gene expression, for segmenting sputum color images and analyzing miRNA expression [6, 7, 8, 13].
A recent study demonstrated that more than 50% of miRNA were located in cancer-associated genomic regions or in fragile sites [10], suggesting that miRNAs may play a more important role in the pathogenesis of a limited range of human cancers than previously thought [5].
The hsa-miR-29 family of microRNAs (miRNAs) was recently reported to be aberrantly expressed in multiple cancers. Increasing evidence shows that the abnormal expression of the miR-29 family is associated with tumorigenesis and cancer progression [13]. In the present study, hsa-miR-29c was found to be over-expressed in Healthy Tissues and under expressed in samples with GC, which corroborates the results obtained by [6 and 7].
Another important miRNA described in the literature was hsamiR- 21. This miRNA was over-expressed in most tumor types, and it acts as an oncogene by targeting many tumor suppressor genes related to proliferation, apoptosis, and invasion. Therefore, hsa-miR-21 was associated with a wide variety of cancers including those of breast, ovaries, cervix, colon, lung, liver, brain, prostate, pancreas, and thyroid [2, 9, 10, 14, 15]. The over expression of hsa-miR-21 has been reported in many malignancies, all of which contain constitutively activated STAT3, or even rely on STAT3 for cell survival or growth.
Therefore, aberrantly expressed may result in many malignancies by blocking the expression of critical apoptosisrelated genes [15]. In vivo and in vitro studies suggest that hsamiR- 21 may serve as a diagnostic and prognostic marker for human malignancies [15]. The findings described above strengthen the results obtained in the study that showed that hsa-miR-21 was over-expressed in the GC samples.
Members of the microRNA-148 (hsa-miR-148) family, which include microRNA-148a (hsa-miR-148a) and microRNA-148b (hsa-miR-148b), were expressed differently in Tumor and Healthy Tissues and have been involved in the genesis and development of disease [6]. Studies have reported the downregulation of the expression of hsa-miR-148a in various types of cancer such as colorectal [5], pancreatic, and hepatocellular carcinoma as well as during cancer metastasis. hsa-miR-148a was identified as a tumor metastasis suppressor in GC [5]. hsa-miR- 148a was suppressed by more than 4-fold in GC Tissues compared with the corresponding Adjacent Tissues, and the down-regulation of hsa-miR-148a was significantly associated with the TNM stage and lymph node metastasis [5]. The overexpression of hsa-miR-148a suppressed GC cell migration in vitro, suppressed lung metastasis formation in vivo, and reduced the mRNA and protein levels. Thus hsa-miR-148a functions as a tumor metastasis suppressor in gastric cancer, and the down-regulation contributes to gastric cancer lymph node metastasis and progression. It may have therapeutic potential to suppress gastric cancer metastasis [5], and also found to be under-expressed in the heathy samples of gastric tissue. The other miRNAs that participated in the molecular signature (hsa-mir-141, hsa-let-7b, hsa-mir-31, hsa-mir-451, and hsa-mir-192) in the present study were also associated with different types of cancer [16, 17].
Several papers reinforce the importance of applying computer intelligence techniques, such as ANNs, for medical diagnosis, breast cancer diagnosis, diagnosing cancer, classifying cancer cells classifying and analyzing brain cancer and predicting distant metastasis [6, 7, 8, 13]. In addition, ANNs were already used to analise miRNA data for signature analysis in colorectal cancer [15], breast cancer analysis, and gastric cancer [13]. There are also papers reporting the use of ANNs to identify miRNA expression patterns in different stages of GC [15]. The main innovation of this study consisted of the methodology used to develop a SOM ANN that processes the differential expression of miRNAs to classify (cluster) Tissues with or without GC.
Conclusion
In the present study, a SOM neural network was created to identify a differential expression profile of nine specific miRNAs (molecular signature): hsa-mir-21, hsa-mir-29a, hsamir- 29c, hsa-mir-148a, hsa-mir-141, hsa-let-7b, hsa-mir-31, hsamir- 451 and hsa-mir-192. The ANN clustered the samples of different gastric tissues into two distinct groups: Healthy Gastric Tissue and Gastric Cancer Tissue. Therefore, this ANN can be used as an important tool for gastric cancer risk factor or risk marked analysis.
Supplementary material
Acknowledgments
This work was part of the Rede de Pesquisa em Genômica Populacional Humana (supported by CAPES/Biologia Computacional) and Rede Paraense de Genômica e Proteômica (supported by FAPESPA). Financial support; Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES; Governo do Pará /SEDECT /FAPESPA, PROPESP /UFPA-FADESP. LARISSA LUZ GOMES supported by IESAM; FABIANO CORDEIRO MOREIRA supported by CESUPA; IGOR G. HAMOY is supported by Pós-Doc Junior (PDJ) fellowship from CNPq/Brazil (162605/2011- 0); SIDNEY SANTOS supported by CNPq/Produtividade; ÂNDREA RIBEIRO-DOS-SANTOS supported by CNPq/Produtividade. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Citation:Gomes et al, Bioinformation 10(5): 246-250 (2014)
References
- 1.Wu WK, et al. Oncogene. 2010;29:5761. doi: 10.1038/onc.2010.352. [DOI] [PubMed] [Google Scholar]
- 2.Lee YS, Dutta A. Annu Rev Pathol. 2009;4:199. doi: 10.1146/annurev.pathol.4.110807.092222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iorio MV, et al. Cancer Res. 2005;65:7065. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 4.George GP, Mittal RD. Indian Journal of Clinical Biochemistry. 2010;25:4. doi: 10.1007/s12291-010-0008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang B, et al. Dev Biol. 2007;302:1. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 6.Ribeiro-dos-Santos A, et al. PLoS One. 2010;5:e13205. doi: 10.1371/journal.pone.0013205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreira FC, et al. PLoS One. 2014 [Google Scholar]
- 8.Gao Z, et al. J Biol Chem. 2005;280:38271. doi: 10.1074/jbc.M506488200. [DOI] [PubMed] [Google Scholar]
- 9.Iorio MV, et al. Cancer Res. 2007;67:8699. doi: 10.1158/0008-5472.CAN-07-1936. [DOI] [PubMed] [Google Scholar]
- 10.Lu J, et al. Nature. 2005;435:834. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 11.Schickel R, et al. Oncogene. 2008;27:5959. doi: 10.1038/onc.2008.274. [DOI] [PubMed] [Google Scholar]
- 12.Murakami Y, et al. Oncogene. 2006;25:2537. doi: 10.1038/sj.onc.1209283. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, et al. Genome Med. 2013;5:91. doi: 10.1186/gm495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volinia S, et al. Proc Natl Acad Sci. 2006;103:2257. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan JA, et al. Cancer Res. 2005;65:6029. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 16.Ming G, et al. World J Gastroenterol. 2013;19:2019. [Google Scholar]
- 17.Nishizawa T, Suzuki H. Int J Mol Sci. 2013;14:9487. doi: 10.3390/ijms14059487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.