

Abstract
Familial hypercholesterolemia (FH) is a genetic disorder of lipoprotein metabolism resulting in elevated serum low-density lipoprotein (LDL) cholesterol levels leading to increased risk for premature cardiovascular diseases (CVDs). The diagnosis of this condition is based on clinical features, family history, and elevated LDL-cholesterol levels aided more recently by genetic testing. As the atherosclerotic burden is dependent on the degree and duration of exposure to raised LDL-cholesterol levels, early diagnosis and initiation of treatment is paramount. Statins are presently the mainstay in the management of these patients, although newer drugs, LDL apheresis, and other investigational therapies may play a role in certain subsets of FH, which are challenging to treat. Together these novel treatments have notably improved the prognosis of FH, especially that of the heterozygous patients. Despite these achievements, a majority of children fail to attain targeted lipid goals owing to persistent shortcomings in diagnosis, monitoring, and treatment. This review aims to highlight the screening, diagnosis, goals of therapy, and management options in patients with FH.
Keywords: Familial hypercholesterolemia, heterozygous familial hypercholesterolemia, homozygous familial hypercholesterolemia, low-density lipoprotein receptor mutation
INTRODUCTION
Familial hypercholesterolemia (FH) is a genetic disorder of lipoprotein metabolism characterized by highly elevated plasma total-cholesterol levels with detrimental cardiovascular consequences that commence in childhood. Although atherosclerosis due to FH manifests primarily in adulthood, it has a precocious inception as early as the 1st decade of life.[1] That early treatment of risk factors can reverse the atherosclerotic changes in the arterial system[2] underscores the need for prompt detection and treatment of children with this condition. Fagge identified this disorder more than a century ago as a skin ailment,[3] but its correlation with atherosclerosis was first recognized in 1939 by Norwegian physician Carl Muller.[4] The past decade saw a flurry of research in this disease with respect to its genetic basis and therapy. However, FH remains underdiagnosed till late due to the lack of awareness among pediatricians and the general public and the diagnosis is often arrived at only after the irreversible consequences of atherosclerosis have been established. This review describes the current status of the diagnosis, screening, and management of this malady.
GENETICS OF FH
’Familial hypercholesterolemia’ represents the phenotypic manifestation of abnormal lipoprotein metabolism caused by a variety of genetic abnormalities. After the seminal discovery by Brown and Goldstein that mutations in the low-density lipoprotein receptor (LDLR) was the cause of monogenetic FH, over 1,500 mutations of this gene have been detected[5,6] and these account for more than 80% of cases of monogenetic FH.[7] Heterozygous FH (HeFH) is not an uncommon disorder in children, with an estimated prevalence of 1 in 500 in the western world.[8] Homozygous FH (HoFH), although uncommon (prevalence is less than one per million in the general population), is a critical condition which commences in the first few years of life.[9] It is principally noted in countries such as Lebanon, Canada, and South Africa possibly because of the founder mutations and isolation of population.[10] In addition to the LDLR defect, two other sets of autosomal dominant mutations play a central role in the pathogenesis of FH; one, a defective apo-B100 component of LDL, known as familial defective apoB-100 (clinically indistinguishable from heterozygous LDLR mutations).[11,12,13] Secondly, a gain of function mutation affecting proprotein convertase subtilisin/kexin 9 (PCSK9) encoded by chromosome 1 has also been shown to trigger FH by negatively modulating LDL receptor expression.[14] Although the rare autosomal recessive form of FH called autosomal recessive hypercholesterolemia has been described in a few families,[15,16] in clinical practice, monogenetic hypercholesterolemia is primarily an autosomal dominant disorder with greater than 90% penetrance.
Though single gene disorders play a crucial role in the etiology of FH, linkage studies have exposed that the majority of cases of FH are caused by numerous unexceptional genetic variations.[11] An interplay of these polygenic variations together with environmental factors remains the leading cause for hypercholesterolemia in the general population.[17,18] However, a monogenetic etiology is usually the reason for more severe forms of LDL elevation and also for phenotypic expression of FH in the 1st decade of life.
SCREENING FOR FH
The ideal strategy to screen for FH is currently a controversial issue. Former lipid guidelines advocated ‘targeted screening’, which comprised a fasting lipid profile test in children with risk factors for FH such as a family history of premature cardiovascular diseases (CVDs), dyslipidemia, or obesity.[19] However, despite its cost effectiveness, this approach entailed the risk of missing 30-60% of affected patients.[20] An alternative approach to screening is termed ‘cascade screening’,[21,22] wherein health workers actively screen for disease among the first and second degree relatives of patients diagnosed by targeted screening. Although this method is associated with improved detection rates, there remains a considerable risk of missing affected individuals. This shortcoming has prompted some of the recent guidelines to recommend a strategy of universal lipid screening.[23,24] However, the cost effectiveness or utility of universal screening as well as the psychological impact on the children and the parents are not well-studied. Furthermore, a minority of patients of FH (7%) may have a normal lipid profile at the time of screening,[25] thus, facing the risk of missing the diagnosis in some despite screening of the entire population.
An equally important question is what to screen — lipids or genes? Genetic screening strategy involves searching for the common genes causing FH among suspected children and their close relatives. Recent National Institute for Health and Care Excellence (NICE) guidelines recommend a DNA testing on all patients diagnosed with FH and a subsequent genetic screening among their close relatives in order to augment case detection rates.[26] Although intuitively attractive, a significant number of patients clinically diagnosed with FH are negative for mutations conventionally tested for by genetic screening, probably due to polygenic inheritance.[27] In such patients, genetic cascade testing is expected to have a very low yield and is unlikely to be cost effective.[17] Hence, genetic cascade screening is likely to benefit only probands where a definite mutation is identified; in others, a strategy of lipid profile-based cascade screening is preferable.
The ideal age of lipid screening among children is also a keenly debated issue. The normal cord blood levels of LDL-cholesterol ranges from 35 to 70 mg/dl.[28] Although cord blood LDL levels for screening for FH is an appealing concept, studies have shown significant overlap in these levels between neonates with and without HeFH,[29] thus precluding this as a screening strategy. The Lipid Research Clinics prevalence studies demonstrated that by the age of 2 years, the serum lipid level reached that of young adults,[30] while the National Health and Nutrition Examination Surveys (NHANESs)[31], reported that the peak lipid levels are reached by the age of 9-11 years. Therefore, universal screening is best performed between 9 and 11 years of age, whereas a screening at any time after the age of 2 years is preferred in those who are candidates of targeted screening.[11,32,33] Table 1 summarizes national lipid association guidelines for screening children for FH.
Table 1.
