

Abstract
Phenethyl isothiocyanate (PEITC)—a naturally occurring isothiocyanate in cruciferous vegetables—has been extensively studied as a chemopreventive agent in several preclinical species and in humans. Pharmacokinetic features of unchanged PEITC are (I) linear and first-order absorption, (II) high protein binding and capacity-limited tissue distribution, and (III) reversible metabolism and capacity-limited hepatic elimination. Membrane transport of PEITC is mediated by BCRP, multidrug resistance-associated protein (MRP) 1, and MRP2 transporters belonging to the ATP-binding-cassette (ABC) family. PEITC is metabolized by glutathione S-transferase (GST) in the liver, with the glutathione conjugate of PEITC undergoing further conversion to mercapturic acid by N-acetyl transferase in rats and humans. PEITC modulates the activity and expression of numerous phase I and phase II drug-metabolizing enzymes and can inhibit the metabolism of procarcinogens to form carcinogens and increase carcinogen elimination. In recent years, several in vitro and in vivo studies have elucidated molecular mechanisms underlying the pharmacodynamics of PEITC in breast cancer that include cancer cell apoptosis by upregulation of apoptotic genes, cell cycle arrest at G2/M phase by generation of reactive oxygen species and depletion of intracellular glutathione, downregulation of the estrogen receptor, decrease in sensitivity to estrogen, and inhibition of tumor metastasis. Inhibition of angiogenesis is one of the recently reported mechanisms of breast cancer prevention by PEITC. Complex pharmacokinetics and pharmacodynamics of PEITC necessitate a systems-biology approach in parallel with PK/PD modeling to develop PEITC as a therapeutic agent for treating cancers.
KEY WORDS: breast cancer resistance protein (BCRP), glutathione S-transferase, multidrug resistance-associated protein (MRP), PEITC, Phenethyl isothiocyanate
INTRODUCTION
Isothiocyanates (ITCs) are formed by the breakdown of glucosinolates, which are major constituents of cruciferous vegetables (watercress, cabbage, brussels sprouts, cauliflower, etc.) (1). The enzyme myrosinase in plants and the microflora in the gastrointestinal tract are responsible for the release of ITCs from glucosinolates after physical damage to cruciferous vegetables (harvesting, cutting, or chewing) and after dietary ingestion, respectively (1,2). ITCs have drawn the attention of the scientific community since the mid-1980s due to their anti-cancer potential (2–5). ITCs inhibit carcinogenesis by several mechanisms including (I) inhibition of carcinogen metabolism by drug-metabolizing enzymes (DMEs) (6), (II) apoptosis of cancer cells (7), and (III) cell cycle inhibition (8). Recent evidence also suggests that ITCs are anti-angiogenic, and this may contribute to their cancer preventive effects (9).
The two major ITCs present in cruciferous vegetables that have been extensively studied are sulforaphane and phenethyl isothiocyanate. This review will focus on phenethyl isothiocyanate (PEITC) (Fig. 1), which is derived from the hydrolysis of its glucosinolate precursor gluconasturtiin (2). In humans, consumption of dietary glucosinolates is estimated to be about 300 mg/day (2) from various cruciferous vegetables and, for every 56.8 g of watercress consumed, approximately 12 mg of PEITC is released (10). PEITC is a low molecular weight (MW = 163.2 g/mol) compound and is fairly lipophilic (logP = 3.47). Of all the ITCs, PEITC is one of the most comprehensively studied ITCs in various cancers (5,7,11–14) and is currently being evaluated in a National Cancer Institute phase II clinical trial to prevent lung cancer in smokers. The beneficial role of PEITC in lung cancer (6) and prostate cancer (13) has been documented. In the past decade, significant findings have emerged in the area of breast cancer research, which will be reviewed in this publication. Overall, this review will discuss the (I) pharmacokinetics of PEITC, (II) membrane transport of PEITC, and (III) pharmacodynamics of PEITC, including modulation of DMEs and molecular mechanisms underlying anti-cancer effects of PEITC in breast cancer.
Fig. 1.
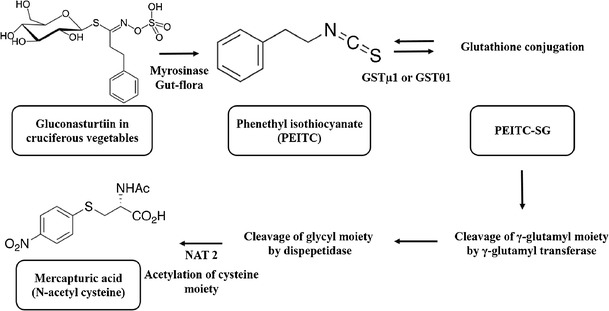
Synthesis and metabolism of phenethyl isothiocyanate in rats and humans
PHARMACOKINETICS OF PEITC
Pharmacokinetic Profiles in Humans and Rats
Ji et al.(15,16) determined the sample stability, plasma protein binding, and dose-dependent pharmacokinetics of PEITC in male Sprague Dawley rats, as well as the pharmacokinetics of PEITC following the ingestion of watercress in humans. For these studies, a validated novel analytical approach was utilized that involved ammonia derivatization of PEITC to phenethylthiourea and liquid chromatography-tandem mass spectrometry (LC/MS/MS) quantitation (15). Prior to the rat and human studies, the temperature- and pH-dependent stability of PEITC in biological samples was evaluated in order to avoid loss of PEITC during sampling or analysis. The first-order degradation half-life (t1/2d) of PEITC decreased from 68.2 to 15.3 h with an increase in the buffer pH from 3.0 to 10.1 at 25°C. At physiological pH of 7.4, PEITC was more stable at refrigerated temperature (t1/2d = 108 h; 4°C) than at room temperature (t1/2d = 56 h; 25°C). Binding of PEITC in rat serum was very high (about 98.1%) and concentration-independent over the range evaluated (10–1,000 μM).
A clinical study was performed in which four healthy subjects were given 100 g of watercress and their plasma and urine samples were collected (at time points up to 24 h) and PEITC plasma concentrations were determined using LC/MS/MS. A one-compartment pharmacokinetic model with first-order absorption and elimination was utilized to describe the pharmacokinetics of PEITC. The average peak plasma concentration (Cmax) was 928.5 nM (SD 250 nM), the time to reach Cmax (tmax) was 2.6 h (SD 1.1 h), the first-order absorption rate constant (ka) was 1.3/h (SD 0.3/h), and half-life (t1/2) was 4.9 h (SD 1.1 h) (15). Absorption of PEITC following watercress ingestion is a two-step process involving enzymatic conversion of gluconasturtiin into PEITC by gut flora and subsequent absorption from the gastrointestinal tract; therefore, the ka value of PEITC would thus be influenced by the slower of these two processes. Chung et al. (10) reported a dose-dependent urinary excretion of PEITC after various watercress doses in humans.
