

Abstract
The P-loop NTPases are involved in diverse cellular functions. Members of the P-loop NTPase superfamily are characterized by presence of a highly conserved sequence pattern GxxxxGKS/T, known as Walker A motif. This motif adopts an archetypal P-loop conformation which allows accommodation of the triphosphate moiety of a bound nucleotide. Despite the presence of Walker A as a common sequence motif, P-loop NTPases exhibit extreme sequence divergence which hampers their phylogenetic or evolutionary classification. Here, we show that P-loop and its flanking region subsequence (termed as “extended-WalkerA motif”) contain distinct signatures that can be utilized to classify NTPase domain of functionally diverse proteins. We find a clearly classified group of diverse NTPases of Conserved Domain Database such as G-proteins, Ylqf, RecA like, DExDc, AAA, CPT, NK, ABC transporter and NifH proteins.
Keywords: P-loop NTPases, Walker A motif, classification
Background
P-loop containing nucleoside triphosphate hydrolase superfamily represent a large protein family that are involved in diverse cellular functions, for example, in translation, transcription, replication, DNA repair, signal transduction, protein transport and localization, signal-sequence recognition, chromosome partitioning, membrane transport and activation of various metabolites. [1–3].
P-loop NTPase folds are α-β proteins that contain regularly recurring α-β units with the five β-strands (β1–β5) and characterized by the presence of sequence patterns such as GxxxxGKS/T (where x is any residue), known as a Walker A motif and a Walker B motif (hhhh[D/E], where h is a hydrophobic residue) [4]. In diverse NTPases, Walker A motif adopts an archetypal loop shape (Figure 1), facilitating the accommodation of phosphate moiety of a bound nucleotide, thereby acquiring the name “P-loop” NTPases [5].
Figure 1.
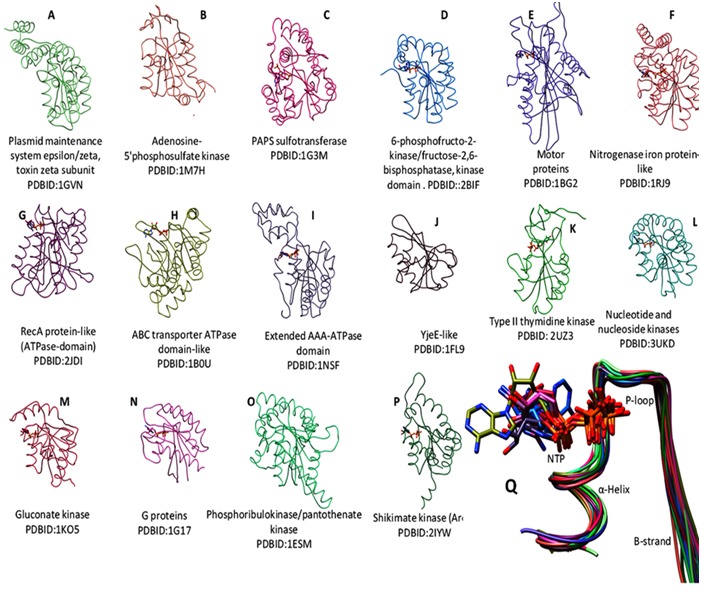
P-loop based superposition of diverse structures of NTPases. (A-P) Tile display of P-loop based superposed representative structures of diverse P-loop NTPases (see methods) using matchmaker tool of UCSF Chimera (http://www.cgl.ucsf.edu/chimera/). Four letter words are the PDBID. Q) Ribbon diagram of typical architecture of P-loop and its flanking region (termed as “extended-WalkerA motif”) with bound nucleotide molecule (stick) of diverse NTPases.