National lipid association key screening recommendations for FH

Clinical features and diagnosis
Patients with HeFH are, by and large, asymptomatic in childhood and adolescence and typically diagnosed by screening methods. Their total and LDL-cholesterol levels are characteristically over the 95th centile of the recommended levels and a strong family history corroborates the diagnosis. Some involved persons may bear peripheral markers of fat deposition such as tendon xanthoma or arcus lipoides.
Homozygous or compound HeFH, on the other hand, presents in the 1st decade of life with a distinctive and severe clinical phenotype. The age at presentation depends on the degree of LDL receptor activity,[16] those with the null phenotype (<2% LDL receptor activity) tend to present earlier, resulting even in intrauterine death. These patients have primarily dermatological and ocular manifestations — tendon xanthomas and interdigital xanthomas are pathognomic of HoFH [Figure 1]. Tendon xanthomas are frequently missed on visual inspection alone and necessitate careful palpation in the Achilles, biceps and triceps tendons for early detection. Although tuberous xanthomas, xanthelasma, and corneal arcus appear in conditions other than FH,[34] their occurrence at a younger age should prompt evaluation for FH. Severe atherosclerosis involving multiple vascular beds, including coronary, cerebral, and peripheral vascular system, manifest in a myriad ways. Though coronary atherosclerosis is frequently the cause of premature death, calcific aortic valve stenosis and aortic root disease, including supravalvular aortic stenosis due to cholesterol and inflammatory cell infiltration, may result in significant morbidity in these patients, often requiring aortic valve and root replacement.[35]
Figure 1.
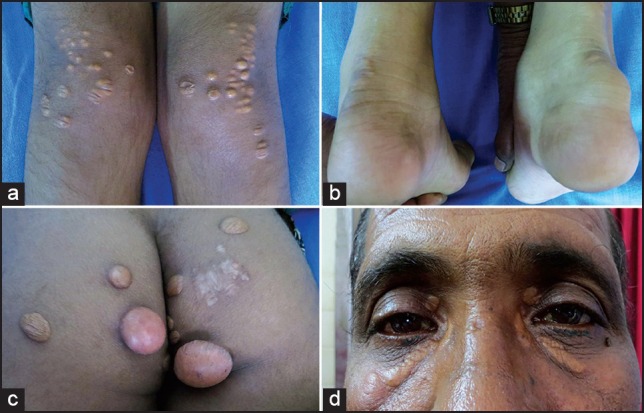
Dermatological manifestations: (a) Eruptive xanthoma, (b) tendon xanthoma, and (c) tuberous xanthoma in a 12-year-old girl with homozygous familial hypercholesterolemia (FH). (d) Her father who was diagnosed to have heterozygous FH with coronary artery disease had xanthelasma
When FH is suspected based on elevated lipid levels and clinical features, secondary dyslipidemias such as diabetes, endocrine disorders including hypothyroidism, renal disorders, obesity, and incriminating drugs must be ruled out before arriving at the diagnosis. A detailed family history should be taken not only to assess the mode of transmission but also to identify other affected individuals for early commencement of treatment. A comprehensive CVD risk assessment is required in all diagnosed patients and correction of modifiable risk factors must be pursued. The value of CVD risk assessment tools used in adults such as Framingham Risk Score have not been validated in the pediatric and adolescent populations with FH and are liable to underestimate the risk.[11] Ancillary investigations such as carotid intima medial thickness and ankle brachial index, which are usually used in research settings, may be helpful in monitoring the progression of disease in selected cases.
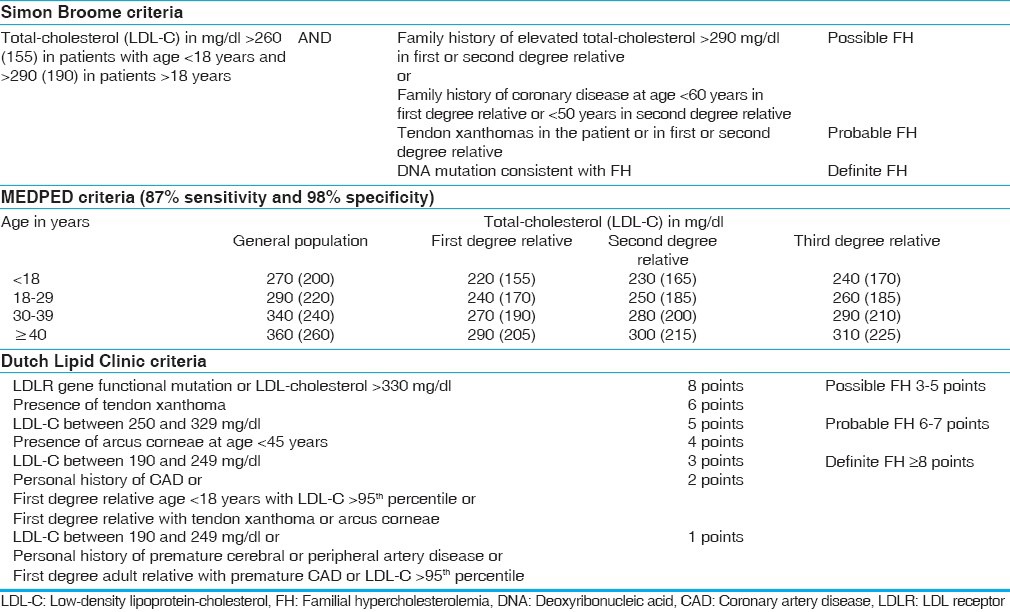
The diagnosis of FH is typically based on elevation of total-, LDL-, and non-HDL-cholesterol above the 95th centile recommended for the age and sex of the patient together with positive family history or identification of a causative mutation. The MEDPED criteria from the United States,[36] the Dutch Lipid Clinic criteria,[37] and the British Simon Broome Registry criteria[38] [Table 2] are validated diagnostic systems in this regard. The first relies solely on the age and the blood lipid levels of the patient, while the latter two require family history and clinical findings as well. These criteria are credited with simplicity and ease of use; however, they may be relatively ineffective at diagnosing index cases. Moreover, these criteria may not be clinically sensitive when applied to mild phenotypes and children in whom phenotypic expression is not yet completed.
Table 2.