For the pharmacokinetic studies in rats (16), PEITC doses were administered intravenously (IV) (2, 10, 100, and 400 μmol/kg) and orally (10 and 100 μmol/kg) and samples were collected at time points up to 96 h. With these increasing IV doses of PEITC, its clearance (CL) (0.70, 0.68, 0.36, and 0.50 L/h/kg) decreased with increasing dose and its volume of distribution (1.94. 3.27, 2.66, and 5.72 L/kg) increased with increasing dose, indicating saturable metabolism and nonlinear tissue distribution. Interactions of ITCs with ATP-binding-cassette (ABC) transporters account for the observed nonlinear distribution of PEITC and have been reviewed (discussed later) (12). The half-life of PEITC was significantly longer for the 400 μmol/kg dose (13.1 h) and the 100 μmol/kg dose (9.19 h) than following the 2 μmol/kg dose (3.52 h), reflecting the dose-dependent disposition of PEITC. Absolute bioavailability (F) was 1.15 and 0.93 for the 10 and 100 μmol/kg oral doses, respectively; thus, the extent of absorption of PEITC is complete in rats. The first-order absorption rate constant was comparable between this study (1.8/h) and the previously described clinical study (1.3/h) (15). The tmax was obtained at 0.44 and 2.0 h at doses of 10 and 100 μmol/kg, respectively, indicating the rapid absorption of PEITC. PEITC plasma concentrations also demonstrated a less-than-dose proportional increase in Cmax and plasma AUC at both oral doses. The data was best fit to a three-compartment model with Michaelis-Menten terms to describe both the central elimination and the tissue distribution terms (Fig. 2a, b).
Fig. 2.
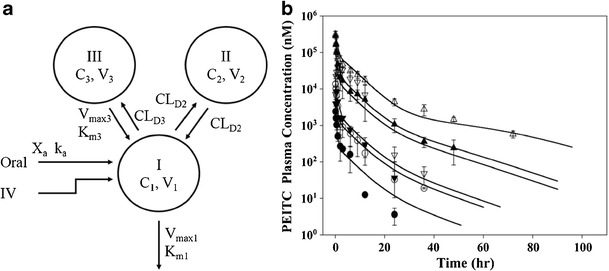
a Mechanistic pharmacokinetic model of unchanged phenethyl isothiocyanate after intravenous and oral administration and b the observed and predicted concentrations of PEITC. a X a represents the drug amount at the absorption site; C 1, C 2, and C3 represent the plasma concentrations of PEITC in central (I) and two peripheral compartments (II and III), respectively; V 1, V 2, and V 3 represent the volume of distribution of PEITC in central (I) and two tissue compartments (II and III), respectively; k a is the absorption rate constant; K m1 and V max1 are the Michaelis-Menten parameters to characterize clearance from the central compartment indicative of capacity-limited metabolism; CL D2 represents distribution clearance between the tissue compartment (II) and central compartment characterized by linear clearance. CL D3 represents distribution clearance from the central compartment to the tissue compartment (III); K m3 and V max3 are the Michaelis-Menten parameters to characterize from the tissue compartment (III) to the central compartment, indicative of transporter-mediated clearance. b Rats were dosed with 2 (closed circles), 10 (open circles), 100 (closed triangles), or 400 mmol/kg (open triangles) of PEITC intravenously or 10 (inverted closed triangles) and 100 mmol/kg (inverted open triangles) orally. Data are expressed as mean ± SD, n = 3 or 4. All the data were fitted simultaneously by ADAPT II, and the lines represent predicted plasma PEITC concentrations using the proposed compartmental model. b Reproduced with permission from Springer from Ji et al. (16)
In another study (17), male Wistar rats (250 ± 10 g; n = 4 per group) were given 0.5, 1.0, and 5.0 mg/kg (3.1, 6.2, and 15.5 μmol/kg) of PEITC (in 1% DMSO) by gastric intubation as a single dose, and as multiple doses, once a day for 4 days, to examine the effect of lower oral doses and multiple dosing of PEITC. A dose of 0.5 mg/kg (3.1 μmol/kg) PEITC was given by IV administration to obtain F and for comparison of pharmacokinetic parameters between the two routes of administration. Samples were collected on the first and last days of PEITC administration, and concentrations were measured using LC/MS. Noncompartmental analysis was used to obtain absorption, distribution, and elimination parameters for single and repeated dosing regimens. Bioavailability decreased with an increase in oral single doses: 0.77 ± 0.13 (0.5 mg/kg oral dose), 0.61 ± 0.11 (1 mg/kg), and 0.23 ± 0.05 (5 mg/kg). These estimates are dependent on the assumption of similar clearance values following the oral doses and that following the 0.5-mg/kg IV dose. With repeated dosing regimens, estimated F values tended to be higher than after single doses. The absorption rate constant was not dose-dependent indicating first-order absorption. PEITC was rapidly absorbed as the tmax was less than an hour; however, values of Cmax increased less-than-dose proportionally for all single and repeated dosing regimens. Taken together, these two studies (15,17) suggest that PEITC demonstrates dose-dependent pharmacokinetics, where absorption does not contribute to its observed nonlinear behavior. The differences in bioavailability reported in the two studies require further study.
Metabolism of PEITC
PEITC is predominantly cleared by metabolism in humans and animals. In humans and rats, glutathione S-transferases (GST) (18) catalyze the conversion of PEITC to its glutathione conjugate (PEITC-SG) in the liver. γ-Glutamyl and glycyl moieties of PEITC-SG are cleaved by γ-glutamyl transferase and dipeptidase, respectively. The resulting cysteine conjugate undergoes N-acetylation of its cysteine moiety by N-acetyltransferase 2 (NAT2) and is excreted as mercapturic acid in the urine (18,19). Formation of mercapturic acid conjugates by NAT2 can occur in the kidneys although metabolism has also been demonstrated in the liver and the location is likely species-dependent (20). PEITC is metabolized by GST isoforms, GSTμ1 and GSTθ1 (17), which are present in the intestine, liver, and kidney. Conjugation of PEITC with glutathione would depend on intracellular concentrations of glutathione (21) and on genetic variants and polymorphisms in GST isoforms, and both of these factors may be responsible for the observed differences in plasma concentrations (22). In fact, increased chemopreventive effects have been associated with GST polymorphisms (23) that result in decreased metabolism of PEITC and therefore higher plasma concentrations. GSTs also catalyze the reversible metabolism of PEITC/PEITC-SG (24). The PEITC-SG conjugate is labile and can spontaneously convert to PEITC and GSH at physiological pH with an estimated t1/2 of 44 min. This reversible metabolism adds to the complex PK of PEITC observed in studies.
