

Abstract
Although global proteomics has shown promise for discovery of many new proteins, biomarkers, protein modifications, and polymorphisms, targeted proteomics is emerging in the proteomics research field as a complement to untargeted shotgun proteomics, particularly when a determined set of low-abundance functional proteins need to be measured. The function and expression of proteins related to drug absorption, distribution, metabolism, and excretion (ADME) such as cytochrome P450 enzymes and membrane transporters are of great interest in biopharmaceutical research. Since the variation in ADME-related protein expression is known to be a major complicating factor encountered during in vitro–in vivo and in vivo–in vivo extrapolations (IVIVE), the accurate quantification of the ADME proteins in complex biological systems becomes a fundamental element in establishing IVIVE for pharmacokinetic predictions. In this review, we provide an overview of relevant methodologies followed by a summary of recent applications encompassing mass spectrometry-based targeted quantifications of membrane transporters.
KEY WORDS: drug absorption, distribution, metabolism, and excretion (ADME); drug transporters; in vitro–in vivo and in vivo–in vivo extrapolations (IVIVE); LC-MS/MS; quantitative targeted proteomics
INTRODUCTION
Drug transporters along with other ADME-related proteins, such as cytochrome P450 and phase II enzymes, play a pivotal role in defining the disposition of xenobiotics and their metabolites (1). Prediction of human pharmacokinetics (PK) remains an active and challenging area in drug discovery and development, and, as a result, various in vitro and in vivo preclinical models, such as physiological-based pharmacokinetics (PBPK) modeling, have been investigated for their capability to predict human parameters. In vitro systems including isolated primary hepatocytes and heterogeneous gene overexpressed cell lines have been widely used to investigate transporter-involved drug disposition; however, translation of in vitro kinetics parameters obtained from different systems or different laboratories to in vivo is not always straightforward. For example, through a collaborative effort between 23 pharmaceutical, academic, and contract research laboratories, a minimum of 20- and 24-fold deviation was found to exist between the lowest and highest P-glycoprotein (P-gp/ABCB1) IC50 values for sertraline and isradipine, respectively, to a maximum of 407- and 796-fold deviation for telmisartan and verapamil (2). The presence of digoxin uptake transporters and their differences in expression in in vitro systems used in different laboratories is considered as the major contributor to the variability of IC50 values (2,3). Moreover, IVIVE of biliary elimination that involves drug transporter-mediated processes is not well established from preclinical species to human. Either due to the lack of clinical bile samples, the IVIVE is further complicated by biliary secretion models, which are highly dependent upon in vitro and preclinical data (4,5). Collectively, the quantitative drug transporters expression is the basis to extrapolate from preclinical models to human when coupled with kinetic parameter determinations.
Proteomics, as defined by its name, is an interdisciplinary science that involves the study of large numbers of proteins. It has rapidly advanced and made tremendous impact on a variety of biological fields. Recently, mass spectrometry (MS)-based targeted proteomics with selected reaction monitoring (SRM) has become a promising tool for relative and absolute quantification of proteins (6–13). Traditionally, this SRM technique was used in conjunction with liquid chromatography (LC) to analyze small molecules such as drugs and their metabolites (14). The technique now can be considered as the mass spectrometry-equivalent of Western blot (15), to selectively target one or more peptides and corresponding transitions to get quantitative results of protein expression. In addition, the SRM approach offers several advantages over traditional antibody-based assays such as no need of specific antibodies, shorter time for method development, and higher specificity. The principle of LC-MS-based targeted proteomics is quite similar to the small molecule analysis when performed on a triple–quadrupole (QQQ) instrument. The first quadrupole selects the peptides precursor ions, which are usually doubly or triply charged protonated molecules; the selected protonated ions are then subjected to collision induced dissociation (CID) in the second quadrupole to produce peptide fragment ions; the third quadrupole selects one or several fragment ions produced from the second quadrupole; finally, the mass detector counts the number of these fragment ions that reach the detector resulting in a chromatographic trace (16). As depicted in Fig. 1, the general procedure for quantitative targeted proteomics includes the following steps: (a) selection of suitable targeted peptides that represent the protein of interest; (b) selection of SRM transitions for the targeted peptides; (c) selection of internal standards, which usually are the same peptides with heavy isotope labels; (d) LC-MS method development including optimization and validation of the selected SRM transitions and chromatography settings; (e) sample preparation, e.g., membrane protein extraction, protein enrichment and digestion; and (f) data acquisition and processing (Fig. 1). In this review, we detail the workflow of targeted quantitative proteomics and discuss the recent applications with an emphasis on the quantitative targeted proteomics reported for membrane transporters that are of particular relevance to drug disposition.
Fig. 1.
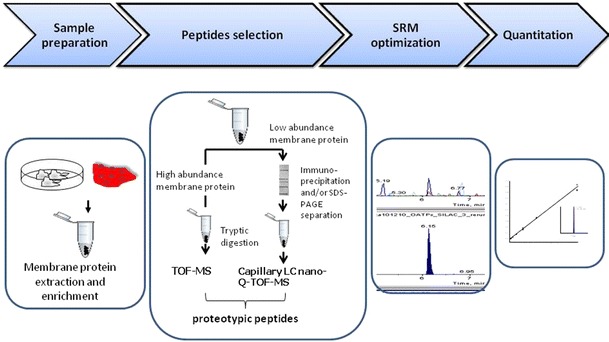
Workflow of targeted quantitative proteomics.
SELECTION OF SURROGATE TARGET PEPTIDES FOR PROTEIN QUANTIFICATION
Because peptides can only serve as surrogates of a particular target protein, selection of target peptides that achieve the highest quantification reliability and sensitivity is the first step of targeted proteomics quantification. These peptides, also named “signature peptides,” are unique to the target protein that can be determined through genome-wide blast. The peptides for quantification are easily detectable in an instrument representing the highest responding peptides for the target protein (17). The selection can be accomplished through the use of either empirical experimental tools in a high-resolution mass spectrometry analysis (18), such as time of flight (TOF) and Orbitrap, or in silico predictive tools (7,19) as discussed below. Protein digestion to release peptides can be achieved by a protease reaction with biological samples. Among the several proteases available for the reaction, trypsin is an ideal initial choice as it often produces fragments that are amenable to detection by MS in terms of size and amino acid composition. In one early study (18), multidrug resistance-associated protein 2 (MRP2), was immunoprecipitated and separated by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis, followed by in-gel digestion. Then, nano-electrospray ionization (nano-ESI) quadrupole TOF (Q-TOF) was utilized to select the target proteotypic peptide with the best detection sensitivity (18). On the other hand, understanding of the specific protease cleavage sites can facilitate the probability of de novo sequencing for a given protein, to predict the peptides that would result from protease digestion of the protein through an in silico digestion. Under the current practice, the combination of in silico analysis and subsequent lab experiments is widely accepted as an effective approach for developing methods to quantify membrane transporters (12,20–22).
