

Abstract
VGF (non-acronymic) is a granin-like protein that is packaged and proteolytically processed within the regulated secretory pathway. VGF and peptides derived from its processing have been implicated in neuroplasticity associated with learning, memory, depression, and chronic pain. In sensory neurons, VGF is rapidly increased following peripheral nerve injury and inflammation. Several bioactive peptides generated from the C-terminus of VGF have pro-nociceptive spinal effects. The goal of the present study was to examine the spinal effects of the peptide TLQP-21 and determine whether it participates in spinal mechanisms of persistent pain. Application of exogenous TLQP-21 induced dose-dependent thermal hyperalgesia in the warm water immersion tail withdrawal test. This hyperalgesia was inhibited by a p38 MAPK inhibitor as well as inhibitors of cyclooxygenase and lipoxygenase. We used immunoneutralization of TLQP-21 to determine the function of the endogenous peptide in mechanisms underlying persistent pain. In mice injected intradermally with complete Freund’s adjuvant, intrathecal treatment with anti-TLQP21 IgG immediately prior to or 5 h after induction of inflammation dose-dependently inhibited tactile hypersensitivity and thermal hyperalgesia. Intrathecal anti-TL21 administration also attenuated the development and maintenance of tactile hypersensitivity in the spared nerve injury model of neuropathic pain. These results provide evidence that endogenous TLQP-21 peptide contributes to the mechanisms of spinal neuroplasticity after inflammation and nerve injury.
INTRODUCTION
VGF (non-acronymic) is a granin-like neuropeptide precursor whose expression is robustly regulated by neuronal lesions and growth factors [5,15,26]. VGF and peptides derived from its proteolytic processing have been implicated in the control of energy balance and metabolism as well as in neuroplasticity associated with learning, memory, depression, and chronic pain [9,19,20,22,28,35,43].
Peptides containing the C-terminus of VGF (Fig. 1) potentiate neural activity in cultured hippocampal neurons (i.e. TLQP-62 and AQEE-30 [1]), hippocampal slices, (i.e. TLQP-62 [9]), and dorsal horn neurons (i.e. TLQP-62 [28]). Furthermore, intrathecal administration of C-terminal VGF peptides results in thermal hyperalgesia (i.e. AQEE-30, [35]) as well as mechanical and cold hypersensitivity (i.e. TLQP-62 [28]). VGF is expressed in sensory neurons as well as spinal dorsal horn neurons and is rapidly upregulated following peripheral inflammation and nerve injury [28,35]. The significance of VGF as a contributor to chronic pain is highlighted by a meta-analysis of twenty independent microarray studies of neuropathic and inflammatory pain models [25]. Out of 2254 genes considered, VGF was among 7 found to be upregulated in more than 1/3 of the twenty studies. Of those 7 genes, VGF was 1 of 3 (along with Neuropeptide Y and Cathepsin S) that were linked to pain mechanisms by causational evidence. Taken together, these observations suggest that VGF and VGF-derived peptides are likely to contribute critically to the mechanisms underlying persistent pain states.
Fig. 1.
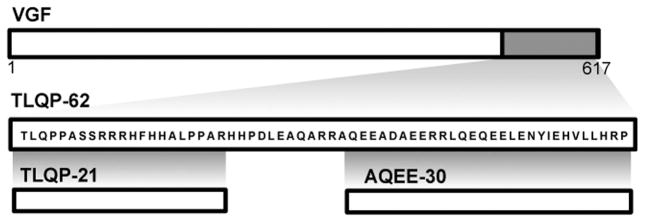
Diagram of VGF and the peptides TLQP-62, TLQP-21, and AQEE-30, which are generated from proteolytic processing of the C-terminus of VGF.
In addition to the C-terminal peptides, processing of TLQP-62 can yield the peptide TLQP-21 (Fig. 1). Following its identification from brain extracts [3], TLQP-21 has been associated with regulation of a number of physiological processes and behaviors, including feeding, energy expenditure, lipolysis, pancreatic hormone secretion, stress, and neuroprotection [3,4,31,32,34,37,39]. Furthermore, exogenous intradermal and intracerebroventricular injection of TLQP-21 respectively increased or decreased nocifensive behavior in the formalin test [36]. However, it is unknown whether the TLQP-21 peptide participates in pain signaling at the level of the spinal cord. Furthermore, the role of endogenously produced TLQP-21 has not been evaluated. We hypothesized that exogenous TLQP-21 has spinal pro-nociceptive effects and that the endogenous peptide contributes to the spinal mechanisms underlying persistent pain.
METHODS
All work with animals adhered to the guidelines of the Committee for Research and Ethical Issues of IASP and was approved by the Institutional Animal Care and Use Committees of the University of Minnesota and Mount Sinai School of Medicine.
Anti-TLQP21 antiserum
The generation of anti-TLQP21 antisera (anti-TLQP21) has been previously described [32]. Briefly, TLQP-21 was conjugated to bovine thyroglobulin (Sigma, St. Louis, Missouri) using glutaraldehyde. The conjugate (1 mg/mL) was emulsified with an equal volume of Freund’s adjuvant (Difco, Detroit, MI) and injected into female New Zealand rabbits (n=3, Harlan, Indianapolis) at two-week intervals (1 mg peptide for initial and 0.5 mg peptide for subsequent immunizations). Protein A purification of the serum was carried out using Pierce Protein A columns according to manufacturer’s instructions (Thermo Fisher Scientific, Rockford, IL). The protein concentration of the resultant IgG fraction was determined using the BCA assay (Thermo Fisher Scientific). Please refer to Results and Fig. 3 for details on the characterization of anti-TLQP21 IgG.
Fig. 3.