P-loop NTPases are divided into two major structural classes: the KG (kinase-GTPase) class, where the β-strand leading to the P-loop and the Walker B strand are direct neighbors. This group includes Ras-like GTPases and its circularly permutated YlqF-like; and the second class ASCE (characterized by an additional β-strand inserted between the P-loop strand and the Walker B strand) which includes ATPase Binding Cassette (ABC), DExD/H-like helicases, 4Fe-4S iron sulfur cluster binding proteins of NifH family, RecA-like F1-ATPases, and ATPases Associated with a wide variety of Activities (AAA). Also included in this group are the diverse sets of nucleotide/nucleoside kinase (NK) families [6–8].
Despite the presence of Walker A as a common sequence motif and P-loop as common conserved structural feature, the P-loop NTPases exhibit extreme sequence as well as structural divergence (Figure 1). Evolutionary classification of highly diverse groups of P-loop NTPases may provide insights into their diverse physiological processes. Earlier, attempts have been made for constructing an evolutionary classification for closely related superclass such as P-loop GTPases and GTPase-related proteins [9] and classification of P-loop kinases and related proteins [10]. However, a clear understanding of the phylogenetic relationships taking all the diverse groups of P-loop NTPases is not yet accomplished, as it is hampered by enormous sequence diversity present within the groups of P-loop NTPases. In the present study, an attempt is made to identify the unique features that classify the diverse groups of P-loop NTPases. We find that the sequence features of the P-loop and its flanking region, termed as “extended Walker A motif”, are distinct for each of the NTPase domain containing groups of functionally diverse proteins. Based on our analysis, we propose that identified features can be utilized to annotate hypothetical proteins as well as classify the proteins with deviant walker A motifs in their respective NTPase group. This study extends the evolutionary information in addition to already known sequence-structure features.
Methodology
Selection of sequences and structures of P-loop containing NTPases:
Conserved domain database at NCBI (CDD; http: //www.ncbi.nlm.nih.gov /Structure/cdd) and protein databank (http://www.rcsb.org/pdb) were utilized to retrieve the sequences and representative high resolution X-ray crystallographic structures of diverse domains of P-loop containing NTPases, respectively. Diverse set of sequences of Ras-like G-proteins, circularly permutated YlqF-like, ATPase Binding Cassette (ABC), DExD/H-like helicases, 4Fe-4S iron sulfur cluster binding proteins of NifH family, RecA-like F1- ATPases, and ATPases Associated with a wide variety of Activities (AAA), and nucleotide/nucleoside kinase (NK) families were retrieved from CDD database of NCBI and utilized for the analysis.
P-loop based superimposition was applied to overlay the highly diverse groups of X-ray crystallographic structures of representatives of P-loop NTPases (Figure 1 A-P). Structural analysis and image snapshots were generated by using chimera tool, version 1.8.1 ( http://www.cgl.ucsf.edu/chimera/).
Classification of diverse P-loop NTPase:
Full length multiple sequence alignment (MSA) of diverse P-loop NTPase sequences was retrieved from CDD database of NCBI. MSA of NTPase domain sequences of diverse groups were then pooled to form one MSA file, and subjected to realignment of multiple sequence. As expected, due to extreme sequence divergence pooled and realigned MSA showed poor and meaningless alignment to draw phylogenetic conclusions. Therefore, we set out to choose a subsequence of a structural component that is maximally and contiguously aligned for all the P-loop NTPases. Walker A motif sequence (8 residues) and flanking region sequences comprising of 5 residues from the N-terminus α-helix and 5 residues from the C-terminus β-strand region were emerged out to be such a structural component (Figure 1Q). We termed this structural component as “extended-WalkerA motif”. Further, we utilized this extended-Walker A motif to construct a neighbour joining tree. Subsequence of P-loop and flanking region was trimmed using Jalview tool [11] and multiple sequence alignment was done using ClustalX tool [12], using its default parameters. MEGA5.0 [13], was used to calculate the distance matrix and neighbour-joining tree were constructed by taking extended Walker A motif subsequence. Tree was visualized using MEGA5.0 software tool [13].
