

Abstract
Curcumin (CUR), a non-toxic polyphenol from Curcuma longa, has been investigated as a potential therapy with anti-inflammatory and anti-oxidative effects for Alzheimer’s disease (AD), which depicts features of chronic inflammatory environment resulting in cellular death. However, it remains largely unknown whether the anti-inflammatory effect of CUR in AD is associated with its property of CpG demethylation, which is another function of CUR with the most research interest during recent years. Neprilysin (NEP, EP24.11), a zinc-dependent metallopeptidase expressed relatively low in the brain, is emerging as a potent inhibitor of AKT/Protein Kinase B. In addition, hypermethylated promoter of NEP has been reported to be associated with decreases in NEP expression. In the present study, using bisulfite-sequencing PCR (BSP) assay, we showed that the CpG sites in NEP gene were hypermethylated both in wild-type mouse neuroblastoma N2a cells (N2a/wt) and N2a cells stably expressing human Swedish mutant amyloid precursor protein (APP) (N2a/APPswe) associated with familial early onset AD. CUR treatment induced restoration of NEP gene via CpG demethylation. This CUR-mediated upregulation of NEP expression was also concomitant with the inhibition of AKT, subsequent suppression of nuclear transcription factor-κB (NF-κB) and its downstream pro-inflammatory targets including COX-2, iNOS in N2a/APPswe cells. This study represents the first evidence on a link between CpG demethylation effect on NEP and anti-inflammation ability of CUR that may provide a novel mechanistic insight into the anti-inflammatory actions of CUR as well as new basis for using CUR as a therapeutic intervention for AD.
KEY WORDS: AKT/NF-κB signaling, Alzheimer’s disease, Curcumin, DNA methylation, Neprilysin
INTRODUCTION
Alzheimer’s disease (AD), the most common dementia that accounts for an estimated of 60–80% of cases, is one of the leading causes of morbidity and death worldwide (1). Accumulating evidence has shown that excessive and dysregulated inflammation is implicated in the progression of AD, contributing to the cellular death and associated neurodegeneration (2,3). AKT/nuclear transcription factor-κB (NF-κB) signaling is a critical cell signaling pathway that plays a major role in various biological functions including inflammation, cell growth, and differentiation (4,5). It is reported that aberrant activation of NF-κB induced by amyloid β-peptide (Aβ), a generally accepted contributor to the etiology of AD (6), contributes to the initiation and progression of AD through upregulation of pro-inflammatory proteins such as cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) in experimental models of AD (7,8). All the above reports suggest that targeting AKT/NF-κB proteins may represent a potential therapeutic strategy for AD therapy.
Neprilysin (NEP, EP24.11), a zinc-dependent metallopeptidase expressed relatively low in the brain, has been characterized as capable of reducing neurotoxicity through degradation of Aβ in AD (9,10). Increasing evidence also indicates an important function of its inhibition in AKT activation (11,12). However, NEP, which alters with age, chronically decreases in AD brains (13). DNA methylation is known to be involved in gene suppression (14). Recent studies have demonstrated that hypermethylation of CpG islands within NEP gene is an important mechanism to inactive NEP expression (15,16).
Curcumin (CUR) is a natural and non-toxic polyphenol of turmeric, a yellow spice isolated from Curcuma longa (17). With anti-inflammatory, anti-oxidative, and anti-carcinogenic properties by targeting various molecules such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), CUR has been extensively used as a food preservative in India (18–20). Now, it is showing promising results in clinical trials as a potential therapy for inflammatory and malignant diseases (21,22). Besides the roles of CUR in anti-inflammation and anti-oxidative stress, accumulating evidence indicates that CUR is a potential epigenetic inhibitor, particularly possessing inhibitory effects on DNA methyltransferases (DNMTs) with low concentration (23,24). Moreover, the potential of CUR analogues EF31 and UBS109 as epigenetic modifier and anti-carcinogenic agent is also being delineated (25,26). To date, little is known about the effect of CUR on the CpG methylation of potential genes in AD and whether this hypomethylating activity of CUR is associated with its role of anti-inflammation.