Criteria for diagnosis of familial hypercholesterolemia
Management
Lipid targets
Recommendations differ with respect to target lipid levels in pediatric and adolescent patients. National Lipid Association guidelines recommend a target LDL level of <130 mg/dl or >50% reduction from baseline values.[24] More rigorous targets are proposed in patients with additional risk factors such as diabetes, obesity, and a family history of CVD. Belgian multisocietal guidelines, on the other hand, recommend age-specific targets.[33] In children aged 10-14 years, an LDL level of <160 mg/dl or >30% reduction from baseline levels is targeted. A rigorous target lipid level of <130 mg/dl is recommended in children between the ages of 14 and 18 years. In patients older than 18 years, a lipid target of <100 mg/dl is deemed appropriate. It should be noted that, a recent cross-sectional study in the Netherlands showed that no more than 21% of HeFH patients realized their lipid goals despite the recent advances in therapy.[39] Among patients who failed to achieve LDL-cholesterol goal, only 21% were on maximal dose of approved drugs, suggesting shortcomings in adequate monitoring and implementation of therapy.[39]
Lifestyle changes
Therapeutic lifestyle adjustments forman important part in the management of FH. This encompasses specific dietary manipulations, physical activity, limitation of alcohol intake, and total avoidance of tobacco products. Recent guidelines recommend a low calorie diet with a total fat intake of ≤3% of the total dietary intake including <8% of saturated fat and <75 mg/1,000 kcal cholesterol for these patients.[33] However, dietary restrictions are noted to have a modest effect in lowering lipid levels,[40] with unproven long-term clinical benefits.[41] Consequently, a concurrent drug therapy is indicated in patients with severe hypercholesterolemia. Dietary supplementation of phytosterol esters and stanol esters is controversial: Although a few recent studies have demonstrated a reduction of LDL levels in children with FH,[42] there are concerns regarding their accumulation in atheromas[43] and lowering of serum levels of lipid soluble vitamins.[44] Similarly, dietary supplementation of soy proteins and polyunsaturated fatty acids in this population is not substantiated by clinical evidence and is, hence, not currently recommended.[33]
Drug therapy-when to start?
The former guidelines issued by National Heart, Lung, and Blood Institute (NHLBI) advised treatment with bile acid sequestrants, the lowest age recommended for initiation being 10 years.[19] This was based on the excellent long-term safety profile of this group of drugs owing to lack of their systemic absorption. However, modest efficacy[45] and poor tolerability of these drugs resulted in alterations in the recent expert opinions and consensus papers.[23,24,33] In a recent statement by the American Heart Association,[46] later endorsed by the American Academy of Pediatrics,[20] statins were proposed as first-line drugs and the age of initiation of therapy was lowered to 8 years.
Bile acid Sequestrants
Formerly, this class of drugs was deemed the first-line of therapy of FH in children owing to their lack of systemic uptake. They bind to bile acids in the intestine, thereby, preventing their systemic absorption; this results in a greater conversion of cholesterol to bile acids and an enhanced production of LDL receptors by the liver. Cholestyramine and colestipol were the most frequently used drugs in this class; however, they fell out of favor due to their modest efficacy (10-20% LDL reduction) and gastrointestinal intolerance. Of late, a novel drug in this class, colesevelam hydrochloride, has been studied in HeFH patients. A short-term, randomized trial showed good tolerability and efficacy of colesevelam alone and in combination with statins leading to a renewed interest in this class of drugs.[47]
Statins
Statins (3-hydroxy-3methyl-glutaryl-CoA reductase inhibitors) are currently the first line of drugs in the treatment of FH in children and adolescents. They inhibit the rate-limiting step in cholesterol synthesis, thus, increasing the expression of LDL receptors, resulting in the rapid clearance of LDL from the blood. However, they have a restricted role in patients of HoFH with null phenotype in view of the need for receptor production for their action. Among the various generic statins available, the Food and Drug Administration (FDA) has approved of pravastatin in children over 8 years of age and lovastatin, atorvastatin, and simvastatin above the age of 10 years.[48]
The prepubertal commencement of statin therapy remains controversial,[49] as this can potentially hamper the production of steroid hormones in the body. Moreover, their effects on muscles and the liver are still an issue of grave concern. A recent Cochrane review[50] and two meta-analysis[51,52] of placebo-controlled trials on statins in children and adolescents with FH showed no major side effects with regard to growth, sexual development, muscle, and liver toxicity. Concurrently, they showed excellent efficacy in lipid lowering with a 26.5% mean relative reduction in LDL-cholesterol levels. The apprehension regarding growth disruption by statins at puberty was allayed, in part, by the paradoxical finding of increased growth in the children treated with the drug.[51,52] However, it is noteworthy that all the trials included in these meta-analyses studied only short-term outcomes; the long-term safety of statins in this population is unknown. The longest follow-up data on the effects of statin therapy in pediatric population is a retrospective study over a 7-year period in 185 children with FH treated with pravastatin, which revealed minor side effects in 13% of patients and myopathy in four patients.[53] Modern trends of drug usage among children indicate that the utilization of statins in the pediatric population is in the upswing,[54] despite the aforementioned concerns in relation to long-term safety.
There are specific recommendations on the subject of monitoring of patients commencing statin therapy. Creatine phosphokinase (CK) to assess muscle toxicity and aspartate amino transferase (AST), and alanine amino transferase (ALT) to monitor liver toxicity are mandatory prior to initiation of statins. Follow-up measurements must be done 1-3 months after starting the drug and yearly thereafter. Drug therapy should be interrupted when CK levels reach five times and AST and ALT three times over the upper limit of normal; the same drug at a lower dose or a different statin may be introduced after a drug-free interval of 3 months. Other drugs may be tried if the patient does not tolerate statins despite these measures.[33]
Ezetimibe
Ezetimibe is a new class of cholesterol absorption inhibitors that acts on the brush border of the small intestinal epithelium. The specific site of its action is believed to be the epithelial cell Niemann — Pick C1-like protein.[55] As their mechanism of action is not based on the expression of LDL receptors, they are especially beneficial in the management of HoFH. Clinical trials have displayed their efficacy in reducing LDL levels when used alone[56] or in combination with statins.[57,58] However, the initial fervor over their use was dampened by the largest prospective trial (ENHANCE trial) on cholesterol absorption inhibitors until this time, which demonstrated that ezetimibe added to high dose simvastatin failed to lessen carotid intima medial thickness in spite of a significant diminution in LDL levels.[59] Lastly, the discovery of a small but significant rise in the incidence of cancer in patients treated with ezetimibe patients in the Simvastatin and Ezetimibe in Aortic Stenosis (SEAS) trial[60] is a cause for concern in view of the need for lifelong therapy required in patients with FH. Therefore, additional data is required on clinically significant outcomes as well as safety endpoints before their widespread adoption in pediatric practice. Although US Food and Drug Administration (FDA) has approved of ezetemibe therapy in children over the age of 10years, current guidelines recommend drug initiation before 18 years of age only in patients intolerant to statins and in patients who fail to realize lipid goals with statin monotherapy.[11,33]
Therapeutic options in patients who failed to attain lipid targets despite maximal medical therapy
Newer drugs
Mipomirsen, an antisense oligonucleotide that targets apoB-100 mRNA in the liver, is presently under investigation in the therapy of FH. This drug significantly lowered LDL and lipoprotein (a) levels in adults with heterozygous[61] and homozygous[62] hypercholesterolemia in recent phase 3 trials. Although the mean LDL reduction with 200 mg of subcutaneous mipomirsen administered weekly was significant in patients with HoFH (-24.7% in treatment group and -3.3% in the placebo group, P = 0.0003), the response to therapy was inconsistent and compounded by a significant number of nonresponders.[62] The most frequent side effects of mipomirsen include reactions at the site of injection and flu-like symptoms, but apprehension regarding their hepatic toxicity, especially steatosis, still remains. Moreover, as they have not been studied in the pediatric population in a prospective clinical trial, their safety profile in this group of patients is not defined.
