

Abstract
Background
Prolonged stress leads over time to allostatic load on the body and is likely to exacerbate a disease process. Long-term of stress exposure is one of a risk factor for metabolism-related diseases such as obesity and type 2 diabetes. However, the relationship between chronic stress and non-alcoholic fatty liver disease (NAFLD) remain unknown.
Methods
To address the hypothesis that chronic stress associate to NAFLD development, we subjected C57bl/6 mice to electric foot shock and restraint stress for 12 weeks to set up chronic stress model. Then the serum and hepatic triglyceride (TG), total cholesterol (TC) were measured. Hepatic HE and Oil red O staining were used to specify the state of the NAFLD. To investigate whether inflammation takes part in the stress-induced NAFLD process, related visceral fat, serum and hepatic inflammatory factors were measured.
Results
We observed that chronic stress led to an overall increase of hepatic triglyceride and cholesterol while decreasing body weight and visceral fat mass. Microvesicular steatosis, lobular inflammation and ballooning degeneration were seen in stress liver section. This effect was correlated with elevated hepatic and serum inflammatory factors. Although the amount of visceral fat was decreased in stress group, various adipocytokines were elevated.
Conclusions
We showed that chronic stress is associated to NAFLD and chronic inflammation in visceral fat, though food intake and visceral fat mass were decreased. These results may contribute to better understanding of the mechanism from steatosis to steatohepatitis, and propose a novel insight into the prevention and treatment of NAFLD.
Keywords: Chronic stress, Homeostasis, Steatosis, Inflammation, Visceral fat
Background
Non-alcoholic fatty liver disease (NAFLD), characterized by the accumulation of large droplets of triglyceride within hepatocytes in the absence of chronic alcohol consumption, is a leading cause of hepatic dysfunction. NAFLD represents a wide spectrum of diseases, ranging from simple steatosis, through steatosis with inflammation (non-alcoholic steatohepatitis, NASH) to cirrhosis. Although simple hepatic steatosis is a slowly-developed and asymptomatic disease, the next stage NASH is more likely to cause progressive cirrhosis, hepatocellular carcinoma and increased mortality [1]. The prevalence of NAFLD has increased dramatically over the last three decades, at nearly 15-30% in the general population in western countries and approximate 15% in big cities of China [2,3]. Despite its high prevalence, factors leading to progression from NAFLD to NASH remain obscure and there is no pharmacological agent approved for the treatment of NAFLD [4].
One major concern is chronic inflammation plays a key role in the pathogenesis of NAFLD [5]. IL-6 and TNF-α, two important inflammatory cytokines, profoundly increased in human patients with NAFLD [6]. Other studies also found a clear correlation between liver IL-6 and TNF-α expression level and NAFLD disease severity [7,8]. The absence of either IL-6 or TNF receptor 1 (TNFR1), decreased high-fat diet induced liver lipid accumulation and liver inflammation as assessed by reduced infiltration with macrophages and neutrophils [9].
Stress systems play a crucial role in maintaining homoeostasis to adapt to the environmental demands imposed by change [10]. However, strong and long-lasting stress stimuli could disturbances in the internal environment, thereby increasing the risk of various health problems. In modern society, which is characterized by a rapid pace of life, individuals are continuously confronted with an increasing number of stressful stimuli, such as emotional stimuli and social stress. It had been validated by clinical trials that chronic stress from work and low quality of life may be the risk factor of obesity and metabolism syndrome [11,12].
Since chronic stress could lead to systematic inflammation [13], we assume that chronic stress may take part in the process of NAFLD. Yet the relationship between chronic stress and NAFLD has not been clarified. Thus, the overall objective of this study was to demonstrate whether chronic stress could lead to NAFLD.
Methods
Experimental animals
Twenty female C57bl/6 mice (8 weeks of age; Sino-British SIPPR/BK Lad Animal Ltd, Shanghai, China) were allowed to acclimate to the environment for 7 days before the experiments began. Then they were randomly assigned to the control or stress group (n = 10, respectively). Control mice were left undistributed, while stress mice were suffered from chronic stress for 12 weeks. The detail process was described in next paragraph. Food intake was measured by weighing the pellets once per week. All animals were maintained on a 12-hour light/12-hour dark cycle with food and water freely available. The temperature of the colony room was maintained at 22°C to 23°C. Animal protocols were approved by the Animal Care and Use Committee of the Second Military Medical University.