Empirical Experimental Tools for the Selection of Quantitative Peptides
With the increasing capability of the cutting-edge instruments such as high-resolution mass spectrometers and the associated powerful processing software, the empirical approach for targeted protein quantification has become a simpler approach. As shown in Fig. 2, the general workflow of the empirical approach includes (1) peptide profiling of protein fragments, (2) data processing and database searching to identify positive hits, (3) surrogate peptide probe selection, and (4) fine tuning of multiple reaction monitoring (MRM) transitions. Peptide profiling is usually done during the same analytical run using cutting-edge instruments where multiple precursors are selected and fragmented for MS/MS acquisition (Fig. 2). It is critical that the high-quality MS/MS spectra are collected for the peptides of interest since it is inevitable for the peptides to co-elute with others especially for a complex sample such as tissue lysate. Target protein enrichment or purification would achieve the best peptides coverage for the target proteins (18), and the fast acquisition rate is a key as the possibility of detecting a specific peptide goes up with the total number of spectra that one can collect within each cycle. For this reason, many instrument vendors strive to improve the acquisition rate with promising results. For example, as many as 50 high-quality MS/MS spectra (with mass resolution above 15 k) can be routinely generated within 1-s cycle time using AB Sciex Triple-TOF 5600 system, and as many as 5,000 distinct peptides could be identified within 15 min of run time. Raw MS data can be exported and searched by public search engines like Mascot and Sequest. Many vendors also have their own processing software such as ProteinPilot of AB Sciex (Fig. 3), ProteinLynx of Waters, and Proteome Discoverer of Thermo Scientific. As exemplified by the ProteinPilot of AB Sciex, the software offers better working interfaces and data visualization and can be used by personal preference (Fig. 3). Followed by peptide profiling, the output results would yield a list of peptide fragments that are generated from the digested protein of interest. Next, surrogate peptide probes for target protein can be selected for quantification based on the protein profiling data, especially if such identification was achieved using protein digests of biological samples to be quantified. The criteria of the in silico approaches as described below can be further applied to narrow down the lists. Software such as Skyline by University of Washington and MRMPilot from AB Sciex can take the profiling results and automatically generate the MRM transitions accordingly. The digested samples can also be reinjected with targeted MRM transitions being monitored to further evaluate the sensitivity and chromatography aspects for higher confidence. The extracted ion chromatograms (XICs) can be plotted for each proposed MRM transition for sensitivity of detection, peak shape, and reproducibility (Fig. 4). The peptide fragments associated with the best MRM transition signal and least standard deviation would naturally be the best candidates of surrogate peptides for target protein quantification. The candidate peptides can be synthesized, and MRM transition settings can further be fine-tuned to improve sensitivity. Modern Qtrap instruments such as AB Sciex 5500 or 6500 Qtrap that offer fast scan speed and scheduled MRM capability, 5 different CE values can be fine-tuned for up to 100 different SRM transitions within one injection.
Fig. 2.
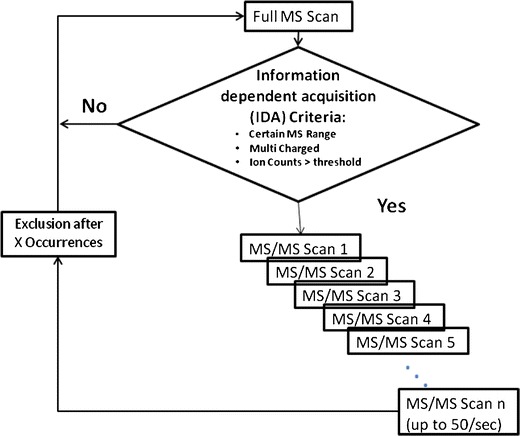
Peptide profiling of protein fragments.
Fig. 3.
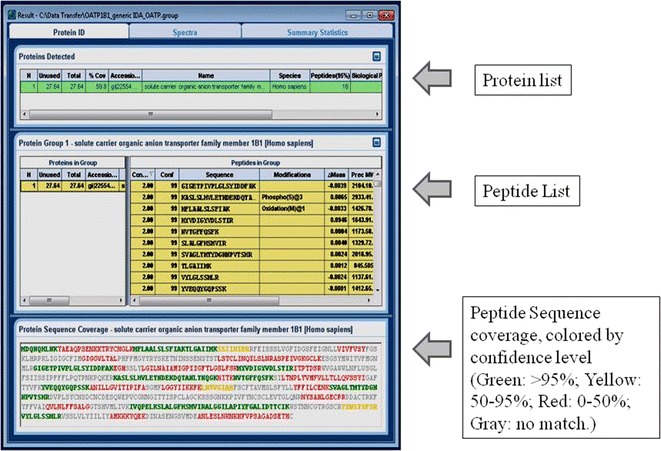
Example of MS data processing and data base searching using Proteinpilot (AB Sciex).
Fig. 4.
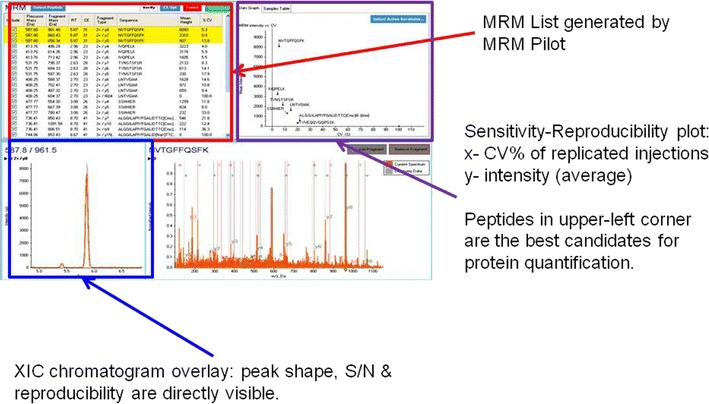
Example of peptide selection and fine tuning of MRM transition in MRM pilot from AB Aciex. MRM multiple reaction monitoring, XIC extracted ion chromatogram, S/N signal to noise ratio.
As all the peptides selected have already been detected in real digested samples during protein profiling and following validation experiments, the empirical experimental approach can provide high confidence in the selection of surrogate peptides. The limitations of this approach include the need for high end instruments and potential loss of protein mapping coverage due to low abundance of membrane protein expression. As mentioned above, upfront sample enrichment and use of nano-flow LC instead of regular LC could improve protein mapping coverage as well as peptide detection sensitivity (18,23).
In Silico Tools for the Selection of Quantitative Peptides
The proteotypic peptides generated through the digestion of intact protein by different proteases can be predicted through online predictive tools (7). Although there are lots of proteases available, trypsin is most commonly used because it can produce peptides with appropriate amino acid length and composition being amenable for LC-MS detection. Software including PeptideMass, PeptideAtlas (http://www.peptideatlas.org/), PeptideCutter (http://expasy.org), ProteinProspector (http://prospector.ucsf.edu/prospector/mshome.htm), and Skyline (https://skyline.gs.washington.edu) provide “in silico” digestion for planning experiments. Input of protein sequence to the software will generate peptide lists created by commonly used proteolytic enzymes. As any protein can be digested to tens or even hundreds of possible peptides, it is not feasible to synthesize all of them for further evaluation and method development. Therefore, it is desirable to narrow down a small panel of peptides, which are likely to be detected and unique for the protein of origin. Fusaro et al. (24) described an enhanced signature peptide predictor (http://www.broadinstitute.org/cancer/software/genepattern/modules/ESPPredictor.html) to predict high-responding peptides from a given protein based on the criteria of peptide selection. In general, peptides that are suitable for target protein quantitation should be between 8 and 20 amino acids long and avoid long hydrophobic and short hydrophilic ones. Peptides containing cysteine, methionine, tryptophan, and N-terminal glutamine residues should be excluded because of their potential for modification. The continuous glutamic acid and aspartic acid residues or lysine and arginine residues in the adjacent sequence, which might hinder the digest efficiency of trypsin, should be avoided. The peptides selected should be in the exposed and soluble domain of the protein. If possible, the peptides in the transmembrane domains should be excluded because of their hydrophobic nature (25,26). The selection of post-translational modification, natural variant amino acid, and single nucleotide polymorphism sites should also be considered to meet the purpose of experiments. In general, multiple peptides should be considered for each protein, and for each peptide, multiple MRM should be monitored to gain reliable quantification information.
Selection of MRM Transitions for Quantitative Peptides
Fragment ions for each peptide precursor ion should provide the highest signal intensity and lowest interference. Commonly three to five MRM transitions should be selected for each peptide. The selection of peptide MRM transitions can be accomplished by shotgun experiments of samples on high resolution MS instruments such as Q-TOF or linear ion trap-orbitrap (LTQ-orbitrap) (27). It is worth noting that the peptide transitions obtained from these high-resolution instruments have different modes of CID, which may generate patterns of fragmentation that differ from QQQ-type instruments, especially LTQ-orbitrap (25,28,29). Additionally, the low abundance proteins may cause problems for MRM selection and optimization. Alternatively crude synthetic peptide libraries can be used for batch analysis to select and optimize MRM transitions on a QQQ-type instrument (16). This method allows extraction and validation of optimal MRM transitions for ∼100 peptides per hour with high confidence (14).
