

Abstract
Principles of dissolution science have been applied to allow waiver of in vivo bioequivalence studies for oral immediate release solid dosage forms, providing certain stipulations are met. This approach reduces regulatory burden without sacrificing product quality and performance requirements that assure continuing equivalence. These principles are broadly applicable to other dosage forms and routes of administration. In this article, we postulate a further opportunity, which relies on a determination of “optimal performance” for nonsolution orally administered drug products. The determination can be applied to certain highly soluble and rapidly dissolving drug products without further study, paving the way possibly for even further reductions in regulatory burden.
KEY WORDS: BCS, dissolution, drug release, optimum product performance, regulatory burden
INTRODUCTION
For the last 50 years and more, manufacturers and regulatory agencies in the USA and elsewhere have worked to create a system of interchangeable medicines, wherein efficacy and safety outcomes determined during the investigational period for a new drug are maintained over the life cycle of a medicine, irrespective of who is manufacturing it. A key component of this system rests on maintenance of exposure measures, typically determined through bioavailability (BA) and bioequivalence studies (BE). To meet the requirements of such a system, the dissolution or drug release test has become an important and time-honored quality measure to assure continuing consistency in product performance over many years once BA or BE has been established. It is applicable to immediate release and modified release orally administered drugs (1,2). Typically, the dissolution test procedure and acceptance criteria are developed following understanding of in vivo performance. While the dissolution procedure has many limitations and has been challenged on many occasions, it remains widely used, is frequently a regulatory expectation, and has continued to be improved over the years. It might well be argued that application of a dissolution test procedure, particularly for immediate release oral drug products, has had a substantial impact on the success of the US (and the world) multisource system, given that in vivo bioavailability/bioequivalence studies are performed infrequently. Increased knowledge of dissolution science permits the use of dissolution as a surrogate marker for bioequivalence and has been used to waive in vivo bioequivalence requirements (biowaiver) for oral dosage forms in certain cases. Principles of dissolution/drug release are similarly applied for biowaiver of topical dosage forms and dosage forms for other routes of administration as well. This commentary describes various aspects and segments of the drug approval life cycle and links dissolution or drug release as a common denominator that works to progressively reduce regulatory burden and maintain product performance. Beyond product performance, regulatory and compendial paradigms also consider chemistry, manufacturing, and controls, and taken together, these general approaches, when coupled with cGMPs, help assure a good quality medicine that should achieve the safety and efficacy outcomes defined in investigational clinical trials. A key proposal of this commentary is that dissolution approaches can be expanded to allow certain nonsolution orally administered drug products to be designated “optimally performing” through dissolution studies alone and without a requirement for comparison to another product.
DISSOLUTION IN REGULATORY PRACTICE
Dissolution of Oral Drug Products
The dissolution test is the single most important physicochemical test to assess the performance of an immediate release drug product. It has been used in product development and selection of the formulation for drug approval. It has been used with marketing approval studies and to assess batch-to-batch uniformity after marketing approval. Today, with the increased knowledge and understanding of the science undergirding dissolution test methodologies and with the availability of more rugged and reliable instruments, application of dissolution testing can be raised to a higher level. Increasingly, the dissolution test, with adaptable procedures and acceptance criteria in the US Pharmacopeia (USP) General Chapters 701 “Disintegration” and 711 “Dissolution,” may now under certain circumstances be used as a surrogate in vitro bioequivalence test. The value of the dissolution test is significantly enhanced when drug dissolution is evaluated as a function of time, i.e., when the dissolution profile is determined rather than a single point determination. Biowaiver criteria are set based on dissolution profile understanding (3).
Biowaiver for Lower Strengths
A new chemical entity becomes the drug substance that is formulated into one or more drug products for clinical use. It is developed by a first-entry manufacturer with submission of an application to FDA in the USA where regulatory decision-making relative to safety and efficacy may allow market access. An explicit understanding of bioavailability, developed typically as part of ADME studies, for systemically absorbed orally administered drug products elucidates an exposure pattern that should be maintained throughout the shelf life of a product and over the life cycle of a medicine. As a result of US congressional decisions expressed in the Drug Price Competition and Patent Term Restoration Act (Hatch-Waxman/1984), a follow-on generic product manufacturer may file an abbreviated application containing information documenting both pharmaceutical equivalence and bioequivalence between their product and the first-entry product (termed in the US the Reference Listed Drug). In many examples of orally administered immediate release dosage forms, multiple strengths of the product are marketed. In these cases, an in vivo bioequivalence study comparing the test and reference product is conducted only using the highest strength, with lower strengths given a biowaiver providing the additional strengths are formulation proportional and have similar dissolution profiles when compared to the highest strength (3).