Characterization of immunoneutralizing anti-TLQP21 (anti-TL21). A, Intrathecal administration of anti-TLQP21, but not normal rabbit IgG (RbIgG), 5 min prior to TLQP-21 injection attenuated TLQP-21 induced thermal hyperalgesia (*p < 0.05, #p < 0.0001; Two-Way ANOVA for repeated measured, Bonferroni post-hoc test, n = 6 per group). B, Time course of the inhibition of TLQP-21 thermal hyperalgesia by anti-TLQP21. TLQP-21 was injected intrathecally in separate groups of mice at different time points after intrathecal pre-treatment with anti-TLQP21 (5 min, 1 h, 2 h, 4 h, 6 h, 24 h). Thermal hyperalgesia was measured 90 min after TLQP-21 injection (within the period of maximal hyperalgesia). The inhibitory effects of anti-TLQP21 declined rapidly after approximately 3 h. C and D, Anti-TLQP21 pre-treatment did not affect thermal hyperalgesia induced by AQEE-30 (C) or TLQP-62 (D). E and F, Dot blot analysis of the specificity of Protein-A-purified anti-TLQP21 demonstrated that anti-TLQP21 recognizes the cognate peptide TLQP-21, but not the unprocessed C-terminal parent peptide TLQP-62 or its C-terminal product AQEE-30. G and H, Anti-TLQP21 labeled DRG from wild type mice, but not mice expressing C-terminally truncated VGF. A–D, n = 3–6 mice per group.
Dot blot analysis
Peptides were blotted on PVDF-FL membranes. The membranes were allowed to dry completely, then wetted in methanol, rinsed in water and PBS and incubated in blocking buffer (Odyssey, Li-Cor Biosciences) for 1 h, followed by primary antibody overnight, washed 3 × 15 min in phosphate buffered saline (PBS), incubated in secondary antibody for 1 h, (IRDye800 Goat anti-Rabbit, Li-Cor Biosciences), washed 3 × 15 min, and imaged (Odyssey Imaging, System, Li-Cor Biosciences). The relative intensity of labeling was measured using ImageJ.
Transgenic mice
Humanized VGF knock-in mouse lines were generated by modifying a previously described targeting construct [20], using mouse Vgf genomic sequences [20] and human BAC (ImaGenes GmbH clone RZPDB737B0725D; B-Bridge International, Mountain View, CA) and VGF genomic clones (gift of A. Levi, U. of Rome) [10]. The human VGF coding sequence was contained on a single exon flanked by loxP sites. A selectable PGK-neo cassette [flanked by FLP recombinase target (FRT) and loxP sites], derived from plasmid PGKneoF2L2DTA (P Soriano; Addgene), was inserted 1.2 kb 3′ to the Vgf polyadenylation signal. A 2.2 kb Sfi 1-Sph 1 fragment that contained human VGF coding, 5′ UTR, and 3′UTR sequences, replaced comparable mouse Vgf coding, 5′ UTR, and 3′UTR sequences, which comprised a 2.3 kb Kpn 1-Xba1 fragment. Inserted human sequences encoded a truncated human VGF protein (amino acids 1-524), lacking several bioactive C-terminal peptides. This was created by introducing a reported VGF single nucleotide polymorphism (SNP) (rs35400704), which resulted in the formation of a stop codon. Human VGF1-524 (SNP) targeting construct was electroporated into 129Sv/J-derived R1 ES cells by Mount Sinai’s Mouse Genetics and Gene Targeting Core Facility [20]. G418-resistant clones were selected and screened for homologous recombination by Southern analysis. Correctly targeted clones were expanded, and injected into C57BL/6 blastocysts to generate chimeras, and germline transmission was obtained in two founders, each derived from an independent targeted clone. Male chimeras were mated with C57BL6/J females to produce F1 breeders and experiments were performed on N2F1 mixed background mice.
Immunohistochemistry
Mice were deeply anesthetized with isoflurane and perfused via the heart with calcium-free Tyrode’s solution (in mM: 116 NaCl, 5.4 KCl, 1.6 MgCl2·6H20, 0.4 MgSO4·7H2O, 1.4 NaH2PO4, 5.6 glucose, and 26 Na2HCO3) followed by fixative (4% paraformaldehyde and 0.2% picric acid in 0.1 M phosphate buffer, pH 6.9), and finally with 10% sucrose in PBS. Tissues were dissected, incubated in 10% sucrose overnight at 4 °C, and then cryostat-sectioned (14 μm) and thaw-mounted onto gelatin-coated slides. Sections were preabsorbed in blocking buffer (PBS containing 0.3% Triton-X 100, 1% BSA, 1% normal donkey serum) for 30 min, incubated in primary antibodies overnight at 4°C, rinsed with PBS 3 × 10 min, incubated in secondary antisera (1:300, Jackson ImmunoResearch, West Grove, CA) for 1 h at room temperature, washed with PBS, and coverslipped using PBS/glycerol containing 0.1% p-phenylenediamine (Sigma). Primary antibodies: Protein A-purified rabbit anti-TLQP21 (1:1000), rabbit anti-Iba1 (1:1000; Wako Chemicals USA).
Intrathecal injections
Peptides, inhibitors, or IgG were administered intrathecally (i.t.) by direct lumbar puncture in conscious adult male ICR/CD-1 outbred mice (20–25 g) as described [14,23]. The mice were gently gripped by the iliac crest, and a 30-gauge, 0.5 inch needle connected to a 50-μL Luer-hub Hamilton syringe was used to deliver 5 μL of injectate in the subarachnoid space over 1–3 seconds at the level of the cauda equina between the L5/L6 vertebrae. The duration of the procedure was approximately 15–30 s per mouse. All injections were performed by experimenters with fifteen-twenty years of experience with the technique.
Thermal hyperalgesia
Tail flick (TF) latency data in response to warm water immersion (49 °C) were collected on all mice prior to intrathecal injections and at selected time points post-injection. Tail withdrawal latencies were measured with a stopwatch by an experimenter blind to experimental group. Inhibitors or IgG were administered i.t. as a 5 min pretreatment. The data were analyzed by calculating delta TF values (Experimental TF latency - Baseline TF latency). For all experiments n = 4–6 mice/group. The following inhibitors were used: SB202190 for p38 MAPK (0.1 and 1 nmol/5 μL; Tocris); indomethacin for cyclooxygenase (COX) (0.1 and 1 nmol/5 μL; Sigma); AA-861 for 5-lipoxygenase (0.95 and 9.5 1 pmol/5 μL; Enzo Life Sciences). SB202190 was dissolved in DMSO and diluted to a final concentration of 5% DMSO with 0.9% normal saline, pH 7.2. Indomethacin was dissolved in 0.9% NaCl with sodium carbonate (1mg/mL). AA-861 was dissolved in DMSO and diluted to 2% DMSO in 5% beta-cyclodextrin; further dilutions were made in 0.9% NaCl. Inhibitor doses were selected based on published reports [33,35,44]. The animals were randomly assigned to each experimental treatment.