SELECTION OF INTERNAL STANDARD (IS)
MS-based protein quantification requires an internal standard (IS) to compensate variability derived from each step during the analysis (30–33). Among different isotope dilution techniques (31,33), a stable isotope-labeled (SIL) synthetic peptide with identical sequence to the quantification peptide is the most commonly used IS for analyte peak area normalization. Other approaches including chemical derivatization, metabolic incorporation of heavy-labeled amino acids, and protein standard absolute quantification (PSAQ) have been described in the literature (34). The identity and the timing of IS introduction offer different advantages in precision and accuracy in quantification, with respect to errors derived from variability in each step during analysis, such as native membrane protein extraction, denaturation, and digestion (Fig. 5). The stable isotope labeling by amino acids in cell culture (SILAC) or stable Isotope labeling in mammals (SILAM) is one such method that offers a metabolic-labeling strategy for label incorporation in vitro or in vivo (35–37). When SILAM or SILAC is used, both heavy and light proteins can be combined at the beginning of the experiment and digested together, after which the heavy isotope-labeled peptide serves as the co-eluting IS. Thus, SILAM or SILAC is considered to be able to normalize for losses derived from any portion of the workflow and therefore has the potential to be a superior approach for quantitative analysis (35,38–42). Similar in concept to SILAC, PSAQ is the IS derived from a labeled protein, and the concentration is measured by quantitative amino acid analysis of purified material, thereby allowing for better accuracy at the protein level (39,43–45). The SIL IS has the same amino acid sequence as the quantitative peptide selected above except that they have heavy-labeled amino acid and therefore has the same structure as the peptide that co-elutes in an LC and has the same fragmentation pattern to its natural counterpart. In SIL methods such as the absolute quantification (AQUA) method (30), the IS is commonly spiked during or post-digestion and is the most feasible and rapid approach. Indeed, SIL peptides are among the most common IS used to evaluate the expression of drug transporters (5–7,18,19,46–51). A comparison of the SIL peptide with SILAM approach was recently carried out in the LC-MS/MS method development in our laboratory (37). The findings suggested that, under the optimized denaturation and digestion conditions (52), the SIL method provided an accelerated preparation while obtaining comparable measurements similar to the SILAM approach.
Fig. 5.
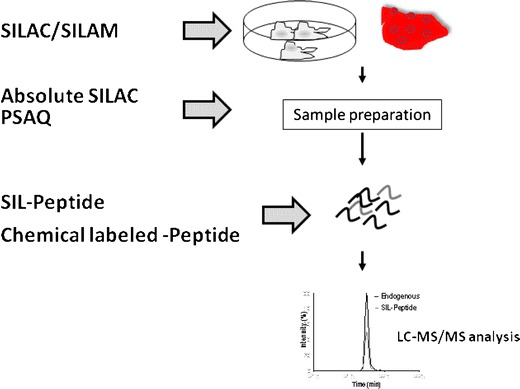
The timing of IS introduction. SILAC stable isotope labeling by amino acid in cell culture, SILAM stable isotope labeling in mammals, SIL stable isotope labeding, PSAQ protein standard absolute quantification.
DENATURATION AND DIGESTION OF SAMPLE PROTEINS
Once the quantitative peptides and SIL ISs are identified and their MRM transitions are optimized, the method can be applied for protein quantification in biological samples. Gel-based techniques including 1-D and 2-D gel electrophoresis and affinity-based techniques including online immunoaffinity column and affinity depletion of high-abundance proteins have been widely utilized prior to LC-MS/MS analysis. Recently, many protein quantification methods especially those for membrane proteins have focused on the in-solution digestion for complex biological samples. For the in-solution digestion, solubilization and denaturation remain obstacles for membrane transporter proteins because they tend to have multiple transmembrane domains, which are not directly accessible for protease enzymes. Most commonly used reagents for protein solubilization involve the ionic or nonionic chemicals including chaotropes such as urea and guanidine, bile acids, organic acids, and various detergents such as Tween, SDS, and Triton (53,54). After protein denaturation, cysteines are blocked by reagents such as iodoacetamide and methyl methanethiosulfonate to prevent disulfide bond reformation. The solubilizing and blocking reagents need to be further diluted for enzyme digestion; otherwise, they may interfere with protease activity. Once the digestion is quenched, LC separation is carried out to separate target peptides and ISs from other interfering substances that are produced through enzyme digestion in biological matrix including cell lysate, tissues, and other different cell compartments.
LC-MS METHOD DEVELOPMENT INCLUDING OPTIMIZATION AND VALIDATION OF THE SELECTED SRM TRANSITIONS, CHROMATOGRAPHY SETTINGS, AND QUALITY CONTROL
Followed by the selection of quantitative targeted peptides, the optimization of SRM, the selection of internal standards, and sample digestions, sample analysis can be conducted on a triple quadrupole mass spectrometer such as API 4000 or 6500 coupled to an LC, ultra performance LC (UPLC), or nano-UPLC system (22,23,37). The LC separation can be achieved in a reverse phase column (e.g., 100 × 3.0 mm particle size 2.6 μm Kinetex® C18 column) with a gradient elution of water with 0.1% formic acid (solvent A) and acetonitrile with 0.1% formic acid (solvent B) as mobile phases. To monitor multiple proteins in a single run, scheduled MRM acquisition method could be constructed using manually optimized declustering potentials, collision energies, as well as collision cell entrance and exit potentials. Three MRMs can be monitored simultaneously for each peptide (22,23). The quality control (QC) should also be conducted for the parameters of bioanalytical method validation guidelines suggested by regulatory agency for peptide stability, selectivity, linearity, reproducibility, and intra-/interday variability (55). Since there is no biological blank matrix available for membrane transporter proteins, digested human serum albumin can be used for preparation of calibration curves and QC samples (22).
EXAMPLES OF QUANTITATIVE TARGETED PROTEOMICS FOR MEMBRANE TRANSPORTERS
Nowadays, various in vitro cell culture systems have been used to assess in vitro transporter kinetics for in vivo extrapolation with drug candidates during drug discovery and development (56). For example, plated hepatocytes or cultured hepatocytes between layers of biomatrix are commonly used for in vitro assessment of liver clearance. Because hepatocytes cultured in a “sandwich” format provide proper orientation and localization of transporters along with the development of intact bile canaliculi (57–60), the tool has become an important system for investigating hepatobiliary transport. However, under the culture conditions including the use of culture media, plate formats, and cell density, the expression of hepatic transporters and other liver specific proteins might be altered (61–63). Furthermore, the modulation of liver specific transporter expressions could also be species specific (64–66), which could limit the predictive power of a model. With the targeted quantitative proteomics methods aforementioned, Li et al. characterized MRP2, bile salt export pump (BSEP), and breast cancer resistance protein (BCRP) expressions in sandwich cultured hepatocytes (SCH) to compare proteins across the cell culture periods and with in vivo expression (6,46,47). They observed a 40% loss of rat Mrp2 protein, but not human MRP2 protein, in cryopreserved hepatocytes as compared to that in the liver. Interestingly, a decrease in BSEP/Bsep and an increase in BCRP/Bcrp were observed in human and rat sandwich cultures, while in the same culture Mrp2 was decreased in rat and MRP2 in human hepatocyte was increased. As a result, following the sandwich culture, species difference between human and rat was diminished for MRP2/Mrp2 and reversed for BCRP/Bcrp (46). Through integrating a scaling factor of hepatobiliary transporter levels between in vitro SCH and in vivo, the prediction of biliary secretion in rats was improved (5). The findings highlighted the importance in understanding in vitro modulation of hepatobiliary transporter expression for in vivo prediction.
In vitro permeability is an important consideration in drug candidate selection for good oral absorption and blood–brain barrier (BBB) permeation. The Madin–Darby canine kidney cell line (MDCK) has been widely used in many companies because of many favorable characteristics including monolayer integrity and the formation of tight para-cellular junctions in a short culture time (∼3–5 days). MDCK wild-type cells (MDCK-WT) overexpressing specific transporters including human P-gp (MDCK-MDR1) have been widely used to characterize active transport and inhibitory effects of drug candidate. Following Kamiie et al. (19) in quantifying mouse P-gp using a single set of sample preparation conditions combined with multiplexed SRM to conduct high-throughput quantifications, Zhang et al. developed a method with the best apparent detection sensitivity applied to quantify the endogenous canine P-gp in MDCK-WT cells, human P-gp protein in MDCK-MDR1 cells, and membrane products from insect cells overexpressing human or mouse P-gp (7). They found that more than an 8-fold overexpression of P-gp in MDCK-MDR1 cells was detected as compared to MDCK-WT cells, which agreed with robust P-gp substrates efflux in MDCK-MDR1 cells over MDCK-WT (67). On the other hand, MDCK-WT cells transfected with human MDR1 gene showed different amounts of endogenous canine P-gp (∼1.5-fold lower than MDCK-WT), which indicates using the net flux ratio approach for determination of human P-gp substrates might not be a valid method to cancel out the endogenous canine transporter activity (67).