Scale-Up and Post-Approval Changes
After the drug product is approved, the manufacturer may adjust its manufacturing process in many ways, including batch size, site change, changes in manufacturing equipment and processing, or in components or composition. The latter includes changes in source of drug substance or excipients. These changes are referred collectively as Scale-up and Post Approval Changes (SUPAC) (4). In many instances, comparison of dissolution profile in suitable media may be used as a measure of equivalence in product performance. In this scenario, dissolution is used as an attribute of product indicating that it will perform in a similar manner in terms of physicochemical properties and is presumed to allow a link back to the batches tested for safety, efficacy, and either bioavailability or bioequivalence. Dissolution is used to requalify the approved product after “change.”
In Vitro-In Vivo Correlation
Development of an in vitro-in vivo correlation (IVIVC) refers to the establishment of a rational relationship between a biological property or a parameter derived from drug plasma concentration (Cmax, or AUC) produced by a dosage form and a physicochemical property or characteristic of the same dosage form (dissolution). The most important use of IVIVC is predictability of in vivo performance of the dosage forms (4). It has also been used to approve SUPAC-related changes, especially for extended release dosage forms (5).
Biopharmaceutics Classification System
The Biopharmaceutics Classification System (BCS) is a scientific framework for classifying drug substances based on their aqueous solubility and intestinal permeability (6). When combined with the dissolution of the dosage form, BCS takes into consideration three important factors, solubility, permeability, and dissolution, that govern the rate and extent of drug absorption (7). High solubility is defined as the highest marketed dose strength soluble in 250 mL of aqueous buffer in the range of pH 1.2–6.8, and high permeability is defined as the drug absorption (including first pass metabolism) to be 85% or greater. Based on the high/low solubility and high/low permeability, the drug substances are classified into four classes—BCS class I, II, III, and IV (6). The dissolution of the tablet or capsule should be carried out in aqueous media (pH 1.2, 4.5, and 6.8) by apparatus 1 (basket method) at 100 rpm or apparatus 2 (paddle method) at 50 rpm [FDA, (7)] or 75 rpm [WHO, (8)]. The dissolution is considered very rapid if the tablet or capsule dosage form dissolves 85% or greater in 15 min, rapid dissolution if it dissolves 85% or greater in 30 min, and slow dissolution if it takes longer than 30 min for 85% of the drug to dissolve. Biowaiver for BCS class I drugs is allowed for immediate release (non-NTI drug) formulations (7). Extending these scientific principles, WHO has suggested biowaiver for BCS class III drugs if they are very rapidly dissolving and to BCS class II weak acids if they dissolve 85% or greater in pH 6.8 (8). For all biowaivers, the dissolution profile of the test product and reference product should be compared in all three media and should meet similarity criteria, f2 (7). Dissolution profile comparison using similarity factor f2 assures product sameness between different strengths from a given manufacturer, between test and reference product (BCS-based biowaivers), and SUPAC-related changed product.
Proposal for an Optimum Product Performance Designation
The USP Medicine Compendium (MC) focuses on providing ingredient and product monographs for medicines legally marketed outside the USA. For the product performance test, MC references General Chapter 12 (applicable only to the MC), which extends the BCS approach beyond current applications and, for certain products, eliminates the need for comparison studies (9). For dosage forms containing a highly soluble drug substance, the general approach is based on the solubility and dissolution characteristics of the dosage form in pH 1.2 and 6.8 media. The basis for the proposal is advanced in the following section of the commentary.
The chain of events necessary for a solid oral dosage form to become bioavailable involves disintegration, dissolution, absorption, and permeation. The first two are assessed by in vitro dissolution studies and conclude with the drug substance in a true solution. They are characteristics of the drug product formulation and thus under the control of a manufacturer. The final two steps are controlled by the absorptive and metabolic pathways of the individual receiving the dosage form. Once the product disintegrates and the drug substance contained therein enters into solution, the rest of the steps for product performance, i.e., absorption, distribution, metabolism, and excretion are beyond the control of the manufacturer. For optimal product performance, the dissolution event should occur in a region of the GI tract that provides a pH at which the drug substance is soluble and where absorption can occur. With these considerations in mind, steps delineated in USP General Chapter 12 to determine whether a dosage form may be optimally performing are:
Determine the solubility of the active pharmaceutical ingredient (API, drug substance) using the highest dose amount in 250 mL of aqueous media pH 1.2 and 6.8. If the API dissolves, it is referred as highly soluble.