In this study, we used wild-type mouse neuroblastoma N2a cells (N2a/wt) and N2a cells stably expressing human Swedish mutant amyloid precursor protein (APP) (N2a/APPswe) associated with familial early onset AD (27) as cell models. Our aim is to explore whether CUR has CpG demethylation and upregulation effects on NEP gene and subsequent regulates NEP-mediated anti-inflammation pathway.
MATERIALS AND METHODS
Cell Culture and Treatment
Wild-type mouse neuroblastoma N2a cells (N2a/wt) and N2a cells stably expressing human Swedish mutant APP (N2a/APPswe) were generously provided by Dr. Huaxi Xu (Burnham Institute for Medical Research, La Jolla, USA). N2a cells at the fourth passage were used. N2a cells were cultured in a 1:1 mixture of DMEM and OPTI-MEM supplemented with 5% fetal bovine serum (Gibico, Carlsbad, CA) at 37°C in 5% CO2, and 150 μg/mL G418 was added in the culture media of N2a/APPswe cells. Cells were seeded for 24 h and then treated with 0.1% DMSO, 2.5 μM CUR, 5 μM CUR for 48 h, 5 μM 5-aza-2′-deoxycytidine (5-aza-CdR) for 72 h, 10 μM LY294002 for 24 h, or combination of 5 μM CUR and 30 μM Thiorphan for 48 h. All the chemicals were obtained from Sigma–Aldrich (St. Louis, MO, USA). Cells were harvested for DNA, RNA, and protein analyses outlined below.
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction
Total RNAs were extracted from the treated cells using Trizol reagent (TaKaRa, Japan). The template cDNA was synthesized from total RNA by reverse transcription (RT) using PrimeScript reverse transcription kit (TaKaRa, Japan), real-time PCR was performed by Thermal Cycler Dice Real Time System (Thermo, USA) using SYBR PrimeScript PCR kit (TaKaRa, Japan) according to the manufacturer’s instructions. Quantitative real-time polymerase chain reaction (qPCR) was performed by an Eppendorf Mastercycler nexus instrument (Eppendorf, Germany) with initial heating at 95°C for 30 s, followed by 40 cycles of denaturation at 95°C for 5 s and annealing plus extension at 60°C for 30 s. The level of gene was normalized to the expression of GAPDH. The following sequences of the primers were used for amplification: NEP forward (5′-CTCTCTGTGCTTGTCTTGCTC-3′) and reverse (5′-GACGTTGCGTTTCAACCAGC-3′) and GAPDH forward (5′-GGTGAAGGTCGGTGTGAACG-3′) and reverse (5′-CTCGCTCCTGGAAGATGGTG-3′) (Sangon Biotech, China). Relative quantification in each sample was performed using the 2−ΔΔCT method.
Preparation of Protein Lyses and Western Blotting
Total proteins, cytoplasm protein fragments, and nuclear protein fragments were extracted, respectively, as described previously (28). The protein concentrations were determined using the bicinchoninic acid (BCA) method (Pierce, Rockford, IL). Equal amounts (50 mg of protein per lane) of proteins from each sample were separated by SDS-PAGE (Bio-Rad, CA, USA). After electrophoresis, the proteins were transferred to the polyvinylidene difluoride (PVDF) membranes (Millipore, USA). After blocking with 5% BSA for 2 h, the PVDF membranes were incubated at 4°C overnight with specified primary antibodies against NEP (Epitomics, USA; 1:1,000) and phosphorylated AKT (p-Akt) (Ser473), AKT, phosphorylated p65 (p-p65) (Ser536), p65 (Cell Signaling Technology, USA; all 1:1,000), or iNOS, COX-2 (Cell Signaling Technology, USA; all 1:500), β-actin (Santa Cruz, CA, USA; 1:1,000), or Lamin A/C (Abcam, USA; 1:500). β-actin or Lamin A/C was used as a loading control. After washing with blocking buffer, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Zhongshan Golden Bridge, Beijing, China; 1:100). Immunoreactive bands were visualized by Chemiluminescence reagents (Beyotime, China). The intensity of each band was measured using Quantity One (Bio-Rad, CA, USA).