Mechanical sensory assessment
Mechanical responses were assessed using an electronic von Frey anesthesiometer (IITC Life Sciences, Woodland Hills, USA). Mice were placed in glass enclosures on an elevated mesh screen and allowed to acclimate for 15–30 minutes. The probe was gently applied to the plantar surface of each hindpaw until a brisk withdrawal response terminated application of pressure, which was recorded by the instrument.
Thermal Sensory Assessment
Mice were placed on a glass platform (Plantar Analgesia Meter, IITC, Life Sciences, Inc. Woodland Hills, CA) in plastic enclosures and allowed to acclimate. Radiant heat was applied to the plantar surface and the latency to hindpaw withdrawal from the heat was recorded by the instrument.
Models of persistent pain
All experiments were conducted by experimenters blinded to treatment and were replicated at least once. Outcomes were consistent. Before behavioral testing, animals were allowed to acclimate to the test environment for 20 min.
Complete Freund’s adjuvant (CFA) inflammation
Mice were injected in the intraplantar region of one hindpaw with 30 μL of a 50% solution of CFA in PBS. Control animals were injected with PBS only. Mechanical thresholds were measured at 1, 2, 3, 4, 5, 6, and 24 h post-CFA. In the same animals, thermal withdrawal latencies were measured at 6 and 24 h post-CFA. Von Frey testing was completed first followed by radiant heat application to the hindpaws.
Spared Nerve Injury (SNI)
SNI was induced in mice according to the method described by Bourquin and colleagues [8]. The left sciatic nerve and its three terminal branches were exposed under isoflurane anesthesia. The common peroneal and tibial nerves were ligated with a 5.0 silk suture and sectioned distal to the ligation, removing 2–4 mm of the distal nerve stump. In sham animals the nerve was exposed but not manipulated. For pretreatment experiments, mechanical withdrawal thresholds were measured before the surgeries and at 2, 6, 14, and 21 days after the surgeries. For post-treatment experiments, mechanical withdrawal thresholds were measured before the surgeries and on day 12 both before and 1, 2, 3, and 24 h after experimental treatments.
Data analysis
Statistical analysis was performed using GraphPad Prism 5 software. Comparisons were made using One-Way ANOVA or Two-Way ANOVA for repeated measures, followed by Bonferroni post-hoc test. The p-values and specific tests used are indicated in the figure legends or the Results section.
RESULTS
Spinal effects of TLQP-21 and characterization of anti-TLQP21 immunoneutralizing antisera
To determine the spinal effects of TLQP-21, the peptide was injected intrathecally in naïve adult mice (Fig. 2). In the warm water (49 °C) tail-immersion assay, TLQP-21 evoked dose-dependent thermal hyperalgesia that peaked 60 to 90 min after the intrathecal injection and persisted for nearly two hours. TLQP-21 evoked hyperalgesia was blocked by inhibition of MAPK p38 as well as the eicosanoid producing enzymes cyclooxygenase and lipoxygenase.
Fig. 2.

Spinal effects of exogenous TLQP-21 peptide. A, Intrathecal injection of TLQP-21 induced dose-dependent thermal hyperalgesia in the warm water immersion tail withdrawal test. B–D, The p38 MAPK inhibitor SB202190 (B), the cyclooxygenase (COX) inhibitor indomethacin (C), and 5-lipoxygenase inhibitor AA861 (D) attenuated thermal hyperalgesia evoked by TLQP2-1 (3 nmol) in a dose-dependent manner (measured 90 min after i.t. injection of TLQP-21; p < 0.05, One-Way ANOVA, Bonferroni post-hoc test). A–D, n = 3–6 mice per group.
TLQP21-evoked thermal hyperalgesia was used as an assay for behavioral characterization of TLQP-21 immunoneutralizing antisera (anti-TLQP21). Equivalent doses of anti-TLQP21 or normal rabbit IgG were injected intrathecally 5 min prior to intrathecal injection of the TLQP-21 peptide (Fig. 3A). Pretreatment with anti-TLQP21, but not normal rabbit IgG, inhibited the development of TLQP-21 thermal hyperalgesia. We assessed the ability of anti-TLQP21 to interfere with pronociceptive effects of other VGF-derived peptides. Pretreatment with anti-TLQP21 did not affect thermal hyperalgesia induced by intrathecally delivered AQEE-30 (Fig. 3C) or the extended peptide TLQP-62 (Fig. 3D), supporting the selectivity of anti-TLQP21 for TLQP-21 over related VGF peptides. In order to rule out a general inhibition of spinal nociception by anti-TLQP21, we assessed its effects on sensory responses evoked by non-VGF related agents. Pretreatment with anti-TLQP21 did not impact thermal hyperalgesia evoked by intrathecally delivered NMDA (0.3 nmol; change in tail withdrawal latency: saline-pretreated mice = −2.92+/−0.16 s; anti-TLQP21 pretreated mice = −3.12+/−0.26 s; n = 4; p > 0.5; t-test) or the behavioral effects of intrathecally injected Substance P (10 nmol; number of nocifensive responses: saline-pretreated mice = 47+/−2; anti-TLQP21 pretreated mice = 45.25+/−2.6; n = 4; p > 0.5; t-test). The duration of the imunoneutralizing activity of intrathecally delivered anti-TLQP21 was evaluated by injecting the TLQP-21 peptide at increasing intervals after IgG pretreatment in separate groups of mice (Fig. 3B). The inhibitory effects of anti-TLQP21 on hyperalgesia induced by a 3 nmol dose of TLQP-21 diminished substantially when the interval between IgG and peptide injection exceeded 4 h. Similar results were obtained with 1 nmol TLQP-21 (data not shown). Finally, to test for a potential impact of anti-TLQP21 on acute nociception, the IgG was delivered prior to application of progressively increasing thermal stimuli. Pretreatment with anti-TLQP21 did not affect tail withdrawal latencies at a range of warm water temperatures (change in tail withdrawal latency of saline-pretreated mice and anti-TLQP21 pretreated mice: 0.01+/−0.2 s and 0.03+/−0.15 s at 49 °C; 0.17+/−0.19 s and 0.16+/−0.15 s at 52.5 °C; 0.16+/−0.12 s and 0.27+/−0.26 s at 55 °C; n = 4; p > 0.5; t-test).