Determine the dissolution of the dosage form in 900 mL of pH 1.2 and/or pH 6.8 dissolution media using basket method at 100 rpm or paddle method at 50 rpm. The dosage form should dissolve 85% in 15 or 30 min as indicated below.
- Case 1. API highly soluble in both pH 1.2 and 6.8.
- Conduct dissolution in pH 1.2 and 6.8.
- Q = 85% in 30 min.
- Case 2. API highly soluble in pH 1.2 only, lowly soluble in pH 6.8.
- Conduct dissolution in pH 1.2.
- Q = 85% in 15 min.
- Conduct dissolution profile in pH 6.8.
- Should meet dissolution profile comparison criteria with a comparator.
- Note: Case 2 can be an example of a weak base. There is a possibility that the dosage form will dissolve in pH 1.2 medium and may precipitate out in higher pH environment. Dosage forms belonging to this class should meet dissolution profile comparison criteria, f2, in pH 6.8 with a comparator for optimally performing designation.
- Case 3. API highly soluble in pH 6.8 only, lowly soluble in pH 1.2.
- Conduct dissolution in pH 6.8.
- Q = 85% in 15 min.
When a drug product fails to meet acceptance criteria or the drug substance is insoluble at either pH, that drug product will need a product-specific dissolution condition, and further evaluation may be necessary. Products that do not meet the acceptance criteria are not necessarily “bad” products. It means that these products will require additional studies to demonstrate acceptable performance. For case 4 drug substances (API lowly soluble at both pH 1.2 and 6.8 conditions), dosage forms would not be eligible to receive an optimally performing designation and a surfactant may be needed in the dissolution method.
Topical Drug Products
Dissolution is used as a quality procedure for orally administered nonsolution dosage forms; similarly, in vitro release testing is used for a topical drug product. The in vitro release test for topical products (also termed semisolids) has been recognized in US FDA’s SUPAC-SS guidance as a measure for equivalence test with regard to product performance after certain manufacturing related changes (10). Recently, the in vitro release test is also recognized as a reasonable and useful test to consider for product release and stability testing (11). When the generic product is qualitatively (Q1) and quantitatively (Q2) similar for excipients, and has the same release characteristics as the innovator product in an in vitro system (specified as Q3), the test is considered to be suitable for biowaiver of acyclovir ointment 5% (12). Scientifically, the same principle of Q1, Q2, and Q3 can be applied to other semisolid dosage forms for biowaiver applications. In the case of nonsolution orally administered dosage forms such as tablets and capsules, lower dosage strengths may have bioequivalence requirement waivers as long as they are formulation proportional and employ the same drug-releasing ingredients as the higher strength. The same principles can be applied for the waiver of lower strength of topical dosage forms. This general similarity between oral and topical dosage forms is shown in Fig. 1.
Fig. 1.
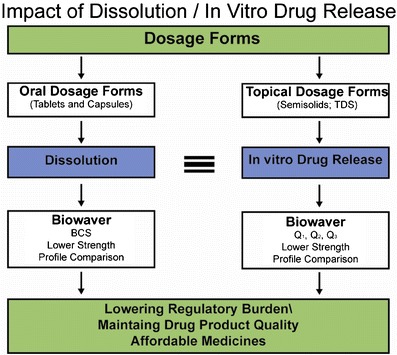
Impact of dissolution/drug release
DISCUSSION
The concept of dissolution relating to its regulatory applications has resulted in progressively reducing regulatory burden while maintaining optimum product performance and drug product quality. After new drug approval and patent expiration, interchangeable multisource generic products are approved based on bioequivalence studies. Subsequently, using dissolution profiles, biowaivers are provided to various dosage forms:
Lower strengths of approved dosage form
BCS class I and III and class II weak acids
SUPAC-related changes
Biowaiver for topical dosage forms based on Q1, Q2, and Q3
Dosage form with optimum performance based on appropriate dissolution tests
Dissolution/in vitro drug release from semisolid dosage forms can be considered for biowaiver of generic semisolid dosage forms as long as they are Q1 and Q2 the same as brand name drug and have the Q3 profile.
A key proposal in this commentary offered for scientific/regulatory consideration is that some nonsolution orally administered dosage forms containing a highly soluble drug substance may be designated optimally performing (OPP) providing they demonstrate rapid dissolution in suitable media. Drug products with an OPP designation, coupled with a sound private or public set of additional quality specifications, support determinations of continuing equivalence in the quality and performance attributes of a medicine, relative to the clinical trial material on which safety and efficacy data were originally based. More than 60% of the conventional release drugs listed under WHO essential medicines list can be eligible for biowaiver based on dissolution performance (13). The overall approach offers remarkable opportunities to create a global system of interchangeable medicines that are safe, effective, and high quality. Each product that is rapidly dissolving, where the drug substance is highly soluble, is as good as it can be and needs no comparative data. The overall paradigm appears in Fig. 2.