Serum PCSK9 are proteins which bind to LDL receptors and promote their degradation, thus, raising LDL levels in the blood. A variety of molecular techniques based on terminating the effect of PCSK9 in order to lower LDL-cholesterol levels is under investigation, including the development of monoclonal antibodies that bind to PCSK9,[63] antisense nucleotide-based therapy,[64] and small interfering RNAs.[65] In a randomized control trial experimenting on monoclonal antibodies in adults with various forms of hypercholesterolemia, the combination of this drug with 10 and 80 mg of atorvastatin was more efficacious than80mg of atorvastatin alone in reducing LDL levels.[66] However, as this antibody requires some residual LDL receptor function to fulfill its function, it is useful only in patients with HeFH and non-null phenotype HoFH.
Lomitapide is a new lipid-lowering agent with a novel method of action: It inhibits the microsomal triglyceride transfer protein(MTP). The role of MTP in the production of LDL involves assisting inthe transfer of triglycerides to apolipoprotein B.[67] The US FDA has approved of its use as an orphan drug in the treatment of HoFH.[68] In a recently published phase 3 dose escalation trial, lomitapide reduced LDL-cholesterol by 50% in HoFH patients with poorly controlled LDL levels.[69] Although this small study showed a satisfactory safety profile of the drug, there are still lingering doubts regarding the hepatic side effects like steatosis and transaminitis owing to their distinctive mechanism of action.
In addition to the aforementioned drugs, other classes of drugs like thyroid mimetics (e.g., eprotirome and sobetirome),[70] HDL-bound enzyme cholesterol ester transfer protein (CETP) inhibitors (e.g., torcetrapib, anacetrapib, and evacetrapib) and reconstituted high-density lipoprotein (rHDL)[71] are currently under research in the treatment of elevated LDL-cholesterol and shows variable efficacy and safety. All the ongoing trials on modern drug therapy of dyslipidemia focuses on adult patients and excludes the pediatric population. Given that a majority of these novel therapies are yet unproven with regard to clinical efficacy and safety endpoints, their role is presently confined to that of a lipid apheresis-sparing therapy in patients with HoFH who have fallen short of their lipid goals. Additional studies in the pediatric population are required prior to their clinical adoption in the treatment of heterozygous patients.
LDL apheresis
Patients with homozygous and compound HeFH frequently have elevated lipid levels in spite of optimal medical therapy. These are fitting candidates for LDL apheresis, which has proved to be a very beneficial treatment option to reducing LDL levels. Numerous studies have affirmed its capability to lower LDL-cholesterol levels by 55-75%.[72] Commonly used techniques of LDL apheresis include heparin-induced extracorporeal LDL-cholesterol precipitation (HELP), dextran sulfate cellulose adsorption (DSA), double filtration plasmapheresis (DFPP), polyacrylate full blood adsorption (PFBA also known as DALI), and immune adsorption. Details on the techniques are beyond the scope of this update and interested readers may consult excellent reviews available on the subject.[73,74,75]
The decline in LDL-cholesterol levels by apheresis is a transitory event and is associated with a rebound escalation of lipid levels after the procedure. This rebound is expeditious in patients without FH, slower in those with HeFH and delayed in patients in HoFH.[76] Weekly to fortnightly sessions are advocated for patients with HoFH, as such episodic sittings have been shown to reduce the degree of rebound and retard the progression of atherosclerosis.[77,78] Regular apheresis therapy along with medications in patients of HoFH has improved the average life expectancy to over 50 years of age compared to the formerly bleak prognosis of death in the2nd or 3rd decade.[79] Despite its established efficacy, lipid apheresis has not yet been widely embraced in clinical practice due to lack of accessibility for the majority of patients, the prohibitive cost involved, the invasive nature of the procedure, and the lack of motivation among patients.
Gene therapy
HoFH was among the first disorders wherein gene therapy was experimented. Contrary to other treatment alternatives, the possibility of a definitive cure by a one-time procedure for a disease that lasts a lifetime renders this an appealing choice. However, due to the problems related to appropriate gene vector, lack of persistent gene expression as well as due to safety concerns,[80] this modality failed to demonstrate substantial clinical efficacy in preliminary trials. Upcoming research should focus on improving gene vectors and transfer techniques, while concurrently reducing their oncogenic dangers before it can be relevant to clinical practice.[81]
Surgical options
In addition to the therapies enumerated prior, surgical options including ileal bypass and portocaval shunt have been tried earlier in refractory cases. Owing to the significant comorbidities involved and the need for treatment before the onset of clinical effects of atherosclerosis, these never became a popular choice of treatment. Recent case reports of successful pediatric liver transplant done for the treatment of HoFH suggest excellent efficacy and good safety profile of this option.[82,83] However, in view of the scarcity of donor liver available and complexities of the transplant and post-transplant management, such a decision should be taken only after carefully assessing the risk benefit ratio.
Natural and modified natural history of FH
The natural history of FH depends primarily on the degree of functional LDL receptor activity present, and in turn, on LDL-cholesterol levels, resulting in widely varying prognosis even among homozygous individuals.[84] Symptom onset is age-dependent and typically occurs in the 2nd decade in homozygous patients. The extent of atherosclerosis is primarily determined by the degree of LDL elevation and its duration, calculated by the cholesterol year score.[85] Precocious onset of clinically significant atherosclerotic changes are very common and involve multiple vascular beds including coronary, cerebral, and peripheral systems.[85] Studies in the pre-statin era indicated poor outcomes in the majority of patients with HoFH, cardiovascular events being the chief cause of morbidity and mortality.[86] Aortic root disease was reported to be the commonest cardiac manifestation followed by coronary artery disease.[86] While some studies in this interval purported a mean survival of 18 years among patients with HoFH,[87] others observed an average survival of 40 years;[86] this variation may be ascribed to the differences in the proportion of receptor-negative patients included in these studies.
It was conventionally believed that modern day drug therapy for HoFH does not alter prognosis owing to the lack of significant reduction in LDL. However, this assumption was challenged by a recent retrospective analysis by Raal et al., involving 149 patients, wherein patients treated with statins had hazard ratios for mortality and cardiovascular events of 0.34 and 0.49, respectively when compared with patients in the pre-statin era, despite achieving only a modest 26% reduction in LDL levels.87 Although this result may be partly influenced by the beneficial effects of cardiovascular preventive drugs such as antiplatelet agents and beta blockers, this study underscores the benefit of statin therapy even in FH homozygous individuals.