Immunocytochemistry Staining
N2a cells were fixed on the glass slides with 4% paraformaldehyde for 10 min at room temperature. After a brief washing in phosphate-buffered saline (PBS), cells were incubated with or without 0.4% Triton for 10 min at room temperature. The cells were blocked with 5% normal goat serum for 2 h and then incubated with anti-NEP (Santa Cruz Biotechnology, USA; 1:50) for 2 h at 37°C or anti-p-p65 (Cell Signaling Technology, USA; 1:100) antibodies overnight at 4°C. After incubation, cells were washed and then incubated with Cy3-conjugated secondary antibody (Beyotime, China; 1:100) for 1.5 h at 37°C and counterstained for nuclei with DAPI (Beyotime, China) for 3 min at room temperature. Cells were finally washed three times with PBS and then analyzed under a laser scanning confocal microscope (A1R, Nikon, Japan).
Bisulfite-Sequencing PCR
Genomic DNA was extracted from the DMSO and CUR-treated N2a cells using the DNeasy Tissue Kit (QIAGEN, Valencia, CA, USA). The bisulfite conversion was carried out with 1 mg of genomic DNA using Methylamp DNA Modification Kit (Epigentek, Brooklyn, NY, USA) according to the manufacturer’s instructions. The bisulfite-modified DNA was amplified by PCR with the primers that amplify the 41 CpGs from −95 to +245 of the murine NEP gene, with the translation start site referenced as +1. The forward and reverse primers were 5′-AATTTTTAGGTTATTTAGGGAATTGT-3′ and 5′-AAACRACTAAACAAACACATCCC-3′, respectively. PCR amplification conditions were as follows: 98°C 4 min, 94°C 45 s, 66°C 45 s, 72°C 1 min, 20 cycles. Then, the first-stage PCR product was amplified at the following conditions: 94°C 45 s, 56°C 45 s, 72°C 1 min, 20 cycles, and a final extension at 72°C for 8 min. PCR products were purified using the QIAquick PCR purification kit (QIAGEN, Valencia, CA, USA) and sequenced by Sangon Biotechnology.
Statistical Analysis
All data were expressed as mean ± SD of at least three separate experiments. Differences between two groups were analyzed using Student’s t test. Differences between multiple groups were analyzed using ANOVA. The CpG methylation rates of NEP gene were analyzed by Fisher’s exact test. p value <0.05 was regarded statistical significance.
RESULTS
CUR Induces Restoration of NEP in N2a/wt and N2a/APPswe Cells
To test whether CUR has an effect on NEP expression, qPCR and Western blot were used to establish the messenger RNA (mRNA) and protein levels of NEP in N2a cells exposed to different concentrations of CUR. We found that 5 μM but not 2.5 μM of CUR significantly increased the mRNA and protein levels of NEP both in N2a/wt and N2a/APPswe cells (Fig. 1a, b). Immunocytochemistry further confirmed that 5 μM of CUR caused more intense staining of NEP in plasma membrane than that from controls (Fig. 1c). Since demethylation of CpG sites is involved in gene reactivation and 5-aza-CdR is a widely used cytosine methylation inhibitor (29), we used 5-aza-CdR-treated sample as a positive demethylation control to test whether NEP transcription is associated with its DNA methylation status in these two N2a cell lines. Our data showed remarkably increased levels of NEP following 5-aza-CdR treatment (Fig. 1a, b).
Fig. 1.