The first metabolic study of PEITC was reported in A/J mice, where mice were dosed with 5 μM (2 μCi/mouse) of [14C] PEITC by oral gavage and organs were collected via terminal sampling at various time points up to 72 h. Since a nonspecific (radioactivity) assay was used to measure PEITC accumulation in the collected organs, these results cannot be used to elucidate the distribution profile of unchanged PEITC. At 72 h postadministration, almost 80% of radioactivity was collected in urine. An HPLC assay was used to identify a cyclic mercaptopyruvic acid conjugate (major metabolite; 25% of the administered dose) and N-acetyl cysteine conjugate (minor metabolite; 10% of the administered dose)—the two metabolites of PEITC in urine (25). In another study, dose-dependent excretion of an N-acetyl cysteine conjugate was measured (using NMR/MS) after ingestion of 30 and 57 g of watercress in four human individuals (10). Unlike mice, only the N-acetyl cysteine conjugate is excreted in humans (10,26) and in rats (19). Figure 1 illustrates the synthesis and metabolism of PEITC in rats and humans.
Membrane Transport of PEITC
Studies have evaluated whether PEITC is a substrate and/or inhibitor of the ABC transporters P-glycoprotein (ABCB1), multidrug resistance-associated proteins 1 and 2 (MRP1, ABCC1; MRP2, ABCC2), and breast cancer-resistant protein (BCRP, ABCG2). The IC50 values of PEITC were similar in MCF-7/Adr—overexpressing P-glycoprotein (P-gp; ABCB1)—and MCF-7/wt cell lines, suggesting that P-gp interactions may not contribute to cytotoxicity in these cell lines through changes in PEITC intracellular concentrations. Accumulation of PEITC was measured in PANC-1 (overexpressing multidrug resistance-associated protein 1, MRP1; ABCC1) and in MCF-7/Adr, MDA435/LCC6, and Caco-2 (all overexpressing P-gp) cell lines at 2 h postincubation (21). In the presence of verapamil, a P-gp inhibitor, accumulation of PEITC remained unchanged in all three P-gp overexpressing cell lines, confirming that PEITC was not a substrate for P-gp. However, accumulation of PEITC significantly increased in PANC-1 cells in the presence of MK571, an inhibitor of MRP1. As MRPs mediate efflux of substrates via conjugation with intracellular glutathione or in association with glutathione, further studies confirmed that PEITC inhibits the MRP1-mediated efflux of daunomycin (DNM) by competing for glutathione and through MRP1 interactions (21). Glutathione depletion, which results in impaired transport by MRPs, was further evaluated using the MDCKII/MRP2 cell line (27); inhibition of glutathione synthesis by buthionine sulfoxinine resulted in increased accumulation of PEITC. In same study, PEITC was reported to be a substrate for MRP2 (ABCC2) (27). Depletion of glutathione at high concentrations of PEITC explains the nonlinear volume of distribution and prolonged tissue binding of PEITC (16), which increases the intracellular residence time of PEITC. Studies have demonstrated that (I) PEITC (10 or 30 μM) competes for transport with mitoxantrone (a BCRP substrate), (II) cellular accumulation of PEITC is significantly lower in MCF-7 cells which overexpress BCRP compared with NCL-H460 cells which are BCRP-negative large cell lung carcinoma cells, and (III) cellular accumulation of PEITC increases in the presence of fumitremorgin C, a BCRP inhibitor (28). PEITC was also reported to be a substrate for BCRP and an inhibitor of BCRP-ATPase activity using inside-out membrane vesicles prepared from MCF-7/MX100 cells (29). Overall, the results from cell culture studies suggest that PEITC and/or PEITC-SG represents an inhibitor and substrate for MRP1 and MRP2 (21,27) and that PEITC itself is also an inhibitor and substrate for breast cancer resistance protein (BCRP; ABCG2) (28). Thus, transport of PEITC or PEITC glutathione conjugates by ABC transporters represents an important feature of PEITC PK/PD.
Overall, the observed pharmacokinetic behavior of PEITC (10,15–17) (Fig. 3) indicates that (I) PEITC is rapidly absorbed by a first-order absorption process, (II) PEITC undergoes saturable metabolism at high doses and potential deconjugation of PEITC-SG; (III) PEITC-SG is converted to mercapturic acid by N-acetylation, which is excreted into urine; (VI) PEITC is highly protein bound; and (V) PEITC, being lipophilic, is rapidly distributed into tissues where it can undergo glutathione (SG) conjugation and both the parent PEITC and the PEITC-SG conjugate are actively effluxed from cells by MRP1/2 (ABCC1/2). Saturation of the efflux transporter or inhibition of transport due to the depletion of intracellular glutathione (21) contributes to the nonlinear increase in volume of distribution with increasing doses.
Fig. 3.
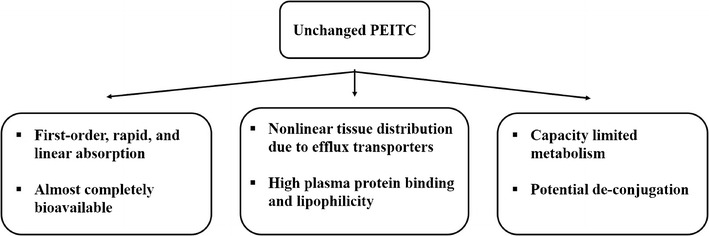
Absorption and disposition of PEITC in vivo
CANCER PREVENTION/TREATMENT EFFECTS OF PEITC
An important pharmacodynamic effect of PEITC is its cancer prevention properties against a broad range of cancers. Epidemiological studies have indicated that the risk of lung, colon, breast, stomach, and prostate cancers is decreased with increasing dietary intake of PEITC. High urinary ITC levels are significantly associated with reduced breast cancer risk in pre- and postmenopausal women (30). Terry et al. (31) reported that consumption of cruciferous vegetables lowers the risk of breast cancer by as much as 20–40% in postmenopausal women.
Modulation of DMEs by PEITC
First reports of the anti-carcinogenic potential of PEITC by inhibition of nitrosamine metabolism occurred in the mid-1980s to early 1990s. Nitrosamines are by-products of industrial waste, which are activated in the liver by CYP450 enzymes (α-hydroxylation) to form diazohydroxide intermediates. They cause alkylation of cellular components, particularly DNA, resulting in the initiation of tumorigenesis (32). Mechanisms of tumor initiation are summarized in a review by Hecht (6). Chung et al. (33) assessed the inhibition of α-hydroxylation of two carcinogenic nitrosamines, N-nitrosodimethylamine (NDMA) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), by PEITC in vitro in liver microsomes of F344 rats and in vivo in F344 rats and found decreased nitrosamine metabolism in vitro and reduced DNA damage in rat livers in vivo with acute (2 h) and chronic (2 weeks) PEITC treatment. Several studies (34–37) further investigated the role of PEITC in alterations of drug-metabolizing enzymes leading to changes in procarcinogen activation and carcinogen elimination.