Among patients with untreated HeFH, coronary artery disease (CAD) develops in about 50% of males by the age of 50 years and 30% of females by 60 years. Although CAD appears 10years later in females compared to males, an accelerated development of CAD is observed after menopause.[88,89] Simon Broome registry data from England in the pre-statin era showed that mortality associated with CAD was increased a 100-fold in the age group of 20-40 years and four-fold in the 40-59 year age group.[38] Among those surviving to the age of 60 years, however, the risk seems akin to that in the general population.[38] The benefits of present day therapeutic advances in this population is confirmed by a large prospective study from the UK, which reveals a 37% relative reduction in standardized mortality rate from 3.4 in the pre-statin era to 2.1 after widespread use of statins.[90] Despite strong association of FH with coronary and peripheral vascular disease, its relation with stroke risk is more controversial. A large prospective registry data from United Kingdom showed that ischemic stroke mortality among treated HeFH patients not to be different from general population.[91] The reason for this difference is presently unknown.
CONCLUSION
FH is a grave ailment with its genesis in early childhood resulting in damaging consequences in later life. Although the need for a screening strategy to detect this disease early is widely accepted, there is no consensus regarding whom and when to screen. Early initiation of lipid-lowering therapy and lifestyle measures might improve the clinical outcome. While such treatment initiatives have notably improved the prognosis of HeFH, the outcomes of familial homozygous hypercholesterolemia remain disappointing. Although most cases may be treated with a combination of statins and cholesterol absorption inhibitors, some will have need of more invasive therapies such as LDL apheresis. The past 2 decades have noted the evolution of novel therapies to lower LDL-cholesterol levels and defer premature atherosclerosis, especially in conjunction with lifestyle modifications. Despite these triumphs, a large majority of children do not attain targeted lipid goals due to shortfalls in diagnosis, monitoring, and treatment. An effective screening strategy together with timely initiation of established therapies would go a long way in reducing the burden of atherosclerosis due to this challenging condition.
ACKNOWLEDGEMENTS
I sincerely thank Prof. SS Kothari for advice in the preparation of this manuscript. I also thank Dr. Riya Jose for editing an earlier version of this manuscript.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Newman WP, 3rd, Freedman DS, Voors AW, Gard PD, Srinivasan SR, Cresanta JL, et al. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N Engl J Med. 1986;314:138–44. doi: 10.1056/NEJM198601163140302. [DOI] [PubMed] [Google Scholar]
- 2.Wiegman A, Hutten BA, de Groot E, Rodenburg J, Bakker HD, Büller HR, et al. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: A randomized controlled trial. JAMA. 2004;292:331–7. doi: 10.1001/jama.292.3.331. [DOI] [PubMed] [Google Scholar]
- 3.Fagge CH. Xanthomatous diseases of the skin. Trans Pathol Soc Lond. 1873:242–50. [Google Scholar]
- 4.Muller C. Angina pectoris in hereditary xanthomatosis. Arch Intern Med. 1939;64:675–700. [Google Scholar]
- 5.Goldstein JL, Brown MS. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem. 1974;249:5153–62. [PubMed] [Google Scholar]
- 6.Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet. 2008;72:485–98. doi: 10.1111/j.1469-1809.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- 7.Soufi M, Kurt B, Schweer H, Sattler AM, Klaus G, Zschocke J, et al. Genetics and kinetics of familial hypercholesterolemia, with the special focus on FH-(Marburg) p. W556R. Atheroscler Suppl. 2009;10:5–11. doi: 10.1016/S1567-5688(09)71802-1. [DOI] [PubMed] [Google Scholar]
- 8.Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Sly WS, Childs B, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill Medical Publishing Division; 2000. pp. 2863–913. [Google Scholar]
- 9.Naoumova RP, Thompson GR, Soutar AK. Current management of severe homozygous hypercholesterolaemias. Curr Opin Lipidol. 2004;15:413–22. doi: 10.1097/01.mol.0000137222.23784.2a. [DOI] [PubMed] [Google Scholar]
- 10.Marais AD. Familial hypercholesterolaemia. Clin Biochem Rev. 2004;25:49–68. [PMC free article] [PubMed] [Google Scholar]
- 11.Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, et al. National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemia: Screening, diagnosis and management of pediatric and adult patients: Clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3 Suppl):S1–8. doi: 10.1016/j.jacl.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Arnett DK, Baird AE, Barkley RA, Basson CT, Boerwinkle E, Ganesh SK, et al. American Heart Association Council on Epidemiology and Prevention; American Heart Association Stroke Council; Functional Genomics and Translational Biology Interdisciplinary Working Group. Relevance of genetics and genomics for prevention and treatment of cardiovascular disease: A scientific statement from the American Heart Association Council on Epidemiology and Prevention, the Stroke Council, and the Functional Genomics and Translational Biology Interdisciplinary Working Group. Circulation. 2007;115:2878–901. doi: 10.1161/CIRCULATIONAHA.107.183679. [DOI] [PubMed] [Google Scholar]
- 13.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: New insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–803. doi: 10.1172/JCI18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen JC, Boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 15.Arca M, Zuliani G, Wilund K, Campagna F, Fellin R, Bertolini S, et al. Autosomal recessive hypercholesterolaemia in Sardinia, Italy, and mutations in ARH: A clinical and molecular genetic analysis. Lancet. 2002;359:841–7. doi: 10.1016/S0140-6736(02)07955-2. [DOI] [PubMed] [Google Scholar]
- 16.Pisciotta L, Priore Oliva C, Pes GM, Di Scala L, Bellocchio A, Fresa R, et al. Autosomal recessive hypercholesterolemia (ARH) and homozygous familial hypercholesterolemia (FH): A phenotypic comparison. Atherosclerosis. 2006;188:398–405. doi: 10.1016/j.atherosclerosis.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 17.Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study. Lancet. 2013;381:1293–301. doi: 10.1016/S0140-6736(12)62127-8. [DOI] [PubMed] [Google Scholar]