As mentioned above, since MDCK-WT cells can form tight para-cellular junctions in a short culture time (∼3–5 days), it is desirable over Caco-2 and PAMPA for measuring passive cellular (epithelial) permeability with high throughput capability. However, the significant drawback of MDCK-WT cells is the expression of functionally active canine P-gp. As a result, the quantitative targeted proteomics method was applied to isolate a subpopulation of MDCK-WT cells, the MDCK-LE (low efflux) cells that express low levels of endogenous canine P-gp (67). The MDCK-LE cells minimize the interference of endogenous canine P-gp on passive permeability measurement and offers benefits over MDCK-WT. In fact, the established MDCK-LE cells were found to exhibit very low and near the detection limit of P-gp expression, which offer a cleaner system to express transporter genes and robust assessment of passive epithelial permeability (67).
The BBB consists of endothelial cells of brain microvessels characterized by the presence of intercellular tight junctions and the polarized expression of numerous transport systems, which display separation of circulating blood from the central nervous system. Recently, the human cerebral microvascular endothelial cell line hCMEC/D3 was established and found to retain most of the morphological and functional characteristics of brain endothelial cells. The cell line is used as an in vitro model to investigate the function and their modulation in human BBB by many laboratories (68–71). A series of investigations demonstrated that the expression pattern of ATP-binding cassette transporter messenger RNAs in hCMEC/D3 cells is, at least qualitatively, similar to that of human brain microvessels (72,73). To understand the suitability and limitations of the hCMEC/D3 cell line being a BBB functional model, Ohtsuki et al. (74) applied quantitative targeted proteomics to determine the protein expression levels of multiple transporters, receptors, and junction proteins in hCMEC/D3 cells and compared the results with isolated human brain microvessels. They found that hCMEC/D3 cells retain protein expression of most transporters including MDR1, BCRP, multidrug resistance-associated protein 4 and receptors such as transferrin and insulin receptors expressed in vivo at the human BBB (74). The quantitative analysis of functionally important transport proteins provides fundamental information in extrapolation of in vitro results to in vivo (74).
In drug discovery, it is a general practice in the pharmaceutical industry to extrapolate preclinical data obtained from in vivo animal models, such as rodents and macaque monkeys, to humans. Understanding the difference of membrane protein expression is the key to bridge the gap between in vitro models and interspecies that underlie interexperimental variation and to extrapolate in vitro data to in vivo (5,6,18,46,47,75). Appreciation of interspecies differences of proteins involved in drug elimination and their expression levels can increase the confidence in data interpretation. Since a major bottleneck encountered in drug discovery and development is the time elapsed between preclinical and clinical studies, quantitative targeted proteomics could serve as a useful tool for rapidly comparing a given protein’s level across species to help reduce the delay of advancing drug candidates. Since the quantitative peptides could be intentionally selected from conserved sequence across species, one of the advantages of quantitative targeted proteomics is to measure the transporter protein across different species in an absolute manner (6,37). For example, the expression of MRP2/Mrp2 protein in livers was found to rank as rat >> monkey > dog ≈ human, where rat Mrp2 was approximately 10-fold higher than human. These results could explain, at least in part, the species difference of biliary excretion for its substrate drugs (76). Similarly, mouse Bcrp protein was found to be about 10-fold higher than human and other species (47), suggesting that mouse is not the best predictive model for Bcrp substrates. The protein quantification using LC-MS targeted proteomics approach can further be amended to a higher-throughput mode to compare the expressions of many proteins in different organs (19,48,49,77,78). For example, Kamiie et al. (19) reported the simultaneous analysis of 36 membrane proteins in mouse BBB, and the follow-up studies extended upon the surrogate peptide library to target an extensive number of membrane proteins in human and monkey brain microvessels (48,50,78). These studies provide valuable information with respect to understand species differences in transporter expression at the BBB (74).
Hepatic sinusoidal uptake could be the rate-determining step in the systemic clearance of drugs. Of all the mechanisms of sinusoidal transport, the organic anion transporting polypeptides (OATPs) are believed to be the most relevant for hepatic uptake of many anionic drugs. In addition to OATPs, The sodium-dependent taurocholate co-transporting polypeptide-mediated transport has been shown to contribute to the hepatic uptake of statins (79–82). The ability to predict uptake clearance and determine the contribution of individual transporters to overall hepatic uptake is therefore critical in assessing the potential pharmacokinetic and pharmacodynamic variability associated with drug–drug interactions and pharmacogenetics. Since hepatic uptake transporters co-located on the sinusoidal membrane of hepatocytes may provide redundant functions in hepatic uptake, there is significant interest in characterizing the expression of co-located transporters with overlapping substrate specificity. Recently, quantitative targeted proteomics was applied to determine the interindividual variability of expression level of hepatic uptake and efflux transporters in a large set of human liver samples to understand the influence of genotype, age and sex on such expression (12,13). It is now known that BCRP expression in the liver with the variant C421A allele was significantly lower than that in the wild-type livers (13). In contrast, OATP1B1 expression in the livers with SLCO1B1 haplotypes *14/*14 and *14/*1a (A388G and C463A variants) was higher than that in the reference haplotype (*1a/*1a) (12), leading to ∼40% decrease in predicted exposure of rosuvastatin or repaglinide in those individuals harboring these variant alleles. Interesting results were also noted as neither age nor sex was associated with expression of hepatic transporters determined (12,13).
Since the studies aforementioned ultimately use the absolute quantification of a surrogate peptide that can be determined with a high level of accuracy to analyze a protein, the true accuracy at the protein level is generally not known and can vary to a significant extent from method to method and from protein to protein (33,43,45,52,83,84). This issue derives from the fact that primary and secondary structural differences among proteins can amount to varying levels of proteolysis under the sample preparation conditions that are compatible with typical studies using trypsin digestion and LC-MS/MS-based peptide detection (52). Only under the assumption that the sample preparation conditions do not have a significant impact on the digestion efficiency for each protein, the quantified peptide concentrations can be used as true surrogate for expression levels of corresponding proteins, and further can be used to compare the abundance of different transporters. For example, as summarized in Table I, the amount of human hepatic transporters detected by different laboratories varies significantly. In particular, OATP1B1 was not detected in 9 of 17 livers (85), which did not agree with the reports obtained between different laboratories (12,65). For the reason just specified, the conclusion of expression “atlas” or expression “profile,” such as “BCRP was expressed 1.6-fold more than MDR1” (50) or “BCRP was the most abundant followed by MDR1” (48), is particularly concerning under the typical enzymatic digestions employed in these types of studies, which require a balance between conditions that enhance proteolysis through denaturation of the target protein, and conditions that will not significantly inhibit the proteolytic activity of the digestion enzyme. In total, to the extent that relative expressions determined under different sets of sample preparation conditions can underlie completely different conclusions. Furthermore, the surrogate peptide-based quantitative targeted proteomics alone is not suitable to draw a pie chart of transporter distribution without protein standards with a known concentrations until further advancements are achieved.
Table 1.