Consistent with the selective inhibition of TLQP-21 evoked hyperalgesia, in dot blot analysis, anti-TLQP21 dose-dependently bound the TLQP-21 peptide, but not the extended peptide TLQP-62 or its C-terminal component AQEE-30 (Fig. 3E and F). Anti-TLQP21 was also characterized by immunohistochemistry using dorsal root ganglia from wild type mice and mice expressing a truncated form of VGF that lacks the C-terminal sequence encoding TLQP-62). Anti-TLQP21 immunolabeling was present in wild-type mouse sensory neurons, but not in sensory neurons of mice expressing C-terminally truncated VGF protein (Fig. 3G and H). Thus the selectivity of anti-TLQP21 was confirmed by behavioral assay, dot blot analysis, and immunohistochemistry.
Immunoneutralization of TLQP-21 attenuates inflammatory hypersensitivity
Based on the pro-nociceptive effects of exogenous TLQP-21, we hypothesized that the endogenous peptide is involved in inflammatory hypersensitivity. To test this hypothesis, mice were injected intrathecally with immunoneutralizing anti-TLQP21, normal rabbit IgG or saline immediately preceding or 5 h after intraplantar injection of Complete Freund’s Adjuvant (CFA) (Fig. 4). CFA-induced tactile hypersensitivity was detected 1 h after the intraplantar injection, and its magnitude remained constant for the duration of the 24 h testing period for the saline- and normal rabbit IgG-pretreated goups (Fig. 4A). While intrathecal pretreatment with anti-TLQP21 had no effect on inflammatory tactile hypersensitivity at 1 h post-CFA, at subsequent time points the magnitude of hypersensitivity of anti-TLQP21 treated mice was significantly reduced compared to the control pretreatments. Significant attenuation of thermal hyperalgesia by anti-TLQP21 pretreatment was observed at 6 and 24 h post-CFA (Fig. 4B). It is noteworthy that thermal hyperalgesia was asessed only at 6 h and 24 h to avoid potential stimulus-induced sensitization and tissue injury (Fig. 4B). The immunoneutralization effects on both tactile and thermal responses were dose-dependent (Fig. 4C and D). We also assessed whether TLQP-21 immunoneutralization inhibits established inflammatory hypersensitivity. When anti-TLQP21 was injected intrathecally 5 h after intradermal CFA injection, tactile hypersensitivity and thermal hyperalgesia were significantly attenuated at the 24 h time point (Fig. 4E and F).
Fig. 4.

Effects of anti-TLQP21 on inflammation-induced hypersensitivity. A and B, Intrathecal pretreatment with anti-TLQP21 attenuated the development of tactile hypersensitivity (A) and thermal hyperalgesia (B) after intraplantar injection of Complete Freund’s Adjuvant (CFA). Mice were injected i.t. with saline, anti-TLQP21 (150 ng/5 μL), or normal rabbit IgG (NR-IgG, 150 ng/5 μL) immediately prior to CFA injection. Control mice injected intradermally with saline received saline intrathecal pretreatment (Saline, Saline). Ipsilateral to CFA, the mechanical withdrawal threshold (A) and thermal withdrawal latency (B) of anti-TLQP21-pretreated CFA mice, but not mice pretreated with normal rabbit IgG, were significantly different from those of saline-pretreated CFA mice (* p < 0.05, ** p < 0.01, # p < 0.001, ## p < 0.0001; Two-Way ANOVA for repeated measures, Bonferroni post-hoc test; mean +/− SE, n = 4 mice per group for NR-IgG group, n = 8 for all other groups). C and D, The inhibition of the development of CFA-induced tactile hypersensitivity (C) and thermal hyperalgesia (D) by anti-TLQP21 was dose-dependent. E and F, Treatment with anti-TLQP21 attenuated established tactile hypersensitivity and thermal hyperalgesia. Five hours (grey arrows) after intraplantar CFA injection, mice were injected intrathecally with saline, anti-TLQP21 (150 ng/5 μL), or normal rabbit IgG (150 ng/5 μL). Ipsilateral to CFA, the mechanical withdrawal threshold (E) and thermal withdrawal latency (F) of anti-TLQP21-treated, but not normal rabbit IgG-treated CFA mice were significantly different from those of saline-treated CFA mice 24 h post-CFA (E: p < 0.0001; F: p < 0.001; Two-Way ANOVA for repeated measures, Bonferroni post-hoc test; mean +/− SE, n = 4 per group).
Immunoneutralization of TLQP-21 attenuates nerve injury-induced hypersensitivity
We used the SNI model of neuropathic pain to evaluate the contribution of endogenous TLQP-21 peptide to the development and maintenance of nerve injury-induced hypersensitivity. Intrathecal administration of anti-TLQP21, but not normal rabbit IgG, within 2 h prior to SNI resulted in significant decrease in the magnitude of nerve injury-induced hypersensitivity at day 2 post-SNI (Fig. 5A). Anti-TLQP21 did not alter the withdrawal thresholds of sham-operated mice. The attenuation of tactile hypersensitivity by the single pretreatment dose persisted for two weeks (Fig. 5B). We also examined the effects of TLQP-21 immunoneutralization on established tactile hypersensitivity. Anti-TLQP21 or normal rabbit IgG were delivered intrathecally at day 12 post-SNI and withdrawal thresholds were measured 1, 2, 3, and 24 h after treatment (Fig. 5C). Tactile hypersensitivity was significantly attenuated 3 h after anti-TLQP21 treatment and was still suppressed at 24 h. Post-treatment with anti-TLQP21 did not affect the thresholds of sham-operated mice (data not shown).
Fig. 5.