Weblogo analysis of extended Walker A motif:
Full lengths of diverse set of P-loop NTPases domain sequences were retrieved from CDD database of NCBI. The subsequence containing the Walker A and flanking region were subjected to Weblogo generation [14]. The consensus sequences are presented as sequence logos generated with WebLogo ( http://weblogo.berkeley.edu/).Weblogo depicts an alignment as a sequence logo, in which each of the position is represented as a stack of one letter amino acid residue. The height of each amino acid letter is proportional to its frequency at that particular position, while the height of the stack, expressed in bits, indicates the mutual information in that position [4]. Mutual information (i.e. stack height) is often interpreted as an indicator of evolutionary conservation at each position.
Results and Discussion
Currently, a total of 832,282 proteins belonging to P-loop containing nucleoside triphosphate hydrolase superfamily (SSF52540;http://supfam.org) have been listed in 3,218 genomes, and they are found in all three major kingdoms of life. At the core of conserved P-loop substructure, these NTPases exhibit huge sequence divergence. Therefore, it is desirable to search for sequence features/profile which can be used to identify, classify and annotate the P-loop NTPases.
P-loop and flanking region subsequence delineate the diverse NTPases:
P-loop of Walker A motif adopts a typical arch shaped conformation in diverse groups of NTP binding proteins. It has a structurally distinct feature consisting of β-strand towards the N-terminus and α-helix at the C-terminus of Walker A motif [4, 5]. Overall structures of P-loop NTPases are highly diverse (Figure 1 A-P) and therefore difficult to multiply align (see methods). However, P-loop based superposition nicely aligns the substructures containing P-loop and flanking N- and C- terminus region, we termed this region as an “extended-Walker A motif” (Figure 1Q). Interestingly, despite the sub-structural similarity, corresponding subsequences are highly diverse, except for the conventional invariant positions such as G, GKS/T of Walker A motif (GXXXXGKS/T, where X is any residue). This incited us to check if the subsequence of such a structurally conserved and well superimposed region is evolutionarily selected for diverse group of NTPases. Secondly, can it be utilized in classifying the highly diverse NTP binding proteins? Intriguingly, when 18 residues spanning of Walker A (8 residues) and flanking region subsequence (5 residues each from N-, and C- terminus of Walker A) from the diverse set of NTPases sequences were utilized to construct an unrooted tree, based on neighbor joining method, a clearly classified group of diverse NTPases such as G-proteins, RecA like, DExDc, AAA, CPT, NK, ABC transporter and NifH proteins emerged out (Figure 2). As shown in Figure 2, NK is linked to G-proteins and CPT group; ABC group is associated with RecA. However, DExDc and NifH clades are clustered separately. Interestingly, Ylqf, circularly permuted GTP binding protein, which otherwise would have not aligned with Ras like GTPases at the full length sequence alignment, due to its circular permutation, showed a close phylogenetic similarity to Ras-like G-proteins. This suggests that extended Walker A motif has an evolutionary feature that relates Ylqf to G-proteins. This strengthens our notion that there are residue positions around P-loop, other than conventionalWalker A motif that are evolutionarily selected within distinct NTPase families.
Figure 2.