Ishizaki et al. (34) showed that PEITC suppressed the activity and expression of CYP2E1 in vitro in rat liver microsomes and in vivo in rats. In liver microsomes isolated from acetone-treated rats, PEITC inhibited CYP2E1-mediated α-hydroxylation of NDMA with an apparent Ki of 1.0 μM. The observed inhibition of NDMA demethylation by PEITC was both time- and concentration-dependent. Immunoblotting studies showed a marked decrease in CYP2E1 expression but an 11-fold increase in the activity of CYP2B1 determined 24 h after a single oral dose of PEITC (1 mmol/kg) in vivo (34). Gao et al. (36) characterized effects of PEITC on phase I and phase II metabolizing enzymes in the liver, lung, and nasal mucosa of F344 rats after a single oral dose of 1 mmol/kg PEITC and found PEITC-mediated modulation of DMEs to be tissue specific. In the liver (L), lung (Lu), and nasal mucosa (N), the activities of DMEs were inhibited (↓), induced (↑), or remained unchanged (↔): NDMA-demethylase (CYP2E1) (L = 80% ↓ at 2 h and remained 40% ↓ at 48 h), pentoxyresorufin O-dealkylase (CYP2B1) (L = 10-fold ↑; Lu = ↔; N = 40–50% ↓), NMK-demethylase (CYP2E1) (L = 60% ↓ at 2 h; Lu = 70% ↓ at 2 h; N = 50% ↓ at 18 h), NAD(P)H quinone oxidoreductase (L = 5-fold ↑; Lu = ↔; N = ↔), GST (L = 1.5-fold ↑; Lu = ↔; N = ↔), and sulfotransferase (L = 32–48% ↓ between 24 and 48 h; Lu = ↔; N = 1.5–2.5-fold ↑ between 24 and 48 h). The activity of UDP-glucuronosyltransferase (UGT) increased moderately at 24 h. The alterations in DMEs by PEITC were reversible in nature (36).
Morse et al. (37) examined the effect of PEITC in NNK-induced lung tumorigenesis using tumor biopsies of A/J mice and F344 rats. Their results were consistent with previous data (33,34), suggesting that the inhibition of lung tumorigenesis by PEITC was a result of inhibition of the NNK metabolism pathway in the liver. Doerr-O’Rourke et al. (35) observed similar inhibitory effects of PEITC on NNK metabolism in cultured rat lung tissues. Moreover, the metabolism of NNK by CYP1A2, CYP2A6, CYP2D6, CYP2E1, and CYP3A4 has been reported in humans (38–40).
Further studies characterized the inhibitory potential of PEITC on both phase I and II enzymes. This was first studied in microsomes from baculovirus-infected insect cells overexpressing human CYP isoforms (41). Inhibition constants (Ki) were measured (mean ± SD) over a wide range of concentrations of PEITC for various probe-substrates of CYP isoforms. PEITC inhibited activities of CYP1A2/phenacetin (4.5 ± 1.0 μM) and CYP2A6/coumarin (18.2 ± 2.5 μM) via competitive inhibition; of CYP2B6/benzyloxyresorufin (1.5 ± 0.0 μM), CYP2C9/S-warfarin (6.5 ± 0.9 μM), CYP2C19/S-mephenytoin (12.0 ± 3.2 μM), CYP2D6/bufuralol (28.4 ± 7.9 μM); and CYP2E1/chlorzoxazone (21.5 ± 3.4 μM) via noncompetitive inhibition; and of CYP3A4/testosterone via both competitive (34.0 ± 6.5 μM) and noncompetitive (63.8 ± 12.5 μM) inhibition. Data suggest that PEITC could alter the pharmacokinetics of these clinically used drugs; however, not all PEITC-drug interactions lead to clinically significant drug-drug interactions (42,43).
Konsue et al. (44) characterized the modulation of the activity and expression of CYPs and phase II enzymes by dietary doses (0.06, 0.6, and 6.0 μmol/g) of PEITC in male Wistar albino rats (180 ± 10 g) and found that changes in activities of DMEs were tissue-specific and dose-dependent. After 14 days of PEITC administration, activities and qualitative changes in protein expression of several DMEs were measured in the rat liver, lungs, and kidneys using probe-substrates and immunoblotting assays, respectively. In the liver, expression of CYP2B1 and CYP3A4 increased and expression of CYP1A1, CYP1A2, CYP1B1, and CYP2E1 decreased in a dose-dependent manner. In contrast to the liver, expression of CYP1A1, CYP1A2, and CYP1B1 increased moderately in a dose-dependent manner in the lungs and CYP2E1 expression showed a significant increase. In the kidneys, expression of these enzymes did not change. In the liver, expression of GSTα, GSTμ, and GSTп increased sharply with dose; however, expression remained unchanged in the lungs and kidneys. Konsue et al. (45,46) further performed in vitro studies using precision-cut rat and human liver slices to assess modulation of DMEs by PEITC (0–50 μM). In rat liver slices, expression of GSTα, GSTμ, GSTп, and NAD(P)H quinone oxidoreductase increased but no conclusions could be drawn for human liver slices due to the high variability observed. In both rat and human liver slices, expression of CYP1A1, CYP1A2, and CYP2B6 decreased and CYP3A4 and CYP3A2 increased in a concentration-dependent manner (46). It was also reported that unchanged PEITC (not its metabolites) modulates the activity and expression of DMEs in rats and humans (47).
Telang et al. (48) evaluated the effect of oral PEITC administration of a 150 μmol/kg dose daily for 5 days on hepatic gene regulation in male Sprague Dawley rats using a gene array of 282 genes and quantitative RT-PCR assays. Gene expression ratios (treatment/control) of Cyp2b15 (3.01) and Ugt1a6 (5.67) indicated strong upregulation of these genes and a strong downregulation of nicotinamide N-methyltransferase (NNMT), an important cancer marker; observed changes in mRNA expression of NNMT and Ugt1a6 were confirmed by quantitative RT-PCR assays. An apoptotic gene Bcl2l2 and several stress regulator genes (Dnajb9, Dnajc5, and Hspb1) were also upregulated in the liver, suggesting alterations in gene transcription by PEITC.
Molecular Mechanisms Underlying Anti-cancer Effects of PEITC in Breast Cancer
Several molecular targets of PEITC for chemoprevention have been extensively reviewed (14) and only those relevant to breast cancer will be discussed in this section. Tseng et al. (49) first reported cytotoxic effects of PEITC and other organic ITCs in human breast cancer MCF-7/Adr, mammary epithelial MCF-12A, and human kidney HK-2 cell lines. After 48 h of exposure, IC50 (mean ± SE) values of PEITC and two conventional anti-cancer drugs, DNM and vinblastine (VBL), were 7.32 ± 0.25, 7.12 ± 0.42, and 0.106 ± 0.004 μM, respectively, in MCF-7/Adr cells. Corresponding IC50 values of PEITC were 7.70 ± 0.07 and 3.27 ± 0.04 μM in MCF-12A and HK-2 cells lines, respectively.