Stress protocol
During the 12 weeks, the mice in stress group were administered electric foot shock and restraint stress every day [14,15]. At 10:00 am, each mouse was placed in a box with a floor composed of stainless grids, and a scrambled electric shock was delivered through the floor grids by a PST-001 AC stimulator (StarMedical, Tokyo, Japan). An interval timer was connected to the stimulator to allow shocks for 7.5-second periods every 2 minutes, the intensity of which was 25v. At 7:00 pm, stressed mice were individually subjected to 2 h of restraint stress as described previously [16]. In brief, restraint stress was applied using a 50-mL conical centrifuge tube with multiple punctures that allowed for a close fit to mice. Control mice were kept isolated from stressed animals and maintained with access to food or water during the same period of stress process to avoid any acoustic or olfactory communication between the groups.
Experimental procedures
After 12 weeks, all mice were fasted overnight then anesthetized using isoflurane. Blood was collected by orbital puncture, followed by sacrifice via cervical dislocation. Then the whole liver and intra-abdominal adipose were removed and weighed. For histology, a small part of liver and visceral fat were immediately fixed in 10% formalin, and processed for paraffin embedding. The rest of the liver was stored at −80°C for biochemical analysis.
Determination of fat cell size
For these studies in fat tissue derived from mice euthanized at 12 week, two sections apart were selected, and the adipocyte cell size in three random fields in each section was determined. Because the cells were not uniformly round, the shortest length between the opposite cell membranes was measured routinely. For each cell, the inter-membrane distance was measured by including the central point in the cell in these planar images, where the lengths were measured in all of the complete cells within the field using Image J. Results were collated for each animal grouping.
Hematoxylin-Eosin (HE) and oil red o staining of liver sections
Following fixation of the livers with10% formalin/phosphate-buffered saline, livers were sliced and stained with HE for histological examination. Liver steatosis was graded semi-quantitatively based on the percentage of hepatocytes according to the following criteria: grade 0, no hepatocytes involved; grade 1, 1% to 25% of hepatocytes involved; grade 2, 26% to 50% of hepatocytes involved; grade 3, 51% to 75% of hepatocytes involved; and grade 4, 76% to 100% of hepatocytes involved. Hepatic lipid content was also determined by staining with Oil Red O (Sigma). The percentage of the area occupied by oil red O-stained lipid droplets was calculated using Image J, averaging 3–5 separate, randomly selected 40× fields.
Serum biochemical analysis
The serum triglyceride (TG), total cholesterol (TC), free fatty acid (FFA), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) were measured by an automatic biochemistry analyzer(Hitachi 7170, Tokyo, Japan).
Measurement of hepatic lipids
Livers were homogenized at 4°C in lysis buffer containing 50 mmol/L Tris (pH8.0), 150 mmol/L NaCl, 1% Triton X-100, and 0.5% sodium deoxycholate. Lipids from the total liver homogenate were extracted using the chloroform/methanol method (2:1), evaporated, and dissolved in 2-propanol. Amounts of TC and TG were measured by an automatic biochemistry analyzer (Hitachi 7170, Tokyo, Japan).
Measurement of cytokines
Serum, adipose and liver samples were profiled with The Bio-Plex mouse Cytokine 17-Plex panel was used with the Bio-Plex Suspension Array System (Bio-Rad, Hercules, CA, USA) to profile expression of 7 inflammatory mediators, including IL-6, TNF-α, IL-1β, IL-18, MCP-1, MIP-2 and PAI-1. The assay was performed according to the manufacturer’s instructions.
Data analysis
The data are presented as means ± SEM. Group means were compared utilizing a paired Student’s t-test or the Student-Newman-Keuls test when appropriate. The analysis of food intake and body weight was performed by ANOVA for repeated measures as appropriate. P < 0.05 was considered statistically significant.