Summary of Expression of Hepatic Transporter Proteins
| Transporter | Tissuesa | Peptide | Protein amount (fmol/μg protein) | Sample preparation method | Sample size | Synthetic peptide as calibration standard (amino acid measured?) | Notes |
|---|---|---|---|---|---|---|---|
| P-gp | Hepatocyte (12) | NTTGALTTR/IATEAIENFR | 0.5 ± 0.1 | Native membrane kit | 12 | Yes | |
| Liver (12) | NTTGALTTR/IATEAIENFR | 0.4 ± 0.2 | Native membrane kit | 64 | Yes | ||
| Liver (85) | EALDESIPPVSFWR | 1.5 ± 0.44 | Ultra centrifuge | 17 | No | ||
| SCHH (86) | EALDESIPPVSFWR | 5.28–30.5 | Ultra centrifuge | 3 lots | No | Matrigel was not used as overlay matrix | |
| BCRP | Hepatocyte (47) | ENLQFSAALR | 0.23 ± 0.02 | Native membrane kit | 3 | Yes | |
| Liver (47) | ENLQFSAALR | 0.147 ± 0.028 | Native membrane kit | 14 | Yes | ||
| Liver (85) | ENLQFSAALR/SSLLDVLAAR | 0.419 ± 0.219 | Ultra centrifuge | 14 | No | ||
| SCHH (86) | ENLQFSAALR/SSLLDVLAAR | 3.9–5.23 | Ultra centrifuge | 3 lots | No | Matrigel was not used as overlay matrix | |
| MRP2 | Hepatocyte (6) | LTIIPQDPILFSGSLR | 0.83 ± 0.05 | Native membrane kit | 3 | Yes | |
| Liver (6) | LTIIPQDPILFSGSLR | 0.63 ± 0.27 | Native membrane kit | 15 | Yes | ||
| Liver (85) | LTIIPQDPILFSGNLR | 1.46 ± 0.65 | Ultra centrifuge | 16 | No | ||
| SCHH (86) | LTIIPQDPILFSGNLR | 6.11–24.05 | Ultra centrifuge | 3 lots | No | Matrigel was not used as overlay matrix | |
| OATP1B1 | Hepatocyte (65) | NVTGFFQSFK | 4.38 ± 1.75 | Native membrane kit | 5 | Yes | |
| Hepatocyte (12) | NVTGFFQSFK/YVEQQYGQPSSK | 2.4 ± 0.5 | Native membrane kit | 12 | Yes | ||
| Liver (65) | NVTGFFQSFK | 4.51 ± 1.30 | Native membrane kit | 9 | Yes | ||
| Liver (85) | LNTVGIAK | 2.74 ± 3.67 | Ultra centrifuge | 8 | No | Not detected in 9 out 17 liver sample | |
| Liver (12) | NVTGFFQSFK/YVEQQYGQPSSK | 2.0 ± 0.9 | Native membrane kit | 64 | Yes | ||
| SCHH (65) | NVTGFFQSFK | 5.76 ± 3.30 | Native membrane kit | 5 | Yes | ||
| SCHH (86) | LNTVGIAK | 4.6–6.83 | Ultra centrifuge | 3 lots | No | Matrigel was not used as overlay matrix | |
| OATP1B3 | Hepatocyte (65) | NVTGFFQSLK | 1.23 ± 0.39 | Native membrane kit | 5 | Yes | |
| Hepatocyte (12) | NVTGFFQSLK/IYNSVFFGR | 0.9 ± 0.5 | Native membrane kit | 12 | Yes | ||
| Liver (65) | NVTGFFQSLK | 1.93 ± 0.44 | Native membrane kit | 9 | Yes | ||
| Liver (85) | IYNSVFFGR | 1.7 ± 0.45 | Ultra centrifuge | 17 | No | ||
| Liver (12) | NVTGFFQSLK/IYNSVFFGR | 1.1 ± 0.5 | Native membrane kit | 64 | Yes | ||
| SCHH (65) | NVTGFFQSLK | 0.84 ± 0.26 | Native membrane kit | 5 | Yes | ||
| SCHH (86) | IYNSVFFGR | 3.95–4.47 | Ultra centrifuge | 3 lots | No | Matrigel was not used as overlay matrix | |
| OATP2B1 | Hepatocyte (65) | SSPAVEQQLLVSGPGK | 1.17 ± 0.43 | Native membrane kit | 5 | Yes | |
| Hepatocyte (12) | VLAVTDSPAR/SSPAVEQQLLVSGPGK | 1.7 ± 0.5 | Native membrane kit | 12 | Yes | ||
| Liver (65) | SSPAVEQQLLVSGPGK | 1.65 ± 0.52 | Native membrane kit | 9 | Yes | ||
| Liver (85) | VLLQTLR | 0.46 ± 0.87 | Ultra centrifuge | 5 | No | Not detected in 12 out 17 liver sample | |
| Liver (12) | VLAVTDSPAR/SSPAVEQQLLVSGPGK | 1.7 ± 0.6 | Native membrane kit | 64 | Yes | ||
| SCHH (65) | SSPAVEQQLLVSGPGK | 1.02 ± 0.18 | Native membrane kit | 5 | Yes | ||
| SCHH (86) | VLLQTLR | 2.63–6.16 | Ultra centrifuge | 3 lots | No | Matrigel was not used as overlay matrix | |
| OCT1 | Liver (85) | LSPSFADLFR | 7.35 ± 3.26 | Ultra centrifuge | 17 | No | |
| SCHH (86) | LSPSFADLFR | 9.1–17.38 | Ultra centrifuge | 3 lots | No | Matrigel was not used as overlay matrix | |
| MATE1 | Liver (85) | HVGVILQR | 1.07 ± 0.35 | Ultra centrifuge | 16 | No | |
| SCHH (86) | HVGVILQR | 2.53–8.52 | Ultra centrifuge | 3 lots | No | Matrigel was not used as overlay matrix | |
| BSEP | Hepatocyte (47) | STALQLIQR | 4.20 ± 0.07 | Native membrane kit | 3 | Yes | |
| Liver (47) | STALQLIQR | 3.40 ± 1.27 | Native membrane kit | 15 | Yes | ||
| Liver (85) | STALQLIQR | 1.48 ± 0.44 | Ultra centrifuge | 17 | No | ||
| SCHH (86) | STALQLIQR | 5.75–7.87 | Ultra centrifuge | 3 lots | No | Matrigel was not used as overlay matrix |
aReference in the parenthesis
PERSPECTIVES
Recently, model-based predictions such as PBPK modeling, have become emerging approaches to predict complex drug disposition in a way of holistic perspectives for the sake of rational drug design (87,88). The model requires analytical tools that can quantitatively determine the components associated with biological processes. As a result, technologies for characterizing membrane proteins have been a growing field, and the quantitative targeted proteomics in particular becomes a key enabling technology and is continuing to evolve rapidly. Quantitative targeted proteomics uses peptides unique to the proteins of interest, which can be readily obtained from commercial sources to serve as surrogate standards to overcome the absence of pure protein as standards. The method can be sensitive and selective and is amenable to a high-throughput mode to quantitatively assess protein expressions in various biological samples (19), and has found its way to broad applications such as those in translational pharmacology. However, significant challenges in quantification of membrane proteins remain, due to relatively low expression levels and often the inclusion of multiple hydrophobic domains that resist exposure to aqueous environment, resulting in limitations of solubilization and denaturation with respect to facilitating protease access and digestion efficiency. Conditions of digestion and membrane solubilization should be optimized, in which the organic solvents, detergents, or chaotropic agents are examined to be compatible with the route of digestion and subsequent LC-MS analysis. Since reliable membrane protein standards are not yet readily available, caution is still warranted in absolute peptide quantifications that may not directly reflect the relative abundance of different proteins. The use of SIL-IS at different level, e.g., SILAC or SILAM, can offer advantages in addressing the incomplete digestion (52,89). Furthermore, experimental strategies are required to improve particularly for the analysis of post-translational modifications in membrane proteins. Equally important, sample preparation needs to be improved in order to differentiate the subcellular components for investigating proteomes of intracellular membranes. The continuous development of methods is central for increasing reproducibility in the rapidly expanding application of quantitative proteomics for membrane transporter proteins. These improvements will undoubtedly continue to make LC-MS/MS based quantifications more reliable and accessible in the ADME community, and the results obtained will facilitate prediction of population-based human transporter-mediated drug disposition, drug–drug interactions, and interindividual variability through mechanistic-based mathematic modeling approach.
ACKNOWLEDGMENTS
We would like to acknowledge Dr. Punit Marathe for her scientific comments and suggestions. We also thank Anthony Marino for the help in editing the manuscript.