Telang et al. (50) compared expression of genes related to apoptosis and cellular proliferation, cell adhesion, estrogen receptor and gene interactions, and prognostic cancer markers following two doses of PEITC (0.3 and 3.0 mM) using an array of 112 genes determined in normal human mammillary epithelial (HME) cells and breast cancer MCF-7 cells. At 0.3 and 3.0 μM concentrations of PEITC, expression levels of 23 and 4 genes were significantly altered in HME cells at these two PEITC concentrations, respectively, and no changes in the expression of genes were detected in MCF-7 cells. Novel findings were upregulation of BCL2-antagonist of cell death (BAD) (24.08-fold ↑; apoptotic gene), estrogen receptor 2 (ERβ) (7.55-fold ↑), p21 (5.28-fold ↑; tumor suppressor), and p27 (14.79-fold ↑; tumor suppressor) at a PEITC concentration of 0.3 μM in HME cells. In an additional study in MCF-7 cells examining other relevant breast cancer genes, upregulation of p57 (5.23-fold ↑), Brca2 (4.22-fold ↑), Il-2 (3.16-fold ↑), Atf-2 (2.6-fold ↑), p53 (2.12-fold ↑), and Hsp27 (1.84-fold ↑) genes was detected following incubation with 3.0 μM of PEITC (51). Taken together (50,51), increases in (I) p21, p27, and p57 result in cell cycle arrest (G2 phase); (II) Il-2 results in cell cycle arrest (G2 phase); (III) Hsp27 and Atf-2 result in increased DNA repair and protection from subsequent DNA damage; and (IV) p53 and Brca2 result in increased DNA repair and apoptosis of cells with damaged DNA.
Studies (52,53) have identified suppression of ERα protein expression by PEITC (0–20 μM) as a potential mechanism of breast cancer inhibition in MCF-7, T-47D, and H3396 cell lines. PEITC (10 μM) was a more potent ERα inhibitor than a conventional inhibitor, ICI 182,780 (10 μM). It caused cell growth inhibition, as a consequence of impaired signaling of mitogenic estrogen by ERα. Interactions of PEITC with B cell lymphoma 2 interacting mediator (Bim) protein (54) led to apoptosis of breast cancer cells and was independent of PEITC-induced p53 upregulation. Knockdown by small interfering RNA of Bim in MCF-7 and MDA-MB-231 cell lines showed partial but significant absence of apoptosis (54). Inhibition of heat shock proteins (HSPs) 27, 70, and 90 and of heat shock factor 1 by PEITC in MDA-MB-231 and MCF-7 cell lines resulted in an impaired cell cycle regulation (downregulation of Cyclin B1, CDK1, Cdc25C, PLK-1), cell cycle arrest (G2/M phase), and apoptosis (activation of caspases 3 and 9) of breast cancer cells (55). A synergistic effect of paclitaxel and PEITC was reported on cell cycle arrest at the G2/M phase that is more pronounced than either paclitaxel or PEITC alone in MCF-7 and MDA-MB-231 cell lines, respectively (56). Cell cycle arrest at the G2/M phase by PEITC in MCF-7 and MDA-MB-231 cell lines has also been attributed to generation of reactive oxidative species (ROS) due to intracellular glutathione depletion (57).
Recently reported studies (9,58–60) have begun to elucidate the role of PEITC in breast cancer in vivo using various animal models. PEITC significantly (I) decreased the protein expression of HER2 receptor and epidermal growth factor receptor (EGFR) and phosphorylation of signal transducer and activator of transcription 3 (STAT3) and (II) increased apoptosis by selectively cleaving caspase 3 and poly-ADP ribose polymerase (PARP) proteins in MCF-7/HH and MDA-MB-231/HH cell lines overexpressing HER2 (HH) in vitro and in female SCID/NOD mice (12 μmol PEITC orally until day 35) implanted with MDA-MB-231/HH cells in vivo. Although authors attributed the resulting cell apoptosis mainly to PEITC-HER2 interactions, cytotoxicity produced by PEITC in MCF-7 (IC50 = 14 μM) and MDA-MB-231 (IC50 = 8 μM) cell lines was similar to those reported by Liu et al. (56), suggesting that cell cycle arrest at G2/M phase could ultimately be responsible for apoptosis. In a follow-up study, Gupta et al. (59) reported that PEITC reduced brain metastasis of MDA-MB-231-BR breast cancer cells (brain seeking) in female athymic nude mice administered cells stably loaded with quantum dots. PEITC (10 μmol) was given orally, 10 days prior to and for 14 days after MDA-MB-231-BR cell injection. Moreover, survival of mice was prolonged by 20.5% and the brain autopsies of the treatment group showed significantly reduced protein expression of HER2 and EGFR after PEITC treatment (59). Administration of PEITC (8 mmol/kg) resulted in 2–3-fold reduction in protein expression of ERα and two transcriptional factors related to estrogen responsiveness of breast cancer cells, FOXA1 and GATA1, in a polyoma middle-T antigen (PyMT) transgenic mice model of type A luminal breast cancer (60).
Aras et al. (9) was first to report the role of PEITC in the initiation and progression of N-methyl nitrosourea (NMU)-induced breast cancer in female rodents. Female Sprague Dawley rats (8 weeks old) were given 50 or 150 μmol/kg PEITC orally, starting prior to NMU administration, and tumor progression was observed for 18 weeks. The PEITC treatment prolonged the tumor-free survival time and decreased the tumor incidence and multiplicity. The time to the first palpable tumor was prolonged from 69 days (control) to 84 days (50 μmol/kg PEITC) and 88 days (150 μmol/kg PEITC). The tumor incidence was 56.6%, 25.0%, and 17.2% in the control, 50, and 150 μmol/kg PEITC-treated groups, respectively, while the tumor multiplicity was 1.03, 0.25, and 0.21, respectively. There were no significant differences between the two doses. The intratumoral capillary density decreased from 4.21 ± 0.30 vessels per field (vpf) (control) to 2.46 ± 0.25 vpf (50 μmol/kg) and 2.36 ± 0.23 vpf (150 μmol/kg) in PEITC-treated animals. Anti-angiogenic effects of PEITC in a chemically induced breast cancer animal model were reported thus for the first time. Anti-angiogenic effects of PEITC have been also reported in a prostate cancer animal model (61). Table I and Fig. 4 summarize studies that have examined the anti-breast cancer effects of PEITC.
Table I.