Results
Chronic stress leads to decreased food intake and body weight gain
To investigate whether chronic stress is linked to NAFLD progression, we made a 12 weeks stress protocol for C57BL/6 mice in order to lead to chronic stress status. During 12 weeks, the food intake and body weight of control group were steadily increased, while stress group didn’t show increased food intake and body weight gain. At the end of the time, both body weight and food intake gain were significantly lower in stress group (t (18) = 6.643, p <0.0001; t (18) = 3.882, p = 0.001) (Figure 1A, B).
Figure 1.
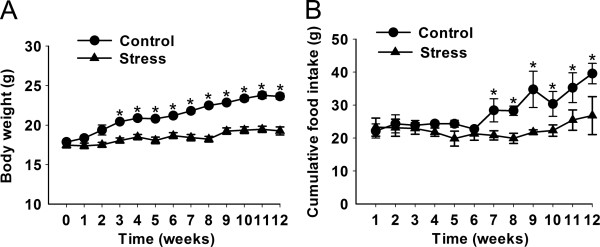
Effects of stress on weight gain and food intake. C57 BL/6 mice were received stress procedure for 12 weeks. (A) Changes in body weight. White squares = control (n = 10), Black squares = stress (n = 10). (B) Food intake during the course of the stress stimulating. Error bars represent standard error (SE). *P < 0.05, **P < 0.01 versus control mice.
Chronic stress decreases visceral fat mass but increases inflammatory cytokines secretion
The amount of visceral adipose and adipocytokines is known to have a closely relationship with the development of NAFLD. Thus, we firstly measured visceral fat mass and serum FFA. The stress mice exhibited remarkable reduction of visceral mass compared to control ones (t (18) = 4.97, p = 0.0001) (Figure 2A), and HE staining showed that adipocytes of stress group were significantly smaller than control ones (t (8) = 3.055, p = 0.0157) (Figure 2B). Consistence with these results, Serum FFA was significantly elevated in stress group (t (18) = 2.846, p = 0.0107) (Figure 2C). These data indicate that chronic stress could lead to decreased amount of visceral fat and increased lipolysis.
Figure 2.
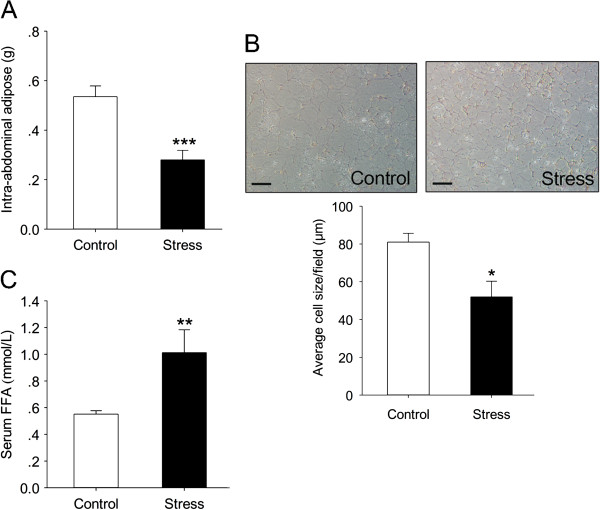
Characterization of intra-abdominal adipose tissue and serum FFA of chronic stress mice. (A) Weight of intra-abdominal adipose tissues from the control and stress mice received 12 weeks stress protocol. Error bars represent standard error (SE) (n = 10). (B) HE staining of intra-abdominal adipose tissue of stress mice received 12 weeks stress protocol. Magnification, ×100. Scale bars: 50 μm. Average cell sizes were calculated by Image J (C) Serum FFA content. Error bars represent standard error (SE). **P < 0.01, ***P < 0.001 versus control mice.
Since adipokine signaling is thought to be the means of NAFLD [17,18], we thought to determine how these molecules differed with chronic stress pattern. Adipose adipokine levels were examined after 12 weeks stress protocol. The results turned out that although the amount of visceral adipose was decreased, a variety of inflammatory factors of visceral adipose were elevated in stress mice. IL-6 and IL-1β were significantly increased (t (18) = 2.800, p = 0.011; t (18) = 2.712, p = 0.014), while others like TNF-α, IL-18 were not changed (t (18) = 0.670, p = 0.508; t (18) = 0.531, p = 0.600). Combined with cytokines, a lot of chemokines such as macrophage inflammatory protein 2 (MIP-2), monocyte chemoattractant protein-1(MCP-1) and PAI-1, related to inflammatory cells gathering were elevated too (t (18) = 2.236, p = 0.049; t (18) = 2.556, p = 0.029; t (18) = 2.756; p = 0.004) (Figure 3).