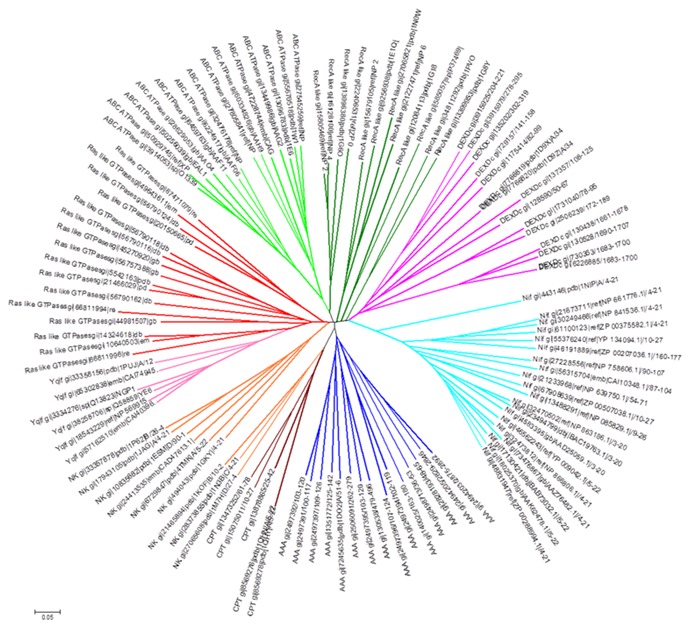
Extended Walker A motif based classification of Diverse group of NTPases. Unrooted neighbour-joining tree constructed by using multiple sequence alignment of extended WalkerA motif containing subsequence of Walker A (8 residues), N- and C-terminus (5residues from each end) of Walker A of diverse P-loop NTPases.Tree was generated using Mega5 software tool. gi number is indicated for each of the diverse sequences utilized in the analysis.
Extended Walker A Motif contains distinct patterns across diverse NTPases:
In order to gain insight into the evolutionary selected residues of P-loop NTPases, we generated and compared the sequence patterns within the P-loop (GX1X2X3XG4KS/T) and its flanking region subsequence (Figure 3). The difference in the overall residue propensity of the P-loop and its flanking region can be analyzed by generating Weblogo [4]. We utilized Weblogo analysis which revealed distinctive sites in the subsequence spanning from P-loop and flanking region (Figure 3). The larger the letter, the more information it provides about the respective position in the protein family [4]. Letters are sorted in descending order depending on their probability. As shown in Figure 3, an informative Asparagine residue was present just adjacent to the Glycine position of Walker A in CPT and lie at X2 and X3 of P-loop of ABC transporters and Ylqf, respectively. In contrast, Glycine as an informative site was observed in different patterns: in DExDc, X3-position; in AAA domains, X3-position; in NifH, X2-X3 positions of P-loop. A deviant Walker A sequence was observed in DExDc and NifH group, where the conserved Glycine is substituted by other amino acids (Figure 3). Such a pattern of evolutionary selected set of residues may distinctively cluster the DExDc and NifH in the N-J tree (Figure 2). Pattern of distinctive signatures were also seen at the N-terminus and C-terminus flanking region of distinct NTPase groups (Figure 3). Convincingly, distinct propensity of amino acid around P-loop and well classified neighbour joining tree demonstrates the evolutionary selected residues.
Figure 3.
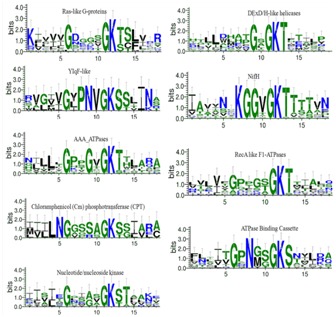
Web logo analysis of P-loop and its flanking region sequence.Graphical sequence logos were created using Weblogo3 for visualization of the conserved sequence pattern. Multiple sequence alignment of diverse P-loop NTPases sequences such as Ras-like G-proteins, circularly permutated YlqF-like, ATPase Binding Cassette (ABC), DExD/H-like helicases, 4Fe-4S iron sulfur cluster binding proteins of NifH family, RecA-like F1-ATPases, and ATPases Associated with a wide variety of Activities (AAA), and diverse set of nucleotide/nucleoside kinase (NK) families are utilized. The overall height of each stack indicates the sequence conservation at that position (measured in bits).