- 18.McGill HC, Jr, McMahan CA, Zieske AW, Malcom GT, Tracy RE, Strong JP. Effects of nonlipid risk factors on atherosclerosis in youth with a favorable lipoprotein profile. Circulation. 2001;103:1546–50. doi: 10.1161/01.cir.103.11.1546. [DOI] [PubMed] [Google Scholar]
- 19.American Academy of Pediatrics. National Cholesterol Education Program: Report of the expert panel on blood cholesterol levels in children and adolescents. Pediatrics. 1992;89:525–84. [PubMed] [Google Scholar]
- 20.Daniels SR, Greer FR Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics. 2008;122:198–208. doi: 10.1542/peds.2008-1349. [DOI] [PubMed] [Google Scholar]
- 21.Hurrell C, Wietlisbach V, Jotterand V, Volet M, Lenain V, Nicod P, et al. High prevalence of major cardiovascular risk factors in first-degree relatives of individuals with familial premature coronary artery disease — the GENECARD project. Atherosclerosis. 2007;194:253–64. doi: 10.1016/j.atherosclerosis.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 22.Bender R, Bell DA, Hooper AJ, Edwards G, van Bockxmeer FM, Watts GF, et al. Screening for familial hypercholesterolaemia. Pathology. 2012;44:122–8. doi: 10.1097/PAT.0b013e32834efa07. [DOI] [PubMed] [Google Scholar]
- 23.Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: Summary report. Pediatrics. 2011;128(Suppl 5):S213–56. doi: 10.1542/peds.2009-2107C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daniels SR, Gidding SS, de Ferranti SD. National Lipid Association Expert Panel on Familial Hypercholesterolemia. Pediatric aspects of familial hypercholesterolemias: Recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3 Suppl):S30–7. doi: 10.1016/j.jacl.2011.03.453. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Garcia AB, Ivorra C, Martinez-Hervas S, Blesa S, Fuentes MJ, Puig O, et al. Reduced penetrance of autosomal dominant hypercholesterolemia in a high percentage of families: Importance of genetic testing in the entire family. Atherosclerosis. 2011;218:423–30. doi: 10.1016/j.atherosclerosis.2011.07.106. [DOI] [PubMed] [Google Scholar]
- 26.DeMott K, Nherera L, Shaw EJ, Minhas R, Humphries SE, Kathoria M, et al. London: National Collaborating Centre for Primary Care and Royal College of General Practitioners; 2008. Clinical Guidelines and Evidence Review for Familial hypercholesterolaemia: The identification and management of adults and children with familial hypercholesterolaemia. [Google Scholar]
- 27.Taylor A, Wang D, Patel K, Whittall R, Wood G, Farrer M, et al. Mutation detection rate and spectrum in familial hypercholesterolaemia patients in the UK pilot cascade project. Clin Genet. 2010;77:572–80. doi: 10.1111/j.1399-0004.2009.01356.x. [DOI] [PubMed] [Google Scholar]
- 28.Forrester JS. Redefining normal low-density lipoprotein cholesterol: A strategy to unseat coronary disease as the nation's leading killer. J Am Coll Cardiol. 2010;56:630–6. doi: 10.1016/j.jacc.2009.11.090. [DOI] [PubMed] [Google Scholar]
- 29.Vuorio AF, Turtola H, Kontula K. Neonatal diagnosis of familial hypercholesterolemia in newborns born to a parent with a molecularly defined heterozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1997;17:3332–7. doi: 10.1161/01.atv.17.11.3332. [DOI] [PubMed] [Google Scholar]
- 30.Tamir I, Heiss G, Glueck CJ, Christensen B, Kwiterovich P, Rifkind BM. Lipid and lipoprotein distributions in white children ages 6-19 yr. The Lipid Research Clinics Program Prevalence Study. J Chronic Dis. 1981;34:27–39. doi: 10.1016/0021-9681(81)90079-5. [DOI] [PubMed] [Google Scholar]
- 31.Hickman TB, Briefel RR, Carroll MD, Rifkind BM, Cleeman JI, Maurer KR, et al. Distributions and trends of serum lipid levels among United States children and adolescents ages 4-19 years: Data from the Third National Health and Nutrition Examination Survey. Prev Med. 1998;27:879–90. doi: 10.1006/pmed.1998.0376. [DOI] [PubMed] [Google Scholar]
- 32.Wierzbicki AS, Humphries SE, Minhas R Guideline Development Group. Familial hypercholesterolaemia: Summary of NICE guidance. BMJ. 2008;337:a1095. doi: 10.1136/bmj.a1095. [DOI] [PubMed] [Google Scholar]
- 33.Descamps OS, Tenoutasse S, Stephenne X, Gies I, Beauloye V, Lebrethon MC, et al. Management of familial hypercholesterolemia in children and young adults: Consensus paper developed by a panel of lipidologists, cardiologists, paediatricians, nutritionists, gastroenterologists, general practitioners and a patient organization. Atherosclerosis. 2011;218:272–80. doi: 10.1016/j.atherosclerosis.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 34.Schaefer EJ, Santos RD. Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K. Dermatology in general medicine. McGraw-Hill Professional; 2012. Xanthomatoses and Lipoprotein Disorders; p. 1601. [Google Scholar]
- 35.Kawaguchi A, Yutani C, Yamamoto A. Hypercholesterolemic valvulopathy: An aspect of malignant atherosclerosis. Ther Apher Dial. 2003;7:439–43. doi: 10.1046/j.1526-0968.2003.00075.x. [DOI] [PubMed] [Google Scholar]
- 36.Williams RR, Hunt SC, Schumacher MC, Hegele RA, Leppert MF, Ludwig EH, et al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72:171–6. doi: 10.1016/0002-9149(93)90155-6. [DOI] [PubMed] [Google Scholar]
- 37.Civeira F. International Panel on Management of Familial Hypercholesterolemia. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004;173:55–68. doi: 10.1016/j.atherosclerosis.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 38.Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ. 1991;303:893–6. doi: 10.1136/bmj.303.6807.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pijlman AH, Huijgen R, Verhagen SN, Imholz BP, Liem AH, Kastelein JJ, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: A large cross-sectional study in The Netherlands. Atherosclerosis. 2010;209:189–94. doi: 10.1016/j.atherosclerosis.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 40.Obarzanek E, Kimm SY, Barton BA, Van Horn L L, Kwiterovich PO, Jr, Simons-Morton DG, et al. Long-term safety and efficacy of a cholesterol-lowering diet in children with elevated low-density lipoprotein cholesterol: Seven-year results of the Dietary Intervention Study in Children (DISC) Pediatrics. 2001;107:256–64. doi: 10.1542/peds.107.2.256. [DOI] [PubMed] [Google Scholar]
- 41.Shafiq N, Singh M, Kaur S, Khosla P, Malhotra S. Dietary treatment for familial hypercholesterolaemia. Cochrane Database Syst Rev. 2010:CD001918. doi: 10.1002/14651858.CD001918.pub2. [DOI] [PubMed] [Google Scholar]
- 42.Guardamagna O, Abello F, Baracco V, Federici G, Bertucci P, Mozzi A, et al. Primary hyperlipidemias in children: Effect of plant sterol supplementation on plasma lipids and markers of cholesterol synthesis and absorption. Acta Diabetol. 2011;48:127–33. doi: 10.1007/s00592-010-0233-1. [DOI] [PubMed] [Google Scholar]