Figure 3.
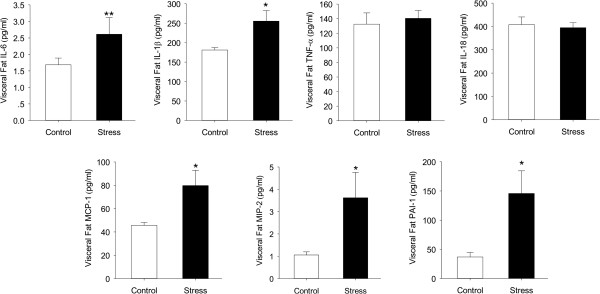
Effects of chronic stress on the release of selected adipokines and cytokines by visceral adipose tissue from C57 BL/6 mice. IL: interleukin, TNF-α: tumor necrosis factor-alpha, MCP-1: monocyte chemoattractant protein-1, MIP-2: macrophage inflammatory protein 2, PAI-1: plasminogen activator inhibitor-1. Error bars represent standard error (SE). *P < 0.05 versus control mice.
Chronic stress induces steatosis
Although stress group had less food intake and weight gain, their hepatic index (HI, the ratio of liver weight to body weight × 100), which usually reflect the fatty liver status, was significantly elevated (t (18) = 0.588, p < 0.001) (Figure 4A). To ensure this phenomenon, Serum lipid metabolism indicators and liver steatosis indexes were evaluated. Serum TC and TG showed increasing tendency but had no statistical significance (t (11) = 1.560, p = 0.147; t (11) = 1.650, p = 0.127) (Figure 4B).Then we evaluated the liver TG and TC concentration and Oil red O staining to certify whether stress cause lipid deposition in liver. Quantitative analysis showed a significant increase in hepatic TG and TC content in stressed mice (t (18) = 2.770, p = 0.013; t (18) = 2.389, p = 0.028) (Figure 4C), which was confirmed by histological staining using Oil red O (t (8) =3.093, p = 0.015) (Figure 4D). Together, these data suggest that chronic stress could induce liver steatosis independent of the dietary factor.
Figure 4.
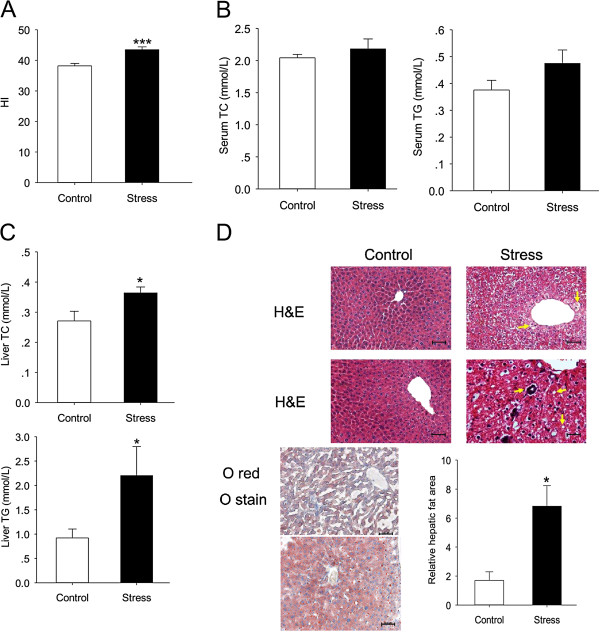
Chronic stress induces hepatic TG and TC accumulation. After mice were anesthetized, whole liver were removed and weighed. Hepatic lipids were extracted, and TG, TC concentrations were measured. (A) Hepatic index = Liver weight (mg)/body weight (g). (B) The levels of serum TC and TG content are represented in bar chart, respectively. (C) Liver TG and TC concentrations in stress group and control group. (D) Representative slides show hematoxylin and eosin (H&E)-stained and Oil red O-stained liver sections from control mice or stress mice for 12 weeks. The percentage of the area occupied by oil red O–stained lipid droplets was calculated using Image J, averaging 3–5 separate, randomly selected 40× fields. HE sections show chronic stress causing hepatocytes ballooning (yellow arrows). In addition, there are foci of inflammatory cell infiltration and lipid deposits (white arrows) Original magnification, ×100. Scale bars: 50 μm. Error bars represent standard error (SE). *P < 0.05, ***P < 0.001 versus control mice.