Summary of In Vitro and In Vivo Studies That Characterized the Pharmacodynamics of PEITC in Breast Cancer
| In vitro studies | |||
| Cell line | PEITC | Summary | Reference |
| HME and MCF-7 cells | 0.3 and 3.0 μM | Comparison of gene expression between normal human mammary and breast cancer cells | (50) |
| HME cells | 3.0 μM | Gene expression in normal mammary cells | (51) |
| MCF-7, T-47D, and H3396 cells | 10 μM | Suppression of ERalpha expression | (52,53) |
| MCF-7 and MDA-MB-231 cells | 2.5 and 5.0 μM | Apoptosis mediated by BIM protein | (54) |
| MCF-7 and MDA-MB-231 cells | 10 μM | Apoptosis and cell cycle arrest at G2/M phase mediated by inhibition of HSP | (55) |
| MCF-7 and MDA-MB-231 cells | 0.1–10,000 nM | Apoptosis and cell cycle arrest at G2/M phase | (56) |
| MCF-7 and MDA-MB-231 | 10 μM | Cell-cycle arrest at G2/M phase mediated by generation of ROS and depletion of intracellular glutathione | (57) |
| MDA-MB-231/HH and MCF-7/HH cells | 1–40 μM | Decrease in expression of EGFR, HER2, and STAT3 and increase in expression of BAX, BIM, and PARP leading to apoptosis; increase in generation of ROS | (58) |
| In vivo studies | |||
| Animal model | PEITC | Summary | Reference |
| SCID/NOD female mice xenograft model (MDA-MB-231/HH cells) | 12 μmol | Decrease in expression of EGFR, HER2, and STAT3 and increase in expression of BAX, BIM, and PARP leading to apoptosis; increase in generation of ROS | (58) |
| Female athymic nude mice xenograft model (MDA-MB-231-BR cells) | 10 μmol | Prevention of metastasis in brain; increase in survival by 20.5%; decrease in expression of EGFR, HER2, and STAT3 | (59) |
| PyMT transgenic mice | 8 mmol/kg | Decrease in expression of ERalpha, FOXA1, and GATA1 | (60) |
| Female Sprague Dawley rats with N-methyl nitrosourea-induced breast cancer | 50 μmol/kg/150 μmol/kg | Decrease in angiogenesis, tumor incidence, and tumor multiplicity; increase in survival time | (9) |
HME human mammillary epithelial cells, PEITC phenethyl isothiocyanate, HSP heat shock proteins, EGFR epidermal growth factor receptor, STAT3 signal transducer and activator of transcription 3, PARP poly-ADP ribose polymerase, ROS reactive oxidative species
Fig. 4.
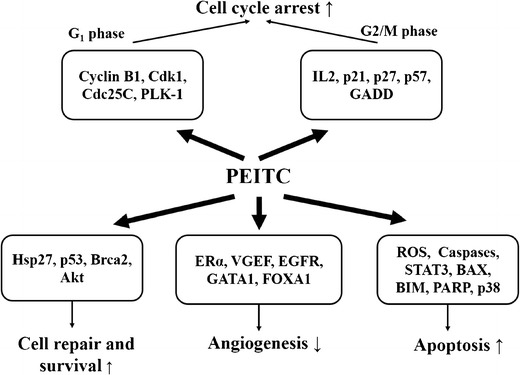
Mechanisms of anti-breast cancer activity of phenethyl isothiocyanate
CONCLUSIONS
Phenethyl isothiocyanate, a naturally occurring ITC, is a component of cruciferous vegetables. It modulates the activity and protein expression of a wide range of phase I oxidative and phase II conjugating DMEs inhibiting the formation of carcinogens and increasing their detoxification. PEITC demonstrates dose-dependent pharmacokinetics with (I) first-order absorption, (II) nonlinear tissue distribution due to interactions with ABC transporters and intracellular glutathione depletion, and (III) capacity-limited elimination due to saturation of GSTs in the liver. Pharmacodynamics of PEITC in breast cancer involves several cell membrane specific, cytosolic, and nuclear mechanisms, ultimately resulting in cell cycle arrest and apoptosis of breast cancer cells. An interesting and recent mechanism described for PEITC is anti-angiogenesis which may contribute to its cancer preventive effects in breast cancer. Since PEITC demonstrates a wide range of pharmacodynamic outcomes at the cellular, organ, and species levels, there is the need for a systems-biology approach in parallel with PK/PD modeling to develop PEITC as a therapeutic agent for treating cancers.
Acknowledgments
This study was supported in part by NIH grants DA023223 and CA121404. RAD was supported in part by a fellowship from the Kapoor Foundation.
REFERENCES
- 1.Fenwick GR, Heaney RK, Mullin WJ. Glucosinolates and their breakdown products in food and food plants. Crit Rev Food Sci Nutr. 1983;18(2):123–201. doi: 10.1080/10408398209527361. [DOI] [PubMed] [Google Scholar]
- 2.Talalay P, Fahey JW. Phytochemicals from cruciferous plants protect against cancer by modulating carcinogen metabolism. J Nutr. 2001;131(11 Suppl):3027S–33. doi: 10.1093/jn/131.11.3027S. [DOI] [PubMed] [Google Scholar]
- 3.Wattenberg LW. Inhibition of carcinogenesis by minor anutrient constituents of the diet. Proc Nutr Soc. 1990;49(2):173–83. doi: 10.1079/PNS19900022. [DOI] [PubMed] [Google Scholar]
- 4.Hecht SS. Chemoprevention by isothiocyanates. J Cell Biochem Suppl. 1995;22:195–209. doi: 10.1002/jcb.240590825. [DOI] [PubMed] [Google Scholar]
- 5.Hecht SS. Inhibition of carcinogenesis by isothiocyanates. Drug Metab Rev. 2000;32(3–4):395–411. doi: 10.1081/DMR-100102342. [DOI] [PubMed] [Google Scholar]
- 6.Hecht SS. Chemoprevention of cancer by isothiocyanates, modifiers of carcinogen metabolism. J Nutr. 1999;129(3):768S–74. doi: 10.1093/jn/129.3.768S. [DOI] [PubMed] [Google Scholar]
- 7.D’Agostini F, Izzotti A, Balansky RM, Bennicelli C, De Flora S. Modulation of apoptosis by cancer chemopreventive agents. Mutat Res. 2005;591(1–2):173–86. doi: 10.1016/j.mrfmmm.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 8.Hasegawa T, Nishino H, Iwashima A. Isothiocyanates inhibit cell cycle progression of HeLa cells at G2/M phase. Anticancer Drugs. 1993;4(2):273–9. doi: 10.1097/00001813-199304000-00021. [DOI] [PubMed] [Google Scholar]
- 9.Aras U, Gandhi YA, Masso-Welch PA, Morris ME. Chemopreventive and anti-angiogenic effects of dietary phenethyl isothiocyanate in an N-methyl nitrosourea-induced breast cancer animal model. Biopharm Drug Dispos. 2013;34(2):98–106. doi: 10.1002/bdd.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung FL, Morse MA, Eklind KI, Lewis J. Quantitation of human uptake of the anticarcinogen phenethyl isothiocyanate after a watercress meal. Cancer Epidemiol Biomarkers Prev. 1992;1(5):383–8. [PubMed] [Google Scholar]
- 11.Hecht SS. Chemoprevention of lung cancer by isothiocyanates. Adv Exp Med Biol. 1996;401:1–11. doi: 10.1007/978-1-4613-0399-2_1. [DOI] [PubMed] [Google Scholar]
- 12.Telang U, Ji Y, Morris ME. ABC transporters and isothiocyanates: potential for pharmacokinetic diet-drug interactions. Biopharm Drug Dispos. 2009;30(7):335–44. doi: 10.1002/bdd.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang LG, Chiao JW. Prostate cancer chemopreventive activity of phenethyl isothiocyanate through epigenetic regulation (review) Int J Oncol. 2010;37(3):533–9. doi: 10.3892/ijo_00000702. [DOI] [PubMed] [Google Scholar]