- 43.Ketomaki A, Gylling H, Miettinen TA. Effects of plant stanol and sterol esters on serum phytosterols in a family with familial hypercholesterolemia including a homozygous subject. J Lab Clin Med. 2004;143:255–62. doi: 10.1016/j.lab.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 44.Amundsen AL, Ntanios F, van der N Put, Ose L. Long-term compliance and changes in plasma lipids, plant sterols and carotenoids in children and parents with FH consuming plant sterol ester-enriched spread. Eur J Clin Nutr. 2004;58:1612–20. doi: 10.1038/sj.ejcn.1602015. [DOI] [PubMed] [Google Scholar]
- 45.Davidson MH. A systematic review of bile acid sequestrant therapy in children with familial hypercholesterolemia. J Clin Lipidol. 2011;5:76–81. doi: 10.1016/j.jacl.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 46.McCrindle BW, Urbina EM, Dennison BA, Jacobson MS, Steinberger J, Rocchini AP, et al. American Heart Association Atherosclerosis, Hypertension, and Obesity in Youth Committee; American Heart Association Council of Cardiovascular Disease in the Young; American Heart Association Council on Cardiovascular Nursing. Drug therapy of high-risk lipid abnormalities in children and adolescents: A scientific statement from the American Heart Association Atherosclerosis, Hypertension, and Obesity in Youth Committee, Council of Cardiovascular Disease in the Young, with the Council on Cardiovascular Nursing. Circulation. 2007;115:1948–67. doi: 10.1161/CIRCULATIONAHA.107.181946. [DOI] [PubMed] [Google Scholar]
- 47.Stein EA, Marais AD, Szamosi T, Raal FJ, Schurr D, Urbina EM, et al. Colesevelam hydrochloride: Efficacy and safety in pediatric subjects with heterozygous familial hypercholesterolemia. (e1-3).J Pediatr. 2010;156:231–6. doi: 10.1016/j.jpeds.2009.08.037. [DOI] [PubMed] [Google Scholar]
- 48.Iughetti L, Bruzzi P, Predieri B. Evaluation and management of hyperlipidemia in children and adolescents. Curr Opin Pediatr. 2010;22:485–93. doi: 10.1097/MOP.0b013e32833ab869. [DOI] [PubMed] [Google Scholar]
- 49.de Ferranti S, Ludwig DS. Storm over statins — the controversy surrounding pharmacologic treatment of children. N Engl J Med. 2008;359:1309–12. doi: 10.1056/NEJMp0805953. [DOI] [PubMed] [Google Scholar]
- 50.Vuorio A, Kuoppala J, Kovanen PT, Humphries SE, Strandberg T, Tonstad S, et al. Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev. 2010:CD006401. doi: 10.1002/14651858.CD006401.pub2. [DOI] [PubMed] [Google Scholar]
- 51.O’Gorman CS, Higgins MF, O’Neill MB. Systematic review and metaanalysis of statins for heterozygous familial hypercholesterolemia in children: Evaluation of cholesterol changes and side effects. Pediatr Cardiol. 2009;30:482–9. doi: 10.1007/s00246-008-9364-3. [DOI] [PubMed] [Google Scholar]
- 52.Arambepola C, Farmer AJ, Perera R, Neil HA. Statin treatment for children and adolescents with heterozygous familial hypercholesterolaemia: A systematic review and meta-analysis. Atherosclerosis. 2007;195:339–47. doi: 10.1016/j.atherosclerosis.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 53.Carreau V, Girardet JP, Bruckert E. Long-term follow-up of statin treatment in a cohort of children with familial hypercholesterolemia: Efficacy and tolerability. Paediatr Drugs. 2011;13:267–75. doi: 10.2165/11591650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 54.Cox ER, Halloran DR, Homan SM, Welliver S, Mager DE. Trends in the prevalence of chronic medication use in children: 2002-2005. Pediatrics. 2008;122:e1053–61. doi: 10.1542/peds.2008-0214. [DOI] [PubMed] [Google Scholar]
- 55.Altmann SW, Davis HR, Jr, Zhu LJ, Yao X, Hoos LM, Tetzloff G, et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303:1201–4. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- 56.Clauss S, Wai KM, Kavey RE, Kuehl K. Ezetimibe treatment of pediatric patients with hypercholesterolemia. J Pediatr. 2009;154:869–72. doi: 10.1016/j.jpeds.2008.12.044. [DOI] [PubMed] [Google Scholar]
- 57.Gagné C, Gaudet D, Bruckert E Ezetimibe Study Group. Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia. Circulation. 2002;105:2469–75. doi: 10.1161/01.cir.0000018744.58460.62. [DOI] [PubMed] [Google Scholar]
- 58.van der Graaf A, Cuffie-Jackson C, Vissers MN, Trip MD, Gagné C, Shi G, et al. Efficacy and safety of coadministration of ezetimibe and simvastatin in adolescents with heterozygous familial hypercholesterolemia. J Am Coll Cardiol. 2008;52:1421–9. doi: 10.1016/j.jacc.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 59.Kastelein JJP, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–43. doi: 10.1056/NEJMoa0800742. [DOI] [PubMed] [Google Scholar]
- 60.Rossebø AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, et al. SEAS Investigators. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–56. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 61.Stein EA, Dufour R, Gagne C, Gaudet D, East C, Donovan JM, et al. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: Results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation. 2012;126:2283–92. doi: 10.1161/CIRCULATIONAHA.112.104125. [DOI] [PubMed] [Google Scholar]
- 62.Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: A randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006. doi: 10.1016/S0140-6736(10)60284-X. [DOI] [PubMed] [Google Scholar]
- 63.Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–18. doi: 10.1056/NEJMoa1105803. [DOI] [PubMed] [Google Scholar]
- 64.Graham MJ, Lemonidis KM, Whipple CP, Subramaniam A, Monia BP, Crooke ST, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. 2007;48:763–7. doi: 10.1194/jlr.C600025-JLR200. [DOI] [PubMed] [Google Scholar]
- 65.Sjouke B, Kusters DM, Kastelein JJ, Hovingh GK. Familial hypercholesterolemia: Present and future management. Curr Cardiol Rep. 2011;13:527–36. doi: 10.1007/s11886-011-0219-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roth EM, McKenney JM, Hanotin C, Asset G, Stein EA. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891–900. doi: 10.1056/NEJMoa1201832. [DOI] [PubMed] [Google Scholar]
- 67.Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–56. doi: 10.1056/NEJMoa061189. [DOI] [PubMed] [Google Scholar]
- 68.FDA Approves Aegerion Pharmaceuticals’ JUXTAPID(TM) (lomitapide) Capsules for Homozygous Familial Hypercholesterolemia (HoFH) (NASDAQ:AEGR) [Internet] [Last cited on 2013 May 29]. Available from: http://ir.aegerion.com/releasedetail.cfm?ReleaseID=728650 .