Applications of Extended Walker A Motif:
The consensus sequence of Walker A (GXXXXGK[S/T], where X is any residue) is often used as a motif for identifying new members of P-loop NTPases [4– 5]. However, with the availability of large number of NTPase sequences, diversity in their conventional conserved motifs is becoming evident. Therefore, the conventional motif based searches limit the NTPases characterization. Also, some of P-loop NTPases have deviant Walker A sequences and there are instances where these Walker A signatures are present in many proteins that do not form P-loop, for example, peroxidases, and enzymes like α- amylase, glutamate dehydrogenase, Taq polymerase, carbonic anhydrase, binding proteins (lectin, trypsin inhibitor), proteases, and others [15]. Consequently, a novel signature is required to identify and classify the P-loop NTPases sequences independent of conventional Walker A and Walker B motif. Therefore, we propose that Walker A and its flanking region subsequence profile, instead of just a Walker A sequence, may be used to identify and classify P-loop NTPases.
Conclusion
Our analysis indicates that patterns of amino acid around and within P-loop region are evolutionary distinct. Such a pattern may provide distinct conformational flexibility and functional diversity during Nucleotide (GTP or ATP) binding and hydrolysis in diverse groups. Earlier, we have identified presence of high density of highly central residue positions in P-loop and its flanking region subsequence [16]. These closeness residues are connected to all other residues in short steps in an amino acid network of protein structure, thereby facilitating a fast dynamic conformational signaling [17]. Taken together, our analysis suggest that that P-loop and its flanking region residues are evolutionarily engineered not only to cater the binding of nucleotide and fine tuning the catalytic activity, but also to disseminate the conformational changes from the nucleotide binding site to distinct sites of diverse P-loop NTPases.
Acknowledgments
Ekta Pathak acknowledges CSIR, Govt. of India for providing the Junior Research Fellowship and Senior Research Fellowship.
Footnotes
Citation:Pathak et al, Bioinformation 10(4): 216-220 (2014)
References
- 1.Koonin EV, Aravind L. Trends Biochem Sci. 2000;25:223. doi: 10.1016/s0968-0004(00)01577-2. [DOI] [PubMed] [Google Scholar]
- 2.Saraste M, et al. Trends Biochem Sci. 1990;15:430. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- 3.Vetter IR, Wittinghofer A. Q Rev Biophys. 1999;32:1. doi: 10.1017/s0033583599003480. [DOI] [PubMed] [Google Scholar]
- 4.Milner-White EJ, et al. J Mol Biol. 1991;221:751. doi: 10.1016/0022-2836(91)80170-y. [DOI] [PubMed] [Google Scholar]
- 5.Walker JE, et al. EMBO J. 1982;1:945. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fröhlich KU, et al. J Cell Sci. 2001;114:1601. doi: 10.1242/jcs.114.9.1601. [DOI] [PubMed] [Google Scholar]
- 7.Frickey T, et al. J Struct Biol. 2004;146:2. doi: 10.1016/j.jsb.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Lupas AN, et al. Curr Opin Struct Biol. 2002;12:746. doi: 10.1016/s0959-440x(02)00388-3. [DOI] [PubMed] [Google Scholar]
- 9.Leipe DD, et al. J Mol Biol. 2002;317:41. doi: 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- 10.Leipe DD, et al. J Mol Biol. 2003;333:781. doi: 10.1016/j.jmb.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 11.Clamp Michele, et al. Bioinformatics. 2004;20:426. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 12.Larkin MA, et al. Bioinformatics. 2007;23:2947. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 13.Tamura, et al. Mol Biol Evol. 2011;28:2731. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider TD, Stephens RM. Nucleic Acids Res. 1990;18:6097. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramakrishnan C, et al. Protein Eng. 2002;15:783. doi: 10.1093/protein/15.10.783. [DOI] [PubMed] [Google Scholar]
- 16.Pathak E, et al. Bioinformation. 2013;19:23. doi: 10.6026/97320630009023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Sol A, et al. Protein Sci. 2006;15:2120. doi: 10.1110/ps.062249106. [DOI] [PMC free article] [PubMed] [Google Scholar]