- 14.Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010;12(1):87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji Y, Morris ME. Determination of phenethyl isothiocyanate in human plasma and urine by ammonia derivatization and liquid chromatography-tandem mass spectrometry. Anal Biochem. 2003;323(1):39–47. doi: 10.1016/j.ab.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Ji Y, Kuo Y, Morris ME. Pharmacokinetics of dietary phenethyl isothiocyanate in rats. Pharm Res. 2005;22(10):1658–66. doi: 10.1007/s11095-005-7097-z. [DOI] [PubMed] [Google Scholar]
- 17.Konsue N, Kirkpatrick J, Kuhnert N, King LJ, Ioannides C. Repeated oral administration modulates the pharmacokinetic behavior of the chemopreventive agent phenethyl isothiocyanate in rats. Mol Nutr Food Res. 2010;54(3):426–32. doi: 10.1002/mnfr.200900090. [DOI] [PubMed] [Google Scholar]
- 18.Wu X, Zhou QH, Xu K. Are isothiocyanates potential anti-cancer drugs? Acta Pharmacol Sin. 2009;30(5):501–12. doi: 10.1038/aps.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mennicke WH, Gorler K, Krumbiegel G. Metabolism of some naturally occurring isothiocyanates in the rat. Xenobiotica Fate Foreign Compd Biol Syst. 1983;13(4):203–7. doi: 10.3109/00498258309052256. [DOI] [PubMed] [Google Scholar]
- 20.Hinchman CA, Matsumoto H, Simmons TW, Ballatori N. Intrahepatic conversion of a glutathione conjugate to its mercapturic acid. Metabolism of 1-chloro-2,4-dinitrobenzene in isolated perfused rat and guinea pig livers. J Biol Chem. 1991;266(33):22179–85. [PubMed] [Google Scholar]
- 21.Hu K, Morris ME. Effects of benzyl-, phenethyl-, and alpha-naphthyl isothiocyanates on P-glycoprotein- and MRP1-mediated transport. J Pharm Sci. 2004;93(7):1901–11. doi: 10.1002/jps.20101. [DOI] [PubMed] [Google Scholar]
- 22.Lampe JW, Peterson S. Brassica, biotransformation and cancer risk: genetic polymorphisms alter the preventive effects of cruciferous vegetables. J Nutr. 2002;132(10):2991–4. doi: 10.1093/jn/131.10.2991. [DOI] [PubMed] [Google Scholar]
- 23.Matic M, Pekmezovic T, Djukic T, Mimic-Oka J, Dragicevic D, Krivic B, et al. GSTA1, GSTM1, GSTP1, and GSTT1 polymorphisms and susceptibility to smoking-related bladder cancer: a case–control study. Urol Oncol. 2013;31(7):1184–92. doi: 10.1016/j.urolonc.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Kolm RH, Mannervik B, Talalay P. Reversible conjugation of isothiocyanates with glutathione catalyzed by human glutathione transferases. Biochem Biophys Res Commun. 1995;206(2):748–55. doi: 10.1006/bbrc.1995.1106. [DOI] [PubMed] [Google Scholar]
- 25.Eklind KI, Morse MA, Chung FL. Distribution and metabolism of the natural anticarcinogen phenethyl isothiocyanate in A/J mice. Carcinogenesis. 1990;11(11):2033–6. doi: 10.1093/carcin/11.11.2033. [DOI] [PubMed] [Google Scholar]
- 26.Adesida A, Edwards LG, Thornalley PJ. Inhibition of human leukaemia 60 cell growth by mercapturic acid metabolites of phenylethyl isothiocyanate. Food Chem Toxicol. 1996;34(4):385–92. doi: 10.1016/0278-6915(96)00124-X. [DOI] [PubMed] [Google Scholar]
- 27.Ji Y, Morris ME. Transport of dietary phenethyl isothiocyanate is mediated by multidrug resistance protein 2 but not P-glycoprotein. Biochem Pharmacol. 2005;70(4):640–7. doi: 10.1016/j.bcp.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 28.Ji Y, Morris ME. Effect of organic isothiocyanates on breast cancer resistance protein (ABCG2)-mediated transport. Pharm Res. 2004;21(12):2261–9. doi: 10.1007/s11095-004-7679-1. [DOI] [PubMed] [Google Scholar]
- 29.Ji Y, Morris ME. Membrane transport of dietary phenethyl isothiocyanate by ABCG2 (breast cancer resistance protein) Mol Pharm. 2005;2(5):414–9. doi: 10.1021/mp050029f. [DOI] [PubMed] [Google Scholar]
- 30.Fowke JH, Chung FL, Jin F, Qi D, Cai Q, Conaway C, et al. Urinary isothiocyanate levels, brassica, and human breast cancer. Cancer Res. 2003;63(14):3980–6. [PubMed] [Google Scholar]
- 31.Terry P, Wolk A, Persson I, Magnusson C. Brassica vegetables and breast cancer risk. JAMA. 2001;285(23):2975–7. doi: 10.1001/jama.285.23.2975. [DOI] [PubMed] [Google Scholar]
- 32.Pegg AE. Metabolism of N-nitrosodimethylamine. IARC Sci Publ. 1980;27:3–22. [PubMed] [Google Scholar]
- 33.Chung FL, Wang MY, Hecht SS. Effects of dietary indoles and isothiocyanates on N-nitrosodimethylamine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone alpha-hydroxylation and DNA methylation in rat liver. Carcinogenesis. 1985;6(4):539–43. doi: 10.1093/carcin/6.4.539. [DOI] [PubMed] [Google Scholar]
- 34.Ishizaki H, Brady JF, Ning SM, Yang CS. Effect of phenethyl isothiocyanate on microsomal N-nitrosodimethylamine metabolism and other monooxygenase activities. Xenobiotica Fate Foreign Compds Biol Syst. 1990;20(3):255–64. doi: 10.3109/00498259009046845. [DOI] [PubMed] [Google Scholar]
- 35.Doerr-O’Rourke K, Trushin N, Hecht SS, Stoner GD. Effect of phenethyl isothiocyanate on the metabolism of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by cultured rat lung tissue. Carcinogenesis. 1991;12(6):1029–34. doi: 10.1093/carcin/12.6.1029. [DOI] [PubMed] [Google Scholar]
- 36.Guo Z, Smith TJ, Wang E, Sadrieh N, Ma Q, Thomas PE, et al. Effects of phenethyl isothiocyanate, a carcinogenesis inhibitor, on xenobiotic-metabolizing enzymes and nitrosamine metabolism in rats. Carcinogenesis. 1992;13(12):2205–10. doi: 10.1093/carcin/13.12.2205. [DOI] [PubMed] [Google Scholar]
- 37.Morse MA, Eklind KI, Hecht SS, Chung FL. Inhibition of tobacco-specific nitrosamine 4-(N-nitrosomethylamino)-1-(3-pyridyl)-1-butanone (NNK) tumorigenesis with aromatic isothiocyanates. IARC Sci Publ. 1991;105:529–34. [PubMed] [Google Scholar]
- 38.Crespi CL, Penman BW, Gelboin HV, Gonzalez FJ. A tobacco smoke-derived nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, is activated by multiple human cytochrome P450s including the polymorphic human cytochrome P4502D6. Carcinogenesis. 1991;12(7):1197–201. doi: 10.1093/carcin/12.7.1197. [DOI] [PubMed] [Google Scholar]