- 69.Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, et al. Phase 3 HoFH Lomitapide Study investigators. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet. 2013;381:40–6. doi: 10.1016/S0140-6736(12)61731-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ladenson PW, Kristensen JD, Ridgway EC, Olsson AG, Carlsson B, Klein I, et al. Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. N Engl J Med. 2010;362:906–16. doi: 10.1056/NEJMoa0905633. [DOI] [PubMed] [Google Scholar]
- 71.Shaw JA, Bobik A, Murphy A, Kanellakis P, Blombery P, Mukhamedova N, et al. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103:1084–91. doi: 10.1161/CIRCRESAHA.108.182063. [DOI] [PubMed] [Google Scholar]
- 72.Thompson GR HEART-UK LDL Apheresis Working Group. Recommendations for the use of LDL apheresis. Atherosclerosis. 2008;198:247–55. doi: 10.1016/j.atherosclerosis.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 73.Mehta PK, Baer J, Nell C, Sperling LS. Low-density lipoprotein apheresis as a treatment option for hyperlipidemia. Curr Treat Options Cardiovasc Med. 2009;11:279–88. doi: 10.1007/s11936-009-0029-1. [DOI] [PubMed] [Google Scholar]
- 74.Lee WP, Datta BN, Ong BB, Rees A, Halcox J. Defining the role of lipoprotein apheresis in the management of familial hypercholesterolemia. Am J Cardiovasc Drugs. 2011;11:363–70. doi: 10.2165/11594970-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 75.Thompson GR. Lipoprotein apheresis. Curr Opin Lipidol. 2010;21:487–91. doi: 10.1097/MOL.0b013e32833e13fd. [DOI] [PubMed] [Google Scholar]
- 76.Kroon AA, van’t Hof MA, Demacker PN, Stalenhoef AF. The rebound of lipoproteins after LDL-apheresis. Kinetics and estimation of mean lipoprotein levels. Atherosclerosis. 2000;152:519–26. doi: 10.1016/s0021-9150(00)00371-3. [DOI] [PubMed] [Google Scholar]
- 77.Mabuchi H, Koizumi J, Shimizu M, Kajinami K, Miyamoto S, Ueda K, et al. Long-term efficacy of low-density lipoprotein apheresis on coronary heart disease in familial hypercholesterolemia. Hokuriku-FH-LDL-Apheresis Study Group. Am J Cardiol. 1998;82:1489–95. doi: 10.1016/s0002-9149(98)00692-4. [DOI] [PubMed] [Google Scholar]
- 78.Matsuzaki M1, Hiramori K, Imaizumi T, Kitabatake A, Hishida H, Nomura M, et al. Intravascular ultrasound evaluation of coronary plaque regression by low density lipoprotein-apheresis in familial hypercholesterolemia: The Low Density Lipoprotein-Apheresis Coronary Morphology and Reserve Trial (LACMART) J Am Coll Cardiol. 2002;40:220–7. doi: 10.1016/s0735-1097(02)01955-1. [DOI] [PubMed] [Google Scholar]
- 79.Thompson GR, Catapano A, Saheb S, Atassi-Dumont M, Barbir M, Eriksson M, et al. Severe hypercholesterolaemia: Therapeutic goals and eligibility criteria for LDL apheresis in Europe. Curr Opin Lipidol. 2010;21:492–8. doi: 10.1097/MOL.0b013e3283402f53. [DOI] [PubMed] [Google Scholar]
- 80.Lehrman S. Virus treatment questioned after gene therapy death. Nature. 1999;401:517–8. doi: 10.1038/43977. [DOI] [PubMed] [Google Scholar]
- 81.Al-Allaf FA, Coutelle C, Waddington SN, David AL, Harbottle R, Themis M. LDLR-Gene therapy for familial hypercholesterolaemia: Problems, progress, and perspectives. Int Arch Med. 2010;3:36. doi: 10.1186/1755-7682-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maiorana A, Nobili V, Calandra S, Francalanci P, Bernabei S, El Hachem M, et al. Preemptive liver transplantation in a child with familial hypercholesterolemia. Pediatr Transplant. 2011;15:E25–9. doi: 10.1111/j.1399-3046.2010.01383.x. [DOI] [PubMed] [Google Scholar]
- 83.Küçükkartallar T, Yankol Y, Kanmaz T, Topaloğlu S, Acarli K, Kalayoglu M. Liver transplantation as a treatment option for three siblings with homozygous familial hypercholesterolemia. Pediatr Transplant. 2011;15:281–4. doi: 10.1111/j.1399-3046.2010.01469.x. [DOI] [PubMed] [Google Scholar]
- 84.Sprecher DL, Schaefer EJ, Kent KM, Gregg RE, Zech LA, Hoeg JM, et al. Cardiovascular features of homozygous familial hypercholesterolemia: Analysis of 16 patients. Am J Cardiol. 1984;54:20–30. doi: 10.1016/0002-9149(84)90298-4. [DOI] [PubMed] [Google Scholar]
- 85.Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: Current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262–8. doi: 10.1016/j.atherosclerosis.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 86.Haitas B, Baker SG, Meyer TE, Joffe BI, Seftel HC. Natural history and cardiac manifestations of homozygous familial hypercholesterolaemia. Q J Med. 1990;76:731–40. [PubMed] [Google Scholar]
- 87.Raal FJ, Pilcher GJ, Panz VR, van Deventer HE, Brice BC, Blom DJ, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124:2202–7. doi: 10.1161/CIRCULATIONAHA.111.042523. [DOI] [PubMed] [Google Scholar]
- 88.Slack J. Risks of ischaemic heart-disease in familial hyperlipoproteinaemic states. Lancet. 1969;2:1380–2. doi: 10.1016/s0140-6736(69)90930-1. [DOI] [PubMed] [Google Scholar]
- 89.Stone NJ, Levy RI, Fredrickson DS, Verter J. Coronary artery disease in 116 kindred with familial type II hyperlipoproteinemia. Circulation. 1974;49:476–88. doi: 10.1161/01.cir.49.3.476. [DOI] [PubMed] [Google Scholar]
- 90.Neil A, Cooper J, Betteridge J, Capps N, McDowell I, Durrington P, et al. Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: A prospective registry study. Eur Heart J. 2008;29:2625–33. doi: 10.1093/eurheartj/ehn422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huxley RR, Hawkins MH, Humphries SE, Karpe F, Neil HA Simon Broome Familial Hyperlipidaemia Register Group and Scientific Steering Committee. Risk of fatal stroke in patients with treated familial hypercholesterolemia: A prospective registry study. Stroke. 2003;34:22–5. doi: 10.1161/01.str.0000047123.14312.3e. [DOI] [PubMed] [Google Scholar]