Effects of anti-TLQP21 on nerve injury-induced hypersensitivity. A, Intrathecal pretreatment with anti-TLQP21 attenuated the development of tactile hypersensitivity after spared nerve injury (SNI). Mice were injected with saline, anti-TLQP21 (anti-TL21, 150 ng/5 μL), or normal rabbit IgG (NR-IgG, 150 ng/5 μL) immediately prior to SNI or sham surgery. On day 2 after surgery, the mechanical withdrawal thresholds of anti-TLQP21 pretreated but not NR-IgG pretreated, SNI mice were significantly different from those of saline pretreated SNI mice. Anti-TLQP21 did not alter the thresholds of sham operated mice compared to saline pretreated sham mice (Saline pretreated SNI vs anti-TLQP21 pretreated SNI, p < 0.0001; Saline pretreated sham vs anti-TLQP21 pretreated sham, p > 0.05; Two-Way ANOVA for repeated measures, Bonferroni post-hoc test; mean +/− SE, n = 7–9 per group). B, Time course of changes in hypersensitivity after pretreatment with anti-TLQP21. Ipsilateral to SNI, the mechanical withdrawal thresholds of anti-TLQP21-pretreated SNI mice, but not mice pretreated with normal rabbit IgG, were significantly different from those of saline-pretreated SNI mice for up to 14 days after nerve injury (Day 2, p < 0.01; Days 6 and 14, p < 0.05; Two-Way ANOVA for repeated measures, Bonferroni post-hoc test; mean +/− SE, n = 4 per group). C, Treatment with anti-TLQP21 attenuated established hypersensitivity. Twelve days after SNI, mice were injected intrathecally with saline, anti-TLQP21 (150 ng/5 μL), or normal rabbit IgG (150 ng/5 μL). The mechanical withdrawal thresholds of anti-TLQP21-treated, but not normal rabbit IgG-treated SNI mice were significantly different from those of saline-treated SNI mice 3 and 24 h after injection (p < 0.0001 and p < 0.05, respectively; Two-Way ANOVA for repeated measures, Bonferroni post-hoc test; mean +/− SE, n = 4 per group).
DISCUSSION
The results from the present study demonstrate spinal pro-nociceptive actions of the VGF-derived peptide TLQP-21 and, importantly, provide evidence that the endogenous peptide participates in mechanisms associated with the development and maintenance of persistent pain. The recent identification of the complement 3a receptor (C3aR1) as a target of TLQP-21 underscores the significance of the current findings [21]. C3AR1 is expressed in a number of immune cells, including microglia, and has been implicated in a spectrum of immunomodulatory processes and in CNS neuroinflammation (e.g. experimental autoimmune encephalitis) [7]. Therefore, increased levels of TLQP-21 following tissue damage may contribute to neuroimmune modulation of spinal neuroplasticity. In addition, there is evidence for C3AR1 expression in spinal neurons [13], suggesting that TLQP-21 may directly influence neuronal activity under conditions of persistent pain. TLQP-21 has also been reported to bind to gC1qR, the globular heads of the complement receptor C1q, and this interaction has been implicated in peripheral mechanisms of nerve injury-induced hypersensitivity [11]. The relative contribution of the two complement receptors, C3aR1 and gC1qR, to the physiological effects of TLQP-21 is presently unclear, and the expression or potential functions of gC1qR in spinal cord or sensory neurons have not been investigated.
Pro-nociceptive spinal effects of exogenous TLQP-21
Similar to the C-terminal VGF peptides AQEE-30 and LQEQ-19 [35], intrathecal injection of TLQP-21 induced p38 MAPK-dependent thermal hyperalgesia in the warm-water tail withdrawal assay. While the magnitude of TLQP-21 hyperalgesia was similar to that of the C-terminal peptides, the time course appeared to be delayed, suggesting perhaps activation of distinct signaling mechanisms upstream of p38. The inhibition of TLQP-21 hyperalgesia by a p38 MAPK inhibitor is consistent with microglial activation [27,40,41]. Furthermore, release of prostaglandin E2 (PGE2) by cultured spinal microglia following stimulation with lipopolysaccharide (LPS) has been shown to require p38 activation [27]. Since TLQP-21 hyperalgesia was inhibited by the cyclooxygenase inhibitor indomethacin, it is possible that the effects of TLQP-21 are mediated by p38-dependent release of prostaglandins from microglia. However, p38, its substrate phospholipase A2 (which generates the substrate of COX, arachidonic acid), as well as COX are also found in dorsal horn neurons; we therefore cannot rule out the possibility that the thermal hyperalgesia is mediated by direct neuronal effects of TLQP-21 [18,24,40]. Finally, our results showing attenuation of hyperalgesia by an inhibitor of 5-lipoxygenase (Fig. 2) suggest that this pathway, which leads to leukotriene production, also contributes to exogenous TLQP-21 pro-nociceptive effects. Leukotrienes have been previously implicated in spinal nociceptive signaling [30].
Immunoneutralization of endogenous TLQP-21
We employed immunoneutralization to examine the role of endogenous spinal TLQP-21 in models of persistent pain. The extensive characterization of anti-TLQP21 strongly supports its specificity. Our working hypothesis is that the IgG neutralizes endogenous TLQP-21 released into the spinal cord parenchyma from primary afferent terminals or intrinsic neurons [28,35]. However, since the expression of the proposed TLQP-21 receptor C3aR1 in spinal cord is not well characterized, we cannot rule out the possibility that anti-TLQP21 acts at other sites. Alternative sites of action include dorsal root ganglia, where VGF peptides may participate in communication between neurons and satellite cells [17,45], or the subdural vasculature, which has been implicated in neuroinflammatory processes [2,38].
Endogenous TLQP-21 contributes to inflammatory hypersensitivity
In the CFA model of inflammatory pain, anti-TLQP21 reduced tactile hypersensitivity and thermal hyperalgesia when administered intrathecally both prior and subsequent to the CFA intraplantar injection. Notably, the magnitude of tactile hypersensitivity was not affected by anti-TLQP21 pretreatment at 1 h post-CFA, but subsequently diminished significantly. Our data also suggest that in this model TLQP-21 may contribute differently to tactile hypersensitivity and thermal hyperalgesia, as the former appeared to be more affected by anti-TLQP21 treatment, potentially reflecting distinct roles of TLQP-21 in the spinal processing of mechanical and thermal stimuli. Taken together these observations support a role of endogenous TLQP-21 in the mechanism underlying persistent inflammatory pain. We have previously reported upregulation of VGF in sensory neurons following CFA inflammation [35]; the source of spinal endogenous TLQP-21 under inflammatory conditions is therefore likely to be sensory neurons. The involvement of p38 and prostanoids in the effects of exogenous TLQP-21 suggests that a p38-dependent increase in prostanoids may contribute to the mechanism by which endogenous TLQP-21 participates in inflammatory hypersensitivity [12,16,29,40–42].