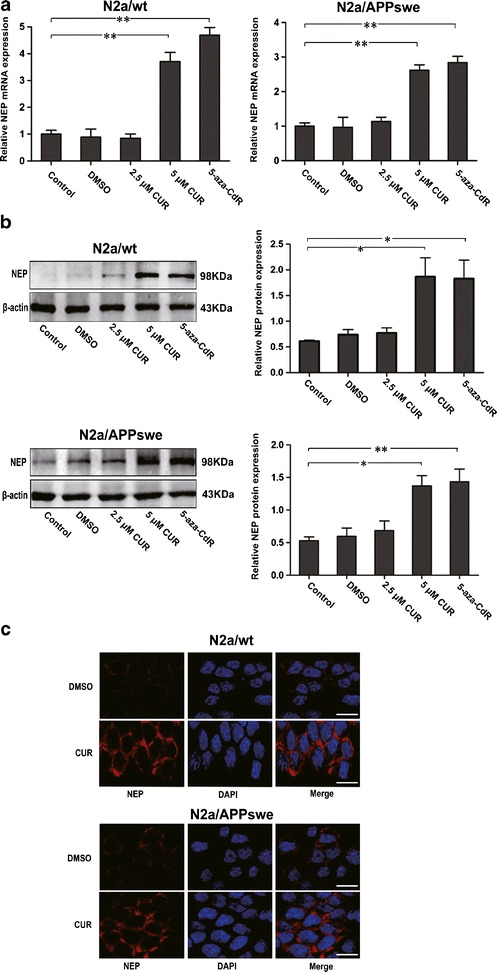
Effects of CUR treatment on NEP expression in N2a/wt and N2a/APPswe cells. N2a cells were incubated with DMSO (0.1%), CUR (2.5 μM, 5 μM) for 48 h, or 5-aza-CdR (5 μM) for 72 h. Cells in each group were then collected; RNA and proteins were assessed. a qPCR analysis of NEP mRNA level. GAPDH was assessed as a loading control. b Western blot analysis of NEP protein level. β-actin was assessed as a loading control. c Immunocytochemistry analysis of NEP protein expression and distribution. N2a cells were treated with 5 μM CUR for 48 h and labeled with anti-NEP antibody (red) and DAPI (blue). Scale bar 100 μm. All data were represented as a mean ± SD of three independent experiments. *p < 0.05, **p < 0.01
CUR Reverses Hypermethylated CpG Sites of NEP Gene in N2a/wt and N2a/APPswe Cells
Forty one CpG sites ranging from −95 to +245 nt relative to the exon 1 (with the translation initiation site designated as +1) in a CpG island were identified on the mouse NEP promoter by sequence analysis (www.genome.ucsc.edu) (Fig. 2a). To investigate whether upregulation of NEP by CUR is associated with modification of DNA methylation of NEP, bisulfite-sequencing PCR (BSP) was performed to quantitatively analyze the methylation levels of CpG sites in NEP gene in N2a/wt and N2a/APPswe cells incubated with or without CUR. We selected ten clones to analyze for each CpG site. BSP revealed that the percentages of total methylated CpG sites in NEP gene in N2a/wt and N2a/APPswe cells were 92.19 and 90.73%, respectively, indicating the baseline hypermethylation level of these regions. Then, 5 μM instead of 2.5 μM of CUR was selected to treat N2a cells due to its significant upregulation effect on NEP expression, which was revealed in Western blot and qPCR. Compared to the control, the average proportion of methylated CpGs decreased to 77.56% (Fisher exact test, p value <0.0001) in N2a/wt cells treated with CUR. Among the whole 41 CpGs, the region from −50 to +185 containing 27 CpG sites was dramatically demethylated (decreased from 95.56 to 68.15%, Fisher exact test, p value <0.0001) (Fig. 2b). Meanwhile, the methylation of CpGs was also reduced by CUR in N2a/APPswe cells (decreased from 90.73 to 81.7%, Fisher exact test, p value <0.0001). The region from −12 to +150 containing 19 CpG sites was particularly demethylated (decreased from 90 to 70.53%, Fisher exact test, p value <0.0001) (Fig. 2c).
Fig. 2.