REFERENCES
- 1.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bentz J, O’Connor MP, Bednarczyk D, Coleman J, Lee C, Palm J, et al. Variability in P-glycoprotein inhibitory potency (IC(5)(0)) using various in vitro experimental systems: implications for universal digoxin drug–drug interaction risk assessment decision criteria. Drug Metab Dispos. 2013;41(7):1347–1366. doi: 10.1124/dmd.112.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellens H, Deng S, Coleman J, Bentz J, Taub ME, Ragueneau-Majlessi I, et al. Application of receiver operating characteristic analysis to refine the prediction of potential digoxin drug interactions. Drug Metab Dispos. 2013;41(7):1367–1374. doi: 10.1124/dmd.112.050542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghibellini G, Johnson BM, Kowalsky RJ, Heizer WD, Brouwer KL. A novel method for the determination of biliary clearance in humans. AAPS J. 2004;6(4):e33. doi: 10.1208/aapsj060433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li N, Singh P, Mandrell KM, Lai Y. Improved extrapolation of hepatobiliary clearance from in vitro sandwich cultured rat hepatocytes through absolute quantification of hepatobiliary transporters. Mol Pharm. 2010;7(3):630–641. doi: 10.1021/mp9001574. [DOI] [PubMed] [Google Scholar]
- 6.Li N, Zhang Y, Hua F, Lai Y. Absolute difference of hepatobiliary transporter multidrug resistance-associated protein (MRP2/Mrp2) in liver tissues and isolated hepatocytes from rat, dog, monkey, and human. Drug Metab Dispos. 2009;37(1):66–73. doi: 10.1124/dmd.108.023234. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Li N, Brown PW, Ozer JS, Lai Y. Liquid chromatography/tandem mass spectrometry based targeted proteomics quantification of P-glycoprotein in various biological samples. Rapid Commun Mass Spectrom. 2011;25(12):1715–1724. doi: 10.1002/rcm.5026. [DOI] [PubMed] [Google Scholar]
- 8.Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell. 2009;138(4):795–806. doi: 10.1016/j.cell.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5(4):573–588. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Achour B, Russell MR, Barber J, Rostami-Hodjegan A. Simultaneous quantification of the abundance of several cytochrome p450 and uridine 5′-diphospho-glucuronosyltransferase enzymes in human liver microsomes using multiplexed targeted proteomics. Drug Metab Dispos. 2014;42(4):500–510. doi: 10.1124/dmd.113.055632. [DOI] [PubMed] [Google Scholar]
- 11.Russell MR, Achour B, McKenzie EA, Lopez R, Harwood MD, Rostami-Hodjegan A, et al. Alternative fusion protein strategies to express recalcitrant QconCAT proteins for quantitative proteomics of human drug metabolizing enzymes and transporters. J Proteome Res. 2013;12(12):5934–5942. doi: 10.1021/pr400279u. [DOI] [PubMed] [Google Scholar]
- 12.Prasad B, Evers R, Gupta A, Hop CE, Salphati L, Shukla S, et al. Interindividual variability in hepatic organic anion-transporting polypeptides and P-glycoprotein (ABCB1) protein expression: quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab Dispos. 2014;42(1):78–88. doi: 10.1124/dmd.113.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prasad B, Lai Y, Lin Y, Unadkat JD. Interindividual variability in the hepatic expression of the human breast cancer resistance protein (BCRP/ABCG2): effect of age, sex, and genotype. J Pharm Sci. 2013;102(3):787–793. doi: 10.1002/jps.23436. [DOI] [PubMed] [Google Scholar]
- 14.Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9(6):555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- 15.Arnott D, Kishiyama A, Luis EA, Ludlum SG, Marsters JC, Jr, Stults JT. Selective detection of membrane proteins without antibodies: a mass spectrometric version of the Western blot. Mol Cell Proteomics. 2002;1(2):148–156. doi: 10.1074/mcp.M100027-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Picotti P, Rinner O, Stallmach R, Dautel F, Farrah T, Domon B, et al. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat Methods. 2010;7(1):43–46. doi: 10.1038/nmeth.1408. [DOI] [PubMed] [Google Scholar]
- 17.Picotti P, Aebersold R, Domon B. The implications of proteolytic background for shotgun proteomics. Mol Cell Proteomics. 2007;6(9):1589–1598. doi: 10.1074/mcp.M700029-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Li N, Nemirovskiy OV, Zhang Y, Yuan H, Mo J, Ji C, et al. Absolute quantification of multidrug resistance-associated protein 2 (MRP2/ABCC2) using liquid chromatography tandem mass spectrometry. Anal Biochem. 2008;380(2):211–222. doi: 10.1016/j.ab.2008.05.032. [DOI] [PubMed] [Google Scholar]
- 19.Kamiie J, Ohtsuki S, Iwase R, Ohmine K, Katsukura Y, Yanai K, et al. Quantitative atlas of membrane transporter proteins: development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm Res. 2008;25(6):1469–1483. doi: 10.1007/s11095-008-9532-4. [DOI] [PubMed] [Google Scholar]
- 20.Uchida Y, Tachikawa M, Obuchi W, Hoshi Y, Tomioka Y, Ohtsuki S, et al. A study protocol for quantitative targeted absolute proteomics (QTAP) by LC-MS/MS: application for inter-strain differences in protein expression levels of transporters, receptors, claudin-5, and marker proteins at the blood–brain barrier in ddY, FVB, and C57BL/6J mice. Fluids Barriers CNS. 2013;10(1):21. doi: 10.1186/2045-8118-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoneyama T, Ohtsuki S, Ono M, Ohmine K, Uchida Y, Yamada T, et al. Quantitative targeted absolute proteomics-based large-scale quantification of proline-hydroxylated alpha-fibrinogen in plasma for pancreatic cancer diagnosis. J Proteome Res. 2013;12(2):753–762. doi: 10.1021/pr3008144. [DOI] [PubMed] [Google Scholar]
- 22.Groer C, Bruck S, Lai Y, Paulick A, Busemann A, Heidecke CD, et al. LC-MS/MS-based quantification of clinically relevant intestinal uptake and efflux transporter proteins. J Pharm Biomed Anal. 2013;85:253–261. doi: 10.1016/j.jpba.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 23.Fallon JK, Neubert H, Hyland R, Goosen TC, Smith PC. Targeted quantitative proteomics for the analysis of 14 UGT1As and -2Bs in human liver using NanoUPLC-MS/MS with selected reaction monitoring. J Proteome Res. 2013;12(10):4402–4413. doi: 10.1021/pr4004213. [DOI] [PubMed] [Google Scholar]
- 24.Fusaro VA, Mani DR, Mesirov JP, Carr SA. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat Biotechnol. 2009;27(2):190–198. doi: 10.1038/nbt.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lange V, Picotti P, Domon B, Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol. 2008;4:222. doi: 10.1038/msb.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kirkpatrick DS, Gerber SA, Gygi SP. The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods. 2005;35(3):265–273. doi: 10.1016/j.ymeth.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 27.Chalkley R. Instrumentation for LC-MS/MS in proteomics. Methods Mol Biol. 2010;658:47–60. doi: 10.1007/978-1-60761-780-8_3. [DOI] [PubMed] [Google Scholar]