Endogenous TLQP-21 contributes to nerve injury-induced hypersensitivity
The present results also provide evidence for a role of endogenous TLQP-21 in both the development and maintenance of nerve injury-induced hypersensitivity. A single pretreatment dose of anti-TLQP21 delayed the full expression of SNI-induced tactile hypersensitivity for more than two weeks. Based on the short duration of anti-TLQP21 immunoneutralizing activity against exogenously delivered peptide (Fig. 3B), this finding suggests that the pretreatment disrupts a critical neuroplasticity event that occurs within the first several hours of nerve injury. However, we cannot rule out the possibility that low levels of anti-TLQP21, sufficient to neutralize endogenously released peptide, persist in the spinal cord parenchyma for an extended period of time. Intrathecal administration of anti-TLQP21 after the establishment of hypersensitivity was also effective in reducing its magnitude, suggesting that TLQP-21-mediated signaling pathways remain engaged during the maintenance of neuropathic pain. The rapid and robust sensory neuron upregulation of VGF, the precursor protein from which TLQP-21 is generated, suggests that TLQP-21 release in dorsal horn may be elevated following nerve injury. The identification of C3aR1 as a receptor for TLQP-21 raises the possibility that TLQP-21 functions as an injury signal that contributes to microglial activation. It remains to be determined whether specific signaling components within activated microglia, such as P2X4, cathepsin S, or BDNF [6], are altered after TLQP-21 immunoneutralization.
Summary
These studies provide the first functional evidence for involvement of an endogenous VGF peptide in mechanisms of persistent pain and support the idea that VGF-derived peptides contribute critically to spinal neuroplasticity after inflammation and nerve injury. Their participation in both the development and maintenance of persistent hypersensitivity is consistent with a role of the VGF signaling system in the transition to chronic pain. Further elucidation of this role requires understanding of the relationship of TLQP-21 to the other C-terminal VGF peptides known to modulate pain processing and characterization of their cellular and molecular targets as well as site(s) of synthesis.
Summary.
The endogenous VGF-derived peptide TLQP-21 contributes to the development and maintenance of inflammatory and nerve injury-induced hypersensitivity.
Acknowledgments
The authors would like to thank Galina Kalyuzhnaya for technical assistance, and Drs. Christopher N. Honda, George Wilcox, and Robert Elde for valuable insights. Grant Support: R01 DE021996 (LV and SRS), T32 RR018719 (JAD), RO1 MH086499 (SRS), R33 MH083496 (SRS), Diabetes Action Research and Education Foundation (SRS), NARSAD van Ameringen Investigator Award (SRS), Hope for Depression Research Foundation (SRS).
Footnotes
There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alder J, Thakker-Varia S, Bangasser DA, Kuroiwa M, Plummer MR, Shors TJ, Black IB. Brain-derived neurotrophic factor-induced gene expression reveals novel actions of VGF in hippocampal synaptic plasticity. J Neurosci. 2003;23(34):10800–10808. doi: 10.1523/JNEUROSCI.23-34-10800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arima Y, Harada M, Kamimura D, Park JH, Kawano F, Yull FE, Kawamoto T, Iwakura Y, Betz UA, Marquez G, Blackwell TS, Ohira Y, Hirano T, Murakami M. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell. 2012;148(3):447–457. doi: 10.1016/j.cell.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 3.Bartolomucci A, La Corte G, Possenti R, Locatelli V, Rigamonti AE, Torsello A, Bresciani E, Bulgarelli I, Rizzi R, Pavone F, D’Amato FR, Severini C, Mignogna19 G, Giorgi A, Schinina ME, Elia G, Brancia C, Ferri GL, Conti R, Ciani B, Pascucci T, Dell’Omo G, Muller EE, Levi A, Moles A. TLQP-21, a VGF-derived peptide, increases energy expenditure and prevents the early phase of diet-induced obesity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(39):14584–14589. doi: 10.1073/pnas.0606102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartolomucci A, Moles A, Levi A, Possenti R. Pathophysiological role of TLQP-21: gastrointestinal and metabolic functions. Eat Weight Disord. 2008;13(3):e49–54. [PubMed] [Google Scholar]
- 5.Bartolomucci A, Possenti R, Mahata SK, Fischer-Colbrie R, Loh YP, Salton SR. The extended granin family: structure, function, and biomedical implications. Endocr Rev. 2011;32(6):755–797. doi: 10.1210/er.2010-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beggs S, Trang T, Salter MW. P2X4R+ microglia drive neuropathic pain. Nature neuroscience. 2012;15(8):1068–1073. doi: 10.1038/nn.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boos L, Campbell IL, Ames R, Wetsel RA, Barnum SR. Deletion of the complement anaphylatoxin C3a receptor attenuates, whereas ectopic expression of C3a in the brain exacerbates, experimental autoimmune encephalomyelitis. J Immunol. 2004;173(7):4708–4714. doi: 10.4049/jimmunol.173.7.4708. [DOI] [PubMed] [Google Scholar]
- 8.Bourquin AF, Suveges M, Pertin M, Gilliard N, Sardy S, Davison AC, Spahn DR, Decosterd I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain. 2006;122(1–2):14, e11–14. doi: 10.1016/j.pain.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 9.Bozdagi O, Rich E, Tronel S, Sadahiro M, Patterson K, Shapiro ML, Alberini CM, Huntley GW, Salton SR. The neurotrophin-inducible gene Vgf regulates hippocampal function and behavior through a brain-derived neurotrophic factor-dependent mechanism. J Neurosci. 2008;28(39):9857–9869. doi: 10.1523/JNEUROSCI.3145-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canu N, Possenti R, Ricco AS, Rocchi M, Levi A. Cloning, structural organization analysis, and chromosomal assignment of the human gene for the neurosecretory protein VGF. Genomics. 1997;45(2):443–446. doi: 10.1006/geno.1997.4945. [DOI] [PubMed] [Google Scholar]