Stress not only leads to steatosis, but also NASH
The former results illustrated that stress could induce liver steatosis and chronic inflammation state. We wanted to further investigate whether the mice encountered stress could develop to NASH. The results showed that stressed mice developed exacerbated NASH compared to wild-type mice as judged by increased levels of serum ALT and AST (t (13) = 3.271, p = 0.006; t (11) = 2.735, p = 0.0194), and NAFLD activity inflammation scores (t (18) = 2.742, p = 0.0134; t (18) = 3.899, p = 0.001) (Figure 5).To figure out which cytokines take part in NASH, we detected the cytokines closely related to NASH in both liver and serum. For IL-6 and TNF-α, stress mice had both higher serum and liver levels than control ones (serum IL-6: t (18) = 2.692, p = 0.0149; liver IL-6: t (18) = 2.784, p = 0.0123; serum TNF-α: t (9) = 3.479, p = 0.007; liver TNF-α: t (18) =3.023, p = 0.007). For IL-1β and MCP-1, serum levels were higher in mice given chronic stress compared with controls (t (18) = 3.365, p = 0.004; t (18) = 2.191, p = 0.042) while liver levels didn’t change (t (18) = 1.784, p = 0.091; t (18) =1.492, p = 0.153). For IL-18, the two groups showed same level in serum and liver after the stress treatment (t (18) = 1.334, p = 0.199; t (18) = 1.140, p = 0.269) (Figure 6).
Figure 5.
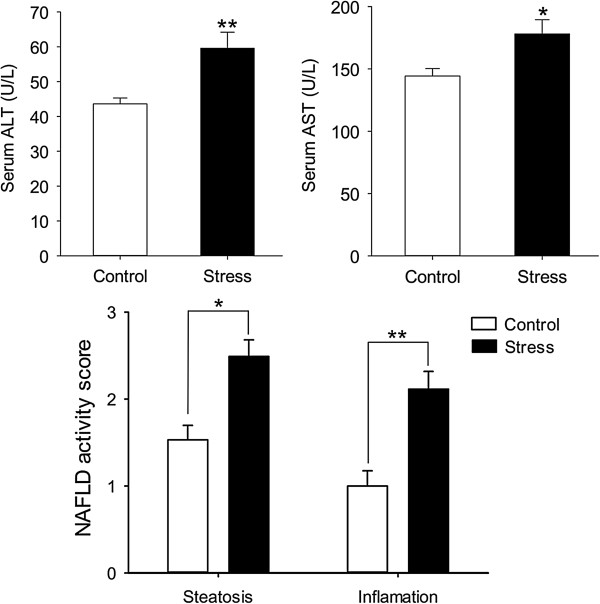
The serum activities of two hepatic injury-associated enzymes, alanine aminotransferase (ALT), and aspartate aminotransferase (AST), at the end of the treatment period. The NAFLD activity score is evaluated by New York NAFLD score system. *P < 0.05, **P < 0.01 versus control mice.
Figure 6.