- 28.Bronstrup M. Absolute quantification strategies in proteomics based on mass spectrometry. Expert Rev Proteomics. 2004;1(4):503–512. doi: 10.1586/14789450.1.4.503. [DOI] [PubMed] [Google Scholar]
- 29.Sherwood CA, Eastham A, Lee LW, Risler J, Vitek O, Martin DB. Correlation between y-type ions observed in ion trap and triple quadrupole mass spectrometers. J Proteome Res. 2009;8(9):4243–4251. doi: 10.1021/pr900298b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A. 2003;100(12):6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barr JR, Maggio VL, Patterson DG, Jr, Cooper GR, Henderson LO, Turner WE, et al. Isotope dilution–mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin Chem. 1996;42(10):1676–1682. [PubMed] [Google Scholar]
- 32.Zhang G, Annan RS, Carr SA, Neubert TA. Overview of peptide and protein analysis by mass spectrometry. Curr Protoc Protein Sci. 2010;Chapter 16:Unit16 1. doi: 10.1002/0471140864.ps1601s62. [DOI] [PubMed] [Google Scholar]
- 33.Brun V, Masselon C, Garin J, Dupuis A. Isotope dilution strategies for absolute quantitative proteomics. J Proteomics. 2009;72(5):740–749. doi: 10.1016/j.jprot.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 34.Elliott MH, Smith DS, Parker CE, Borchers C. Current trends in quantitative proteomics. J Mass Spectrom. 2009;44(12):1637–1660. doi: 10.1002/jms.1692. [DOI] [PubMed] [Google Scholar]
- 35.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–386. doi: 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Rauniyar N, McClatchy DB, Yates JR., 3rd Stable isotope labeling of mammals (SILAM) for in vivo quantitative proteomic analysis. Methods. 2013;61(3):260–268. doi: 10.1016/j.ymeth.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Qiu X, Bi YA, Balogh LM, Lai Y. Absolute measurement of species differences in sodium taurocholate cotransporting polypeptide (NTCP/Ntcp) and its modulation in cultured hepatocytes. J Pharm Sci. 2013;102(9):3252–3263. doi: 10.1002/jps.23582. [DOI] [PubMed] [Google Scholar]
- 38.Geiger T, Wisniewski JR, Cox J, Zanivan S, Kruger M, Ishihama Y, et al. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat Protoc. 2011;6(2):147–157. doi: 10.1038/nprot.2010.192. [DOI] [PubMed] [Google Scholar]
- 39.Hanke S, Besir H, Oesterhelt D, Mann M. Absolute SILAC for accurate quantitation of proteins in complex mixtures down to the attomole level. J Proteome Res. 2008;7(3):1118–1130. doi: 10.1021/pr7007175. [DOI] [PubMed] [Google Scholar]
- 40.Harsha HC, Molina H, Pandey A. Quantitative proteomics using stable isotope labeling with amino acids in cell culture. Nat Protoc. 2008;3(3):505–516. doi: 10.1038/nprot.2008.2. [DOI] [PubMed] [Google Scholar]
- 41.Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat Protoc. 2006;1(6):2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- 42.Ong SE, Mann M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol Biol. 2007;359:37–52. doi: 10.1007/978-1-59745-255-7_3. [DOI] [PubMed] [Google Scholar]
- 43.Brun V, Dupuis A, Adrait A, Marcellin M, Thomas D, Court M, et al. Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol Cell Proteomics. 2007;6(12):2139–2149. doi: 10.1074/mcp.M700163-MCP200. [DOI] [PubMed] [Google Scholar]
- 44.Ishihama Y, Sato T, Tabata T, Miyamoto N, Sagane K, Nagasu T, et al. Quantitative mouse brain proteomics using culture-derived isotope tags as internal standards. Nat Biotechnol. 2005;23(5):617–621. doi: 10.1038/nbt1086. [DOI] [PubMed] [Google Scholar]
- 45.Lebert D, Dupuis A, Garin J, Bruley C, Brun V. Production and use of stable isotope-labeled proteins for absolute quantitative proteomics. Methods Mol Biol. 2011;753:93–115. doi: 10.1007/978-1-61779-148-2_7. [DOI] [PubMed] [Google Scholar]
- 46.Li N, Bi YA, Duignan DB, Lai Y. Quantitative expression profile of hepatobiliary transporters in sandwich cultured rat and human hepatocytes. Mol Pharm. 2009;6(4):1180–1189. doi: 10.1021/mp900044x. [DOI] [PubMed] [Google Scholar]
- 47.Li N, Palandra J, Nemirovskiy OV, Lai Y. LC-MS/MS mediated absolute quantification and comparison of bile salt export pump and breast cancer resistance protein in livers and hepatocytes across species. Anal Chem. 2009;81(6):2251–2259. doi: 10.1021/ac8024009. [DOI] [PubMed] [Google Scholar]
- 48.Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, et al. Quantitative targeted absolute proteomics of human blood–brain barrier transporters and receptors. J Neurochem. 2011;117(2):333–345. doi: 10.1111/j.1471-4159.2011.07208.x. [DOI] [PubMed] [Google Scholar]
- 49.Sakamoto A, Matsumaru T, Ishiguro N, Schaefer O, Ohtsuki S, Inoue T, et al. Reliability and robustness of simultaneous absolute quantification of drug transporters, cytochrome P450 enzymes, and Udp-glucuronosyltransferases in human liver tissue by multiplexed MRM/selected reaction monitoring mode tandem mass spectrometry with nano-liquid chromatography. J Pharm Sci. 2011;100(9):4037–4043. doi: 10.1002/jps.22591. [DOI] [PubMed] [Google Scholar]
- 50.Shawahna R, Uchida Y, Decleves X, Ohtsuki S, Yousif S, Dauchy S, et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm. 2011;8(4):1332–1341. doi: 10.1021/mp200129p. [DOI] [PubMed] [Google Scholar]
- 51.Uchida Y, Ohtsuki S, Kamiie J, Terasaki T. Blood–brain barrier (BBB) pharmacoproteomics: reconstruction of in vivo brain distribution of 11 P-glycoprotein substrates based on the BBB transporter protein concentration, in vitro intrinsic transport activity, and unbound fraction in plasma and brain in mice. J Pharmacol Exp Ther. 2011;339(2):579–588. doi: 10.1124/jpet.111.184200. [DOI] [PubMed] [Google Scholar]
- 52.Balogh LM, Kimoto E, Chupka J, Zhang H, Lai Y. Membrane protein quantification by peptide-based mass spectrometry approaches: studies on the organic anion-transporting polypeptide family. J Proteomics Bioinforma. 2012;S4(003):1–8. [Google Scholar]
- 53.Helenius A, McCaslin DR, Fries E, Tanford C. Properties of detergents. Methods Enzymol. 1979;56:734–749. doi: 10.1016/0076-6879(79)56066-2. [DOI] [PubMed] [Google Scholar]
- 54.Lin Y, Zhou J, Bi D, Chen P, Wang X, Liang S. Sodium-deoxycholate-assisted tryptic digestion and identification of proteolytically resistant proteins. Anal Biochem. 2008;377(2):259–266. doi: 10.1016/j.ab.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 55.FDA_Guidance. Guidance for industry: bioanalytical method validation. http://wwwfdagov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM368107pdf. 2013.