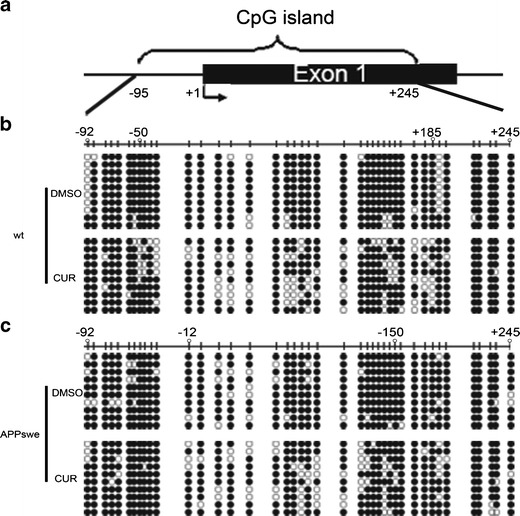
Effects of CUR on DNA methylation of NEP in N2a/wt and N2a/APPswe cells. a Genomic structure of the CpG island in the mouse NEP promoter region. The arrow represents the transcription initiation site (+1). b, c Bisulfite sequencing chromatogram of NEP CpG sits in N2a/wt cells (b) and N2a/APPswe cells (c) treated with or without CUR at 5 μM for 48 h. The vertical bars delineate the position of CpG sits. Each row of circles indicates a single cloned allele. Black and open circles represent methylated and unmethylated cytosines, respectively. Three independent experiments were analyzed for BSP
CUR-Induced NEP Upregulation Suppresses the AKT/NF-κB Pathway in N2a/APPswe Cells
NEP has been demonstrated as an inhibitor of AKT (11). To determine whether upregulation of NEP induced by CUR mediates downregulation of activation of AKT and its downstream target NF-κB, the N2a/APPswe cells, engineered to over-produce Aβ (30), was used. 5 μM of CUR was added to N2a/APPswe cells and incubated for 48 h. Western blot showed that CUR significantly reduced the protein expression of phosphorylated AKT (p-AKT), which indicates the level of AKT activation, although the least effect was observed on AKT levels (Fig. 3a). p65, the subunit of the NF-κB, containing transactivation domain, is phosphorylated and translocated to the nucleus when NF-κB is activated (31). Thus, proteins of nuclear and cytoplasmic extracts were examined for the expression of p65 and phosphorylated p65 (p-p65) to analyze the activation of NF-κB. We found that CUR treatment resulted in remarkable decrease of protein expression of p65 in nuclear and p-p65 both in nucleus and cytoplasm, although synthesis of the cytoplasmic p65 protein remained unaffected (Fig. 3a). To test whether inhibition of NF-κB is associated with loss of AKT activation in this cell line, LY294002 (10 μM), an AKT inhibitor, was also added. We observed the decrease of NF-κB activation following inhibition of AKT by LY294002 (Fig. 3a). Furthermore, the reduced translocation of p-p65 to the nucleus was revealed by immunocytochemistry (Fig. 3b), whereas the highly specific NEP inhibitor, Thiorphan (30 μM), added to N2a/APPswe cells during incubation with CUR, prevented CUR-induced inactivation of AKT and p65 (Fig. 3a, b).
Fig. 3.