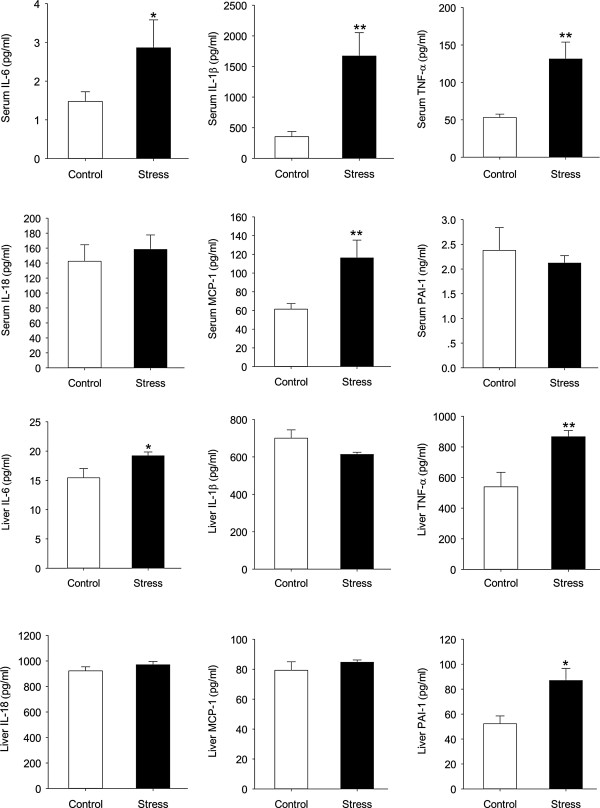
Effects of chronic stress on the release of selected adipokines and cytokines by serum and liver from C57 BL/6 mice. IL: interleukin, TNF-α: tumor necrosis factor-alpha, MCP-1: monocyte chemoattractant protein-1, PAI-1: plasminogen activator inhibitor-1. Error bars represent standard error (SE). *P < 0.05 versus control mice.
Discussion
The key finding in the present study is that mice could develop NASH only in intervention of chronic stress, despite the reduction in food intake and visceral fat, which always recognized as the risk factor of the metabolic disorder [19]. Furthermore, chronic stress could lead to chronic inflammatory state, including high concentration of inflammatory factors, such as TNF-α and IL-6, both in the circulation and liver. While previous studies mostly concentrated on the effect of chronic stress on obesity or metabolic syndrome combined with the high-fat diet [20], this is, to our knowledge, the first study indicating that chronic stress is a contributing factor of NAFLD, since the dietary factor and other related factors could be excluded in this experiment.
In some clinical trials, the amount of visceral fat is highly correlated with liver steatosis and steatohepatitis [21-23]. Obese persons with excess visceral adipose tissue are at higher risk for NAFLD components than those whose fat is predominantly located in the subcutaneous part [24]. Furthermore, weight loss induced by bariatric surgery is suggested to be responsible for the benefits in liver function and steatosis [25]. However, we noticed that, although stressed mice had less visceral fat content, they still exhibited a significantly higher hepatic TG and serum FFA level while the serum TC and TG were unchanged. The volume of fat tissue depends on its content of triglyceride. Therefore, we speculate that increase FFA was caused by lipotropic action in visceral adipose.
Apart from its ability of releasing and delivering FFA into the portal vein, visceral fat could secrete a variety of soluble factors which might be important sources to promote steatosis [26]. Then we detected the inflammation factors in visceral fat. The result showed that a variety of inflammatory cytokines and chemokines, such as IL-6, IL-1β, MIP-2 and MCP-1, elevated in the visceral adipose of stressed group. MIP-2 is a potent chemotactic agent for polymorphonuclear leukocytes, which was major secreted from stimulated macrophages [27]. MCP-1is secreted by a variety of cells as a response to several inflammatory stimuli, and play an important role in the stimulation of the inflammatory infiltrate. It could enhance expression of adhesion molecules in monocytes and promotion of pro-inflammatory cytokine synthesis [28,29]. All of these chemokines have the ability to activate and attract inflammatory cells, thus amplifying the inflammatory cascade. On the basis of these results, we infer that, chronic stress might induce steatosis through two ways: (1) chronic stress promotes visceral adipose lipolysis, thus increasing liver FFA input; (2) chronic stress lead to secretion of inflammatory cytokines and chemokines in visceral adipose tissue. However, the link between inflammatory adipose tissue and steatosis need to be further illustrated.
Conclusions
In conclusion, this study provides a new insight into the pathogenesis of NAFLD. Our findings demonstrate that chronic stress status may be may be an important risk factor of NAFLD, although stressed mice had less food intake and weight gain. Furthermore, visceral adipose and hepatic inflammation may be the crucial mechanism of the stress-induced NAFLD, which need to be further proved. Up to now, there is a common sense that NAFLD is largely a hepatic manifestation of obesity and metabolic syndrome. Therefore, these results may remind us that, those who have normal weight and hip-waist ratio but suffer long-term stress may also have the chance to have NAFLD. To prevent NAFLD, not only should we restrict the calorie properly, we also should concentrate on stress management.