- 56.Brouwer KL, Keppler D, Hoffmaster KA, Bow DA, Cheng Y, Lai Y, et al. In vitro methods to support transporter evaluation in drug discovery and development. Clin Pharmacol Ther. 2013;94(1):95–112. doi: 10.1038/clpt.2013.81. [DOI] [PubMed] [Google Scholar]
- 57.Liu X, Chism JP, LeCluyse EL, Brouwer KR, Brouwer KL. Correlation of biliary excretion in sandwich-cultured rat hepatocytes and in vivo in rats. Drug Metab Dispos. 1999;27(6):637–644. [PubMed] [Google Scholar]
- 58.Liu X, LeCluyse EL, Brouwer KR, Lightfoot RM, Lee JI, Brouwer KL. Use of Ca2+ modulation to evaluate biliary excretion in sandwich-cultured rat hepatocytes. J Pharmacol Exp Ther. 1999;289(3):1592–1599. [PubMed] [Google Scholar]
- 59.Bi YA, Kazolias D, Duignan DB. Use of cryopreserved human hepatocytes in sandwich culture to measure hepatobiliary transport. Drug Metab Dispos. 2006;34(9):1658–1665. doi: 10.1124/dmd.105.009118. [DOI] [PubMed] [Google Scholar]
- 60.Pfeifer ND, Hardwick RN, Brouwer KL. Role of hepatic efflux transporters in regulating systemic and hepatocyte exposure to xenobiotics. Annu Rev Pharmacol Toxicol. 2014;54:509–535. doi: 10.1146/annurev-pharmtox-011613-140021. [DOI] [PubMed] [Google Scholar]
- 61.Reid LM, Jefferson DM. Culturing hepatocytes and other differentiated cells. Hepatology. 1984;4(3):548–559. doi: 10.1002/hep.1840040332. [DOI] [PubMed] [Google Scholar]
- 62.Chandra P, Lecluyse EL, Brouwer KL. Optimization of culture conditions for determining hepatobiliary disposition of taurocholate in sandwich-cultured rat hepatocytes. In Vitro Cell Dev Biol Anim. 2001;37(6):380–385. doi: 10.1007/BF02577575. [DOI] [PubMed] [Google Scholar]
- 63.Turncliff RZ, Tian X, Brouwer KL. Effect of culture conditions on the expression and function of Bsep, Mrp2, and Mdr1a/b in sandwich-cultured rat hepatocytes. Biochem Pharmacol. 2006;71(10):1520–1529. doi: 10.1016/j.bcp.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 64.Tchaparian EH, Houghton JS, Uyeda C, Grillo MP, Jin L. Effect of culture time on the basal expression levels of drug transporters in sandwich-cultured primary rat hepatocytes. Drug Metab Dispos. 2011;39(12):2387–2394. doi: 10.1124/dmd.111.039545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kimoto E, Yoshida K, Balogh LM, Bi YA, Maeda K, El-Kattan A, et al. Characterization of Organic Anion Transporting Polypeptide (OATP) expression and its functional contribution to the uptake of substrates in human hepatocytes. Mol Pharm. 2012;9:3535–3542. doi: 10.1021/mp300379q. [DOI] [PubMed] [Google Scholar]
- 66.Kotani N, Maeda K, Watanabe T, Hiramatsu M, Gong LK, Bi YA, et al. Culture period-dependent changes in the uptake of transporter substrates in sandwich-cultured rat and human hepatocytes. Drug Metab Dispos. 2011;39(9):1503–1510. doi: 10.1124/dmd.111.038968. [DOI] [PubMed] [Google Scholar]
- 67.Di L, Whitney-Pickett C, Umland JP, Zhang H, Zhang X, Gebhard DF, et al. Development of a new permeability assay using low-efflux MDCKII cells. J Pharm Sci. 2011;100(11):4974–4985. doi: 10.1002/jps.22674. [DOI] [PubMed] [Google Scholar]
- 68.Poller B, Drewe J, Krahenbuhl S, Huwyler J, Gutmann H. Regulation of BCRP (ABCG2) and P-glycoprotein (ABCB1) by cytokines in a model of the human blood–brain barrier. Cell Mol Neurobiol. 2010;30(1):63–70. doi: 10.1007/s10571-009-9431-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dickens D, Webb SD, Antonyuk S, Giannoudis A, Owen A, Radisch S, et al. Transport of gabapentin by LAT1 (SLC7A5) Biochem Pharmacol. 2013;85(11):1672–1683. doi: 10.1016/j.bcp.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 70.Poller B, Gutmann H, Krahenbuhl S, Weksler B, Romero I, Couraud PO, et al. The human brain endothelial cell line hCMEC/D3 as a human blood–brain barrier model for drug transport studies. J Neurochem. 2008;107(5):1358–1368. doi: 10.1111/j.1471-4159.2008.05730.x. [DOI] [PubMed] [Google Scholar]
- 71.Weksler B, Romero IA, Couraud PO. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS. 2013;10(1):16. doi: 10.1186/2045-8118-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carl SM, Lindley DJ, Couraud PO, Weksler BB, Romero I, Mowery SA, et al. ABC and SLC transporter expression and pot substrate characterization across the human CMEC/D3 blood–brain barrier cell line. Mol Pharm. 2010;7(4):1057–1068. doi: 10.1021/mp900178j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dauchy S, Miller F, Couraud PO, Weaver RJ, Weksler B, Romero IA, et al. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem Pharmacol. 2009;77(5):897–909. doi: 10.1016/j.bcp.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 74.Ohtsuki S, Ikeda C, Uchida Y, Sakamoto Y, Miller F, Glacial F, et al. Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood–brain barrier model. Mol Pharm. 2013;10(1):289–296. doi: 10.1021/mp3004308. [DOI] [PubMed] [Google Scholar]
- 75.Ohtsuki S, Uchida Y, Kubo Y, Terasaki T. Quantitative targeted absolute proteomics-based ADME research as a new path to drug discovery and development: methodology, advantages, strategy, and prospects. J Pharm Sci. 2011;100(9):3547–3559. doi: 10.1002/jps.22612. [DOI] [PubMed] [Google Scholar]
- 76.Lai Y. Identification of interspecies difference in hepatobiliary transporters to improve extrapolation of human biliary secretion. Expert Opin Drug Metab Toxicol. 2009;5(10):1175–1187. doi: 10.1517/17425250903127234. [DOI] [PubMed] [Google Scholar]
- 77.Niessen J, Jedlitschky G, Grube M, Kawakami H, Kamiie J, Ohtsuki S, et al. Expression of ABC-type transport proteins in human platelets. Pharmacogenet Genomics. 2010;20(6):396–400. doi: 10.1097/FPC.0b013e32833997b0. [DOI] [PubMed] [Google Scholar]
- 78.Ito K, Uchida Y, Ohtsuki S, Aizawa S, Kawakami H, Katsukura Y, et al. Quantitative membrane protein expression at the blood–brain barrier of adult and younger cynomolgus monkeys. J Pharm Sci. 2011;100(9):3939–3950. doi: 10.1002/jps.22487. [DOI] [PubMed] [Google Scholar]
- 79.Bi YA, Qiu X, Rotter CJ, Kimoto E, Piotrowski M, Varma MV, et al. Quantitative assessment of the contribution of sodium-dependent taurocholate co-transporting polypeptide (NTCP) to the hepatic uptake of rosuvastatin, pitavastatin and fluvastatin. Biopharm Drug Dispos. 2013;34(8):452–461. doi: 10.1002/bdd.1861. [DOI] [PubMed] [Google Scholar]
- 80.Ho RH, Tirona RG, Leake BF, Glaeser H, Lee W, Lemke CJ, et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130(6):1793–1806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 81.Fujino H, Saito T, Ogawa S, Kojima J. Transporter-mediated influx and efflux mechanisms of pitavastatin, a new inhibitor of HMG-CoA reductase. J Pharm Pharmacol. 2005;57(10):1305–1311. doi: 10.1211/jpp.57.10.0009. [DOI] [PubMed] [Google Scholar]
- 82.Greupink R, Dillen L, Monshouwer M, Huisman MT, Russel FG. Interaction of fluvastatin with the liver-specific Na+-dependent taurocholate cotransporting polypeptide (NTCP) Eur J Pharm Sci. 2011;44(4):487–496. doi: 10.1016/j.ejps.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 83.Proc JL, Kuzyk MA, Hardie DB, Yang J, Smith DS, Jackson AM, et al. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J Proteome Res. 2010;9(10):5422–5437. doi: 10.1021/pr100656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klammer AA, MacCoss MJ. Effects of modified digestion schemes on the identification of proteins from complex mixtures. J Proteome Res. 2006;5(3):695–700. doi: 10.1021/pr050315j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohtsuki S, Schaefer O, Kawakami H, Inoue T, Liehner S, Saito A, et al. Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: comparison with mRNA levels and activities. Drug Metab Dispos. 2012;40(1):83–92. doi: 10.1124/dmd.111.042259. [DOI] [PubMed] [Google Scholar]
- 86.Schaefer O, Ohtsuki S, Kawakami H, Inoue T, Liehner S, Saito A, et al. Absolute quantification and differential expression of drug transporters, cytochrome P450 enzymes, and UDP-glucuronosyltransferases in cultured primary human hepatocytes. Drug Metab Dispos. 2012;40(1):93–103. doi: 10.1124/dmd.111.042275. [DOI] [PubMed] [Google Scholar]
- 87.Atkinson AJ, Jr, Smith BP. Models of physiology and physiologically based models in clinical pharmacology. Clin Pharmacol Ther. 2012;92(1):3–6. doi: 10.1038/clpt.2012.67. [DOI] [PubMed] [Google Scholar]
- 88.Smith BJ. An industrial perspective on contemporary applications of PBPK models in drug discovery and development. Biopharm Drug Dispos. 2012;33(2):53–54. doi: 10.1002/bdd.1778. [DOI] [PubMed] [Google Scholar]
- 89.Wu CC, Yates JR., 3rd The application of mass spectrometry to membrane proteomics. Nat Biotechnol. 2003;21(3):262–267. doi: 10.1038/nbt0303-262. [DOI] [PubMed] [Google Scholar]