- 11.Chen YC, Pristera A, Ayub M, Swanwick RS, Karu K, Hamada Y, Rice AS, Okuse K. Identification of a receptor for neuropeptide VGF and its role in neuropathic pain. The Journal of biological chemistry. 2013;288(48):34638–34646. doi: 10.1074/jbc.M113.510917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng HT, Suzuki M, Hegarty DM, Xu Q, Weyerbacher AR, South SM, Ohata M, Inturrisi CE. Inflammatory pain-induced signaling events following a conditional deletion of the N-methyl-D-aspartate receptor in spinal cord dorsal horn. Neuroscience. 2008;155(3):948–958. doi: 10.1016/j.neuroscience.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davoust N, Jones J, Stahel PF, Ames RS, Barnum SR. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia. 1999;26(3):201–211. doi: 10.1002/(sici)1098-1136(199905)26:3<201::aid-glia2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 14.Fairbanks CA. Spinal delivery of analgesics in experimental models of pain and analgesia. Adv Drug Deliv Rev. 2003;55(8):1007–1041. doi: 10.1016/s0169-409x(03)00101-7. [DOI] [PubMed] [Google Scholar]
- 15.Ferri GL, Noli B, Brancia C, D’Amato F, Cocco C. VGF: an inducible gene product, precursor of a diverse array of neuro-endocrine peptides and tissue-specific disease biomarkers. J Chem Neuroanat. 2011;42(4):249–261. doi: 10.1016/j.jchemneu.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Fitzsimmons BL, Zattoni M, Svensson CI, Steinauer J, Hua XY, Yaksh TL. Role of spinal p38alpha and beta MAPK in inflammatory hyperalgesia and spinal COX-2 expression. Neuroreport. 2010;21(4):313–317. doi: 10.1097/WNR.0b013e32833774bf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu ES, Zhang YP, Sagen J, Candiotti KA, Morton PD, Liebl DJ, Bethea JR, Brambilla R. Transgenic inhibition of glial NF-kappa B reduces pain behavior and inflammation after peripheral nerve injury. Pain. 2010;148(3):509–518. doi: 10.1016/j.pain.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghilardi JR, Svensson CI, Rogers SD, Yaksh TL, Mantyh PW. Constitutive spinal cyclooxygenase-2 participates in the initiation of tissue injury-induced hyperalgesia. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24(11):2727–2732. doi: 10.1523/JNEUROSCI.5054-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahm S, Fekete C, Mizuno TM, Windsor J, Yan H, Boozer CN, Lee C, Elmquist JK, Lechan RM, Mobbs CV, Salton SR. VGF is required for obesity induced by diet, gold thioglucose treatment, and agouti and is differentially regulated in proopiomelanocortin- and neuropeptide Y-containing arcuate neurons in response to fasting. J Neurosci. 2002;22(16):6929–6938. doi: 10.1523/JNEUROSCI.22-16-06929.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hahm S, Mizuno TM, Wu TJ, Wisor JP, Priest CA, Kozak CA, Boozer CN, Peng B, McEvoy RC, Good P, Kelley KA, Takahashi JS, Pintar JE, Roberts JL, Mobbs CV, Salton SR. Targeted deletion of the Vgf gene indicates that the encoded secretory peptide precursor plays a novel role in the regulation of energy balance. Neuron. 1999;23(3):537–548. doi: 10.1016/s0896-6273(00)80806-5. [DOI] [PubMed] [Google Scholar]
- 21.Hannedouche S, Beck V, Leighton-Davies J, Beibel M, Roma G, Oakeley EJ, Lannoy V, Bernard J, Hamon J, Barbieri S, Preuss I, Lasbennes MC, Sailer AW, Suply T, Seuwen K, Parker CN, Bassilana F. The identification of the C3a Receptor (C3AR1) as the target of the VGF derived peptide TLQP-21 in rodent cells. The Journal of biological chemistry. 2013 doi: 10.1074/jbc.M113.497214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunsberger JG, Newton SS, Bennett AH, Duman CH, Russell DS, Salton SR, Duman RS. Antidepressant actions of the exercise-regulated gene VGF. Nat Med. 2007;13(12):1476–1482. doi: 10.1038/nm1669. [DOI] [PubMed] [Google Scholar]
- 23.Hylden JL, Wilcox GL. Intrathecal substance P elicits a caudally-directed biting and scratching behavior in mice. Brain Res. 1981;217(1):212–215. doi: 10.1016/0006-8993(81)90203-1. [DOI] [PubMed] [Google Scholar]
- 24.Kim DH, Fitzsimmons B, Hefferan MP, Svensson CI, Wancewicz E, Monia BP, Hung G, Butler M, Marsala M, Hua XY, Yaksh TL. Inhibition of spinal cytosolic phospholipase A(2) expression by an antisense oligonucleotide attenuates tissue injury-induced hyperalgesia. Neuroscience. 2008;154(3):1077–1087. doi: 10.1016/j.neuroscience.2008.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaCroix-Fralish ML, Austin JS, Zheng FY, Levitin DJ, Mogil JS. Patterns of pain: meta-analysis of microarray studies of pain. Pain. 2011;152(8):1888–1898. doi: 10.1016/j.pain.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 26.Levi A, Ferri GL, Watson E, Possenti R, Salton SR. Processing, distribution, and function of VGF, a neuronal and endocrine peptide precursor. Cell Mol Neurobiol. 2004;24(4):517–533. doi: 10.1023/B:CEMN.0000023627.79947.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsui T, Svensson CI, Hirata Y, Mizobata K, Hua XY, Yaksh TL. Release of prostaglandin E(2) and nitric oxide from spinal microglia is dependent on activation of p38 mitogen-activated protein kinase. Anesthesia and analgesia. 2010;111(2):554–560. doi: 10.1213/ANE.0b013e3181e3a2a2. [DOI] [PubMed] [Google Scholar]