- 39.Smith TJ, Guo Z, Gonzalez FJ, Guengerich FP, Stoner GD, Yang CS. Metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in human lung and liver microsomes and cytochromes P-450 expressed in hepatoma cells. Cancer Res. 1992;52(7):1757–63. [PubMed] [Google Scholar]
- 40.Patten CJ, Smith TJ, Murphy SE, Wang MH, Lee J, Tynes RE, et al. Kinetic analysis of the activation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by heterologously expressed human P450 enzymes and the effect of P450-specific chemical inhibitors on this activation in human liver microsomes. Arch Biochem Biophys. 1996;333(1):127–38. doi: 10.1006/abbi.1996.0373. [DOI] [PubMed] [Google Scholar]
- 41.Nakajima M, Yoshida R, Shimada N, Yamazaki H, Yokoi T. Inhibition and inactivation of human cytochrome P450 isoforms by phenethyl isothiocyanate. Drug Metab Dispos. 2001;29(8):1110–3. [PubMed] [Google Scholar]
- 42.Caporaso N, Whitehouse J, Monkman S, Boustead C, Issaq H, Fox S, et al. In vitro but not in vivo inhibition of CYP2D6 by phenethyl isothiocyanate (PEITC), a constituent of watercress. Pharmacogenetics. 1994;4(5):275–80. doi: 10.1097/00008571-199410000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Chen L, Mohr SN, Yang CS. Decrease of plasma and urinary oxidative metabolites of acetaminophen after consumption of watercress by human volunteers. Clin Pharmacol Ther. 1996;60(6):651–60. doi: 10.1016/S0009-9236(96)90213-1. [DOI] [PubMed] [Google Scholar]
- 44.Konsue N, Ioannides C. Tissue differences in the modulation of rat cytochromes P450 and phase II conjugation systems by dietary doses of phenethyl isothiocyanate. Food Chem Toxicol. 2008;46(12):3677–83. doi: 10.1016/j.fct.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 45.Konsue N, Ioannides C. Differential response of four human livers to modulation of phase II enzyme systems by the chemopreventive phytochemical phenethyl isothiocyanate. Mol Nutr Food Res. 2010;54(10):1477–85. doi: 10.1002/mnfr.200900598. [DOI] [PubMed] [Google Scholar]
- 46.Konsue N, Ioannides C. Modulation of carcinogen-metabolising cytochromes P450 in human liver by the chemopreventive phytochemical phenethyl isothiocyanate, a constituent of cruciferous vegetables. Toxicology. 2010;268(3):184–90. doi: 10.1016/j.tox.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 47.Konsue N, Ioannides C. Phenethyl isocyanate is not the metabolite of phenethyl isothiocyanate responsible for mechanism-based inhibition of cytochrome P450. Arch Toxicol. 2010;84(10):751–9. doi: 10.1007/s00204-010-0522-z. [DOI] [PubMed] [Google Scholar]
- 48.Telang U, Morris ME. Effect of orally administered phenethyl isothiocyanate on hepatic gene expression in rats. Mol Nutr Food Res. 2010;54(12):1802–6. doi: 10.1002/mnfr.200900607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tseng E, Scott-Ramsay EA, Morris ME. Dietary organic isothiocyanates are cytotoxic in human breast cancer MCF-7 and mammary epithelial MCF-12A cell lines. Exp Biol Med (Maywood) 2004;229(8):835–42. doi: 10.1177/153537020422900817. [DOI] [PubMed] [Google Scholar]
- 50.Telang U, Brazeau DA, Morris ME. Comparison of the effects of phenethyl isothiocyanate and sulforaphane on gene expression in breast cancer and normal mammary epithelial cells. Exp Biol Med (Maywood) 2009;234(3):287–95. doi: 10.3181/0808-RM-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moon YJ, Brazeau DA, Morris ME. Dietary phenethyl isothiocyanate alters gene expression in human breast cancer cells. Evid Based Complement Alternat Med. 2011 doi: 10.1155/2011/462525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kang L, Ding L, Wang ZY. Isothiocyanates repress estrogen receptor alpha expression in breast cancer cells. Oncol Rep. 2009;21(1):185–92. [PMC free article] [PubMed] [Google Scholar]
- 53.Kang L, Wang ZY. Breast cancer cell growth inhibition by phenethyl isothiocyanate is associated with down-regulation of oestrogen receptor-alpha36. J Cell Mol Med. 2010;14(6B):1485–93. doi: 10.1111/j.1582-4934.2009.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hahm ER, Singh SV. Bim contributes to phenethyl isothiocyanate-induced apoptosis in breast cancer cells. Mol Carcinog. 2012;51(6):465–74. doi: 10.1002/mc.20811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarkars R, Mukherjee S, Roy M. Targeting heat shock proteins by phenethyl isothiocyanate results in cell-cycle arrest and apoptosis of human breast cancer cells. Nutr Cancer. 2013;65(3):480–93. doi: 10.1080/01635581.2013.767366. [DOI] [PubMed] [Google Scholar]
- 56.Liu K, Cang S, Ma Y, Chiao JW. Synergistic effect of paclitaxel and epigenetic agent phenethyl isothiocyanate on growth inhibition, cell cycle arrest and apoptosis in breast cancer cells. Cancer Cell Int. 2013;13(1):10. doi: 10.1186/1475-2867-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Syed Alwi SS, Cavell BE, Donlevy A, Packham G. Differential induction of apoptosis in human breast cancer cell lines by phenethyl isothiocyanate, a glutathione depleting agent. Cell Stress Chaperones. 2012;17(5):529–38. doi: 10.1007/s12192-012-0329-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gupta P, Srivastava SK. Antitumor activity of phenethyl isothiocyanate in HER2-positive breast cancer models. BMC Med. 2012;10:80. doi: 10.1186/1741-7015-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gupta P, Adkins C, Lockman P, Srivastava SK. Metastasis of breast tumor cells to brain is suppressed by phenethyl isothiocyanate in a novel metastasis model. PLoS One. 2013;8(6):e67278. doi: 10.1371/journal.pone.0067278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCune K, Mehta R, Thorat MA, Badve S, Nakshatri H. Loss of ERalpha and FOXA1 expression in a progression model of luminal type breast cancer: insights from PyMT transgenic mouse model. Oncol Rep. 2010;24(5):1233–9. doi: 10.3892/or_00000977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hudson TS, Perkins SN, Hursting SD, Young HA, Kim YS, Wang TC, et al. Inhibition of androgen-responsive LNCaP prostate cancer cell tumor xenograft growth by dietary phenethyl isothiocyanate correlates with decreased angiogenesis and inhibition of cell attachment. Int J Oncol. 2012;40(4):1113–21. doi: 10.3892/ijo.2012.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]