Competing interests
The authors disclose no potential competing of interest.
Authors’ contributions
YZL designed and executed the experiments, interpreted data, and wrote the manuscript. JKC, YZ and XW performed molecular biology experiment and animal experiment. SQ and CLJ conceived the study, and participated in its design and helped to draft the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Yun-Zi Liu, Email: yunziliu@hotmail.com.
Ji-Kuai Chen, Email: cjk.smmu@hotmail.com.
Yi Zhang, Email: richard.t.alexander@hotmail.com.
Xia Wang, Email: wangxia8692@gmail.com.
Shen Qu, Email: qushencn@hotmail.com.
Chun-Lei Jiang, Email: cljiang@vip.163.com.
Acknowledgments
This work was supported by NIP (2013YQ190467) and Military Twelfth Five-Year Plan (BWS11J016, AWS12J002).
References
- Adams LA, Lymp JF, St SJ, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol. 2010;53:372–384. doi: 10.1016/j.jhep.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Fan JG, Farrell GC. Epidemiology of non-alcoholic fatty liver disease in China. J Hepatol. 2009;50:204–210. doi: 10.1016/j.jhep.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Lomonaco R, Sunny NE, Bril F, Cusi K. Nonalcoholic fatty liver disease: current issues and novel treatment approaches. Drugs. 2013;73:1–14. doi: 10.1007/s40265-012-0004-0. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Crespo J, Cayon A, Fernandez-Gil P, Hernandez-Guerra M, Mayorga M, Dominguez-Diez A, Fernandez-Escalante JC, Pons-Romero F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34:1158–1163. doi: 10.1053/jhep.2001.29628. [DOI] [PubMed] [Google Scholar]
- Abiru S, Migita K, Maeda Y, Daikoku M, Ito M, Ohata K, Nagaoka S, Matsumoto T, Takii Y, Kusumoto K, Nakamura M, Komori A, Yano K, Yatsuhashi H, Eguchi K, Ishibashi H. Serum cytokine and soluble cytokine receptor levels in patients with non-alcoholic steatohepatitis. Liver Int. 2006;26:39–45. doi: 10.1111/j.1478-3231.2005.01191.x. [DOI] [PubMed] [Google Scholar]
- Manco M, Marcellini M, Giannone G, Nobili V. Correlation of serum TNF-alpha levels and histologic liver injury scores in pediatric nonalcoholic fatty liver disease. Am J Clin Pathol. 2007;127:954–960. doi: 10.1309/6VJ4DWGYDU0XYJ8Q. [DOI] [PubMed] [Google Scholar]
- Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrousos GP. Stressors, stress, and neuroendocrine integration of the adaptive response. The, Hans Selye Memorial Lecture. Ann N Y Acad Sci. 1997;1998(851):311–335. doi: 10.1111/j.1749-6632.1998.tb09006.x. [DOI] [PubMed] [Google Scholar]
- Chandola T, Brunner E, Marmot M. Chronic stress at work and the metabolic syndrome: prospective study. BMJ. 2006;332:521–525. doi: 10.1136/bmj.38693.435301.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moles A, Bartolomucci A, Garbugino L, Conti R, Caprioli A, Coccurello R, Rizzi R, Ciani B, D'Amato FR. Psychosocial stress affects energy balance in mice: modulation by social status. Psychoneuroendocrinology. 2006;31:623–633. doi: 10.1016/j.psyneuen.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, Rizzetto M. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. doi: 10.1053/jhep.2003.50161. [DOI] [PubMed] [Google Scholar]
- Knepel W, Nutto D, Hertting G. Evidence for inhibition by beta-endorphin of vasopressin release during foot shock-induced stress in the rat. Neuroendocrinology. 1982;34:353–356. doi: 10.1159/000123327. [DOI] [PubMed] [Google Scholar]