- 28.Moss A, Ingram R, Koch S, Theodorou A, Low L, Baccei M, Hathway GJ, Costigan M, Salton SR, Fitzgerald M. Origins, actions and dynamic expression patterns of the neuropeptide VGF in rat peripheral and central sensory neurones following peripheral nerve injury. Mol Pain. 2008;4:62. doi: 10.1186/1744-8069-4-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narita M, Shimamura M, Imai S, Kubota C, Yajima Y, Takagi T, Shiokawa M, Inoue T, Suzuki M, Suzuki T. Role of interleukin-1beta and tumor necrosis factor-alpha-dependent expression of cyclooxygenase-2 mRNA in thermal hyperalgesia induced by chronic inflammation in mice. Neuroscience. 2008;152(2):477–486. doi: 10.1016/j.neuroscience.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 30.Okubo M, Yamanaka H, Kobayashi K, Noguchi K. Leukotriene synthases and the receptors induced by peripheral nerve injury in the spinal cord contribute to the generation of neuropathic pain. Glia. 2010;58(5):599–610. doi: 10.1002/glia.20948. [DOI] [PubMed] [Google Scholar]
- 31.Petrella C, Broccardo M, Possenti R, Severini C, Improta G. TLQP-21, a VGFderived peptide, stimulates exocrine pancreatic secretion in the rat. Peptides. 2012;36(1):133–136. doi: 10.1016/j.peptides.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 32.Possenti R, Muccioli G, Petrocchi P, Cero C, Cabassi A, Vulchanova L, Riedl MS, Manieri M, Frontini A, Giordano A, Cinti S, Govoni P, Graiani G, Quaini F, Ghe C, Bresciani E, Bulgarelli I, Torsello A, Locatelli V, Sanghez V, Larsen BD, Petersen JS, Palanza P, Parmigiani S, Moles A, Levi A, Bartolomucci A. Characterization of a novel peripheral pro-lipolytic mechanism in mice: role of VGF-derived peptide TLQP-21. Biochem J. 2012;441(1):511–522. doi: 10.1042/BJ20111165. [DOI] [PubMed] [Google Scholar]
- 33.Rady JJ, Campbell WB, Fujimoto JM. Antianalgesic action of nociceptin originating in the brain is mediated by spinal prostaglandin E(2) in mice. The Journal of pharmacology and experimental therapeutics. 2001;296(1):7–14. [PubMed] [Google Scholar]
- 34.Razzoli M, Bo E, Pascucci T, Pavone F, D’Amato FR, Cero C, Sanghez V, Dadomo H, Palanza P, Parmigiani S, Ceresini G, Puglisi-Allegra S, Porta M, Panzica GC, Moles A, Possenti R, Bartolomucci A. Implication of the VGF-derived peptide TLQP-21 in mouse acute and chronic stress responses. Behav Brain Res. 2012;229(2):333–339. doi: 10.1016/j.bbr.2012.01.038. [DOI] [PubMed] [Google Scholar]
- 35.Riedl MS, Braun PD, Kitto KF, Roiko SA, Anderson LB, Honda CN, Fairbanks CA, Vulchanova L. Proteomic analysis uncovers novel actions of the neurosecretory protein VGF in nociceptive processing. J Neurosci. 2009;29(42):13377–13388. doi: 10.1523/JNEUROSCI.1127-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rizzi R, Bartolomucci A, Moles A, D’Amato F, Sacerdote P, Levi A, La Corte G, Ciotti MT, Possenti R, Pavone F. The VGF-derived peptide TLQP-21: a new modulatory peptide for inflammatory pain. Neurosci Lett. 2008;441(1):129–133. doi: 10.1016/j.neulet.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 37.Severini C, Ciotti MT, Biondini L, Quaresima S, Rinaldi AM, Levi A, Frank C, Possenti R. TLQP-21, a neuroendocrine VGF-derived peptide, prevents cerebellar granule cells death induced by serum and potassium deprivation. Journal of neurochemistry. 2008;104(2):534–544. doi: 10.1111/j.1471-4159.2007.05068.x. [DOI] [PubMed] [Google Scholar]
- 38.Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim KW, Klein E, Kalchenko V, Bendel P, Lira SA, Jung S, Schwartz M. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013;38(3):555–569. doi: 10.1016/j.immuni.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stephens SB, Schisler JC, Hohmeier HE, An J, Sun AY, Pitt GS, Newgard CB. A VGF-Derived Peptide Attenuates Development of Type 2 Diabetes via Enhancement of Islet beta-Cell Survival and Function. Cell Metab. 2012;16(1):33–43. doi: 10.1016/j.cmet.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svensson CI, Fitzsimmons B, Azizi S, Powell HC, Hua XY, Yaksh TL. Spinal p38beta isoform mediates tissue injury-induced hyperalgesia and spinal sensitization. Journal of neurochemistry. 2005;92(6):1508–1520. doi: 10.1111/j.1471-4159.2004.02996.x. [DOI] [PubMed] [Google Scholar]
- 41.Svensson CI, Hua XY, Protter AA, Powell HC, Yaksh TL. Spinal p38 MAP kinase is necessary for NMDA-induced spinal PGE(2) release and thermal hyperalgesia. Neuroreport. 2003;14(8):1153–1157. doi: 10.1097/00001756-200306110-00010. [DOI] [PubMed] [Google Scholar]
- 42.Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86(6):1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- 43.Thakker-Varia S, Krol JJ, Nettleton J, Bilimoria PM, Bangasser DA, Shors TJ, Black IB, Alder J. The neuropeptide VGF produces antidepressant-like behavioral effects and enhances proliferation in the hippocampus. J Neurosci. 2007;27(45):12156–12167. doi: 10.1523/JNEUROSCI.1898-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trang T, Sutak M, Quirion R, Jhamandas K. Spinal administration of lipoxygenase inhibitors suppresses behavioural and neurochemical manifestations of naloxone-precipitated opioid withdrawal. Br J Pharmacol. 2003;140(2):295–304. doi: 10.1038/sj.bjp.0705440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie W, Strong JA, Zhang JM. Early blockade of injured primary sensory afferents reduces glial cell activation in two rat neuropathic pain models. Neuroscience. 2009;160(4):847–857. doi: 10.1016/j.neuroscience.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]