Fig. 2.
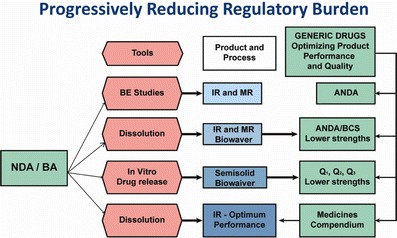
Progressively reducing regulatory burden
In general, an appropriate in vitro approach is more sensitive and reproducible than conducting bioequivalence study. Regulatory requirements of bioequivalence studies in human for the generic drug product approval can be easily avoided. This will reduce regulatory burden without sacrificing drug product quality.
CONCLUSIONS
It has been possible to use appropriate in vitro dissolution/drug release tests for biowaiver. In this commentary, the authors utilize general principles of bioavailability, bioequivalence, and dissolution to advocate a further extension of the BCS approach. The approach is based on an understanding that certain dosage forms can be designated optimally performing provided they contain a highly soluble drug substance and exhibit rapid dissolution in suitable media. The advantages of such an approach would be especially applicable to countries that are moving to an interchangeable system. In this instance, a requirement for the optimally performing designation would be a major advance, given that two optimally performing products would necessarily be equivalent to one another without a direct comparison. The authors realize that the proposal merits careful scientific and regulatory consideration.
References
- 1.FDA Guidance for Industry. 1997. Dissolution testing of immediate release solid dosage forms. http://www.fda.gov/cder/guidance/index.htm. Accessed 14 Jan 2014.
- 2.FDA Guidance for Industry. 1997. Extended release dosage forms: development, evaluation and application of in vitro in vivo correlation. http://www.fda.gov/cder/guidance/index.htm Accessed 14 Jan 2014. [DOI] [PubMed]
- 3.FDA Guidance for Industry. 2003. Bioavailability and bioequivalence studies for orally administered drug products. General considerations. http://www.fda.gov/cder/guidance/index.htm Accessed 14 Jan 2014.
- 4.FDA Guidance for Industry. 1995. SUPAC-IR. Immediate release solid oral dosage forms. Scale-Up and post approval changes. Chemistry, manufacturing and controls. In vitro dissolution testing and in vivo bioequivalence documentation. http://www.fda.gov/cder/guidance/index.htm. Accessed 14 Jan 2014.
- 5.FDA Guidance for Industry. 1997. SUPAC-MR. Modified release solid oral dosage forms. Scale-Up and post approval changes. Chemistry, manufacturing and controls. In vitro dissolution testing and in vivo bioequivalence documentation. http://www.fda.gov/cder/guidance/index.htm. Accessed 14 Jan 2014.
- 6.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biophrmaceutic drug classification system: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 7.FDA Guidance for Industry. 2000. Waiver in vivo bioavailability and bioequivalence studies for immediate release solid oral dosage forms based on biopharmaceutics classification system. http://www.fda.gov/cder/guidance/index.htm. Accessed 14 Jan 2014.
- 8.WHO technical report series 937, Annex 7. Multisource (generic) pharmaceutical products. Guidance on registration requirements to establish interchangeability. Geneva, Switzerland. WHO, 2006. http://apps.who.int/prequal/info_general/documents/TRS937/WHO_TRS_937__annex7_eng.pdf. Accessed 14 Jan 2014.
- 9.USP MC <12>, United States Pharmacopeial Convention, Rockville, MD 20852.
- 10.SUPAC-SS. Nonsterile semisolid dosage forms. Scale-Up and post approval changes. Chemistry, manufacturing and controls. In vitro release testing and in vivo bioequivalence documentation. http://www.fda.gov/cder/guidance/index.htm. Accessed 14 Jan 2014.
- 11.Chang R-K, Raw A, Lionberger R, Yu L. Generic development of topical dermatological products: formulation development, process development and testing of topical dermatological products. AAPS J. 2013;15(1):41–52. doi: 10.1208/s12248-012-9411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.FDA/OGD Draft guidance on acyclovir ointment. March 2012. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm296733.pdf. Accessed 14 Jan 2014.
- 13.WHO technical report series 937, Annex 8. Proposal to waive in vivo bioequivalence requirements for WHO model list of essential medicines immediate-release, solid oral dosage forms. Geneva, Switzerland. WHO, 2006. FDA Guidance for Industry. 1997.