- Blanc G, Herve D, Simon H, Lisoprawski A, Glowinski J, Tassin JP. Response to stress of mesocortico-frontal dopaminergic neurones in rats after long-term isolation. Nature. 1980;284:265–267. doi: 10.1038/284265a0. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Takeshita K, Shimokawa T, Yi H, Isobe K, Loskutoff DJ, Saito H. Plasminogen activator inhibitor-1 is a major stress-regulated gene: implications for stress-induced thrombosis in aged individuals. Proc Natl Acad Sci U S A. 2002;99:890–895. doi: 10.1073/pnas.022608799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrar MH, Baranova A, Collantes R, Ranard B, Stepanova M, Bennett C, Fang Y, Elariny H, Goodman Z, Chandhoke V, Younossi ZM. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2008;27:412–421. doi: 10.1111/j.1365-2036.2007.03586.x. [DOI] [PubMed] [Google Scholar]
- Tilg H, Hotamisligil GS. Nonalcoholic fatty liver disease: cytokine-adipokine interplay and regulation of insulin resistance. Gastroenterology. 2006;131:934–945. doi: 10.1053/j.gastro.2006.05.054. [DOI] [PubMed] [Google Scholar]
- Bergman RN, Kim SP, Catalano KJ, Hsu IR, Chiu JD, Kabir M, Hucking K, Ader M. Why visceral fat is bad: mechanisms of the metabolic syndrome. Obesity (Silver Spring) 2006;14(Suppl 1):16S–19S. doi: 10.1038/oby.2006.277. [DOI] [PubMed] [Google Scholar]
- Czech B, Neumann ID, Muller M, Reber SO, Hellerbrand C. Effect of chronic psychosocial stress on nonalcoholic steatohepatitis in mice. Int J Clin Exp Pathol. 2013;6:1585–1593. [PMC free article] [PubMed] [Google Scholar]
- Eguchi Y, Eguchi T, Mizuta T, Ide Y, Yasutake T, Iwakiri R, Hisatomi A, Ozaki I, Yamamoto K, Kitajima Y, Kawaguchi Y, Kuroki S, Ono N. Visceral fat accumulation and insulin resistance are important factors in nonalcoholic fatty liver disease. J Gastroenterol. 2006;41:462–469. doi: 10.1007/s00535-006-1790-5. [DOI] [PubMed] [Google Scholar]
- Angulo P. NAFLD, obesity, and bariatric surgery. Gastroenterology. 2006;130:1848–1852. doi: 10.1053/j.gastro.2006.03.041. [DOI] [PubMed] [Google Scholar]
- van der Poorten D, Milner KL, Hui J, Hodge A, Trenell MI, Kench JG, London R, Peduto T, Chisholm DJ, George J. Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology. 2008;48:449–457. doi: 10.1002/hep.22350. [DOI] [PubMed] [Google Scholar]
- Hamdy O, Porramatikul S, Al-Ozairi E. Metabolic obesity: the paradox between visceral and subcutaneous fat. Curr Diabetes Rev. 2006;2:367–373. doi: 10.2174/1573399810602040367. [DOI] [PubMed] [Google Scholar]
- Engl J, Sturm W, Sandhofer A, Kaser S, Tschoner A, Tatarczyk T, Weiss H, Tilg H, Patsch JR, Ebenbichler CF. Effect of pronounced weight loss on visceral fat, liver steatosis and adiponectin isoforms. Eur J Clin Invest. 2008;38:238–244. doi: 10.1111/j.1365-2362.2008.01929.x. [DOI] [PubMed] [Google Scholar]
- Choi S, Diehl AM. Role of inflammation in nonalcoholic steatohepatitis. Curr Opin Gastroenterol. 2005;21:702–707. doi: 10.1097/01.mog.0000182863.96421.47. [DOI] [PubMed] [Google Scholar]
- Wolpe SD, Sherry B, Juers D, Davatelis G, Yurt RW, Cerami A. Identification and characterization of macrophage inflammatory protein 2. Proc Natl Acad Sci U S A. 1989;86:612–616. doi: 10.1073/pnas.86.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Beller DI, Frendl G, Graves DT. Monocyte chemoattractant protein-1 regulates adhesion molecule expression and cytokine production in human monocytes. J Immunol. 1992;148:2423–2428. [PubMed] [Google Scholar]
- Devalaraja MN, Mcclain CJ, Barve S, Vaddi K, Hill DB. Increased monocyte MCP-1 production in acute alcoholic hepatitis. Cytokine. 1999;11:875–881. doi: 10.1006/cyto.1999.0495. [DOI] [PubMed] [Google Scholar]