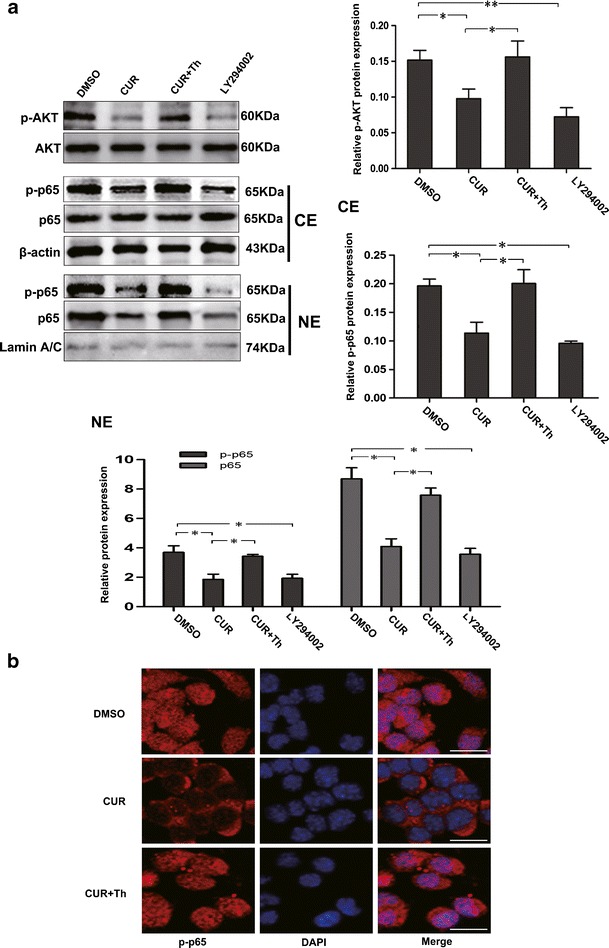
Effects of CUR-induced NEP upregulation on AKT/NF-κB signaling in N2a/APPswe cells. N2a/APPswe cells were treated with DMSO (0.1%), CUR (5 μM), combination of CUR (5 μM) and NEP inhibitor Thiorphan (Th, 30 μM) for 48 h, or AKT inhibitor LY294002 (10 μM) for 24 h. Cells in each group were collected and protein levels were then assessed. a Western blot analysis of the protein levels of p-AKT, AKT, and p-p65, p65 both in nucleus and cytoplasm. β-actin was assessed as a cytoplasmic protein loading control. Lamin A/C was assessed as a nucleus protein loading control. b Immunocytochemistry analysis of p-p65 translocation. N2a/APPswe cells were labeled with anti-p-p65 antibody (red) and DAPI (blue). Scale bar 100 μm. Th Thiorphan, NE nuclear extracts, CE cytoplasmic extracts. All data were represented as a mean ± SD of five independent experiments. *p < 0.05, **p < 0.01
Inhibition of AKT/NF-κB Signaling by CUR Is Associated with Modulation of NF-κB Downstream Gene Products in N2a/APPswe Cells
To test whether alterations in the AKT/NF-κB signaling by CUR lead to changes in downstream targets, the expression of COX-2 and iNOS, the major pro-inflammatory markers of NF-κB targets, was examined. N2a/APPswe cells were treated with CUR at 5 μM for 0, 24, 48, and 72 h. Western blot revealed that CUR led to a decrease in both COX-2 and iNOS expression in a time-dependent manner (Fig. 4).
Fig. 4.

Effects of CUR-induced AKT/NF-κB signaling disruption on NF-κB downstream targets. N2a/APPswe cells treated with CUR at 5 μM for 0, 24, 48, and 72 h. Proteins were then assessed for levels of COX-2 and iNOS. β-actin was assessed as a loading control. All data were represented as a mean ± SD of five independent experiments. *p < 0.05, **p < 0.01
DISCUSSION
DNA methylation, one of epigenetic regulations, refers to the methylation of the fifth cytosine in the pyrimidine ring catalyzed by DNMTs. In general, DNA methylation is associated with blockade of gene expression (14). Several lines of evidence now demonstrate that aberrant DNA methylations of genes in AD are implicated in the initiation and progression of AD (32). Neprilysin (NEP), a zinc-dependent metallopeptidase, serves as the major physiological Aβ-degrading enzyme (9). However, both the expression and activity of NEP decrease with age in AD brains (13,33), and the transcriptional regulatory mechanisms that regulate NEP expression are relatively unknown. In the present study, we observed the DNA methylation status of NEP gene in wild-type N2a cells (N2a/wt), which is devoid of NEP (34), and N2a cells stably expressing human Swedish mutant APP (N2a/APPswe) associated with familial early onset AD (27). Interestingly, the CpG sites in promoter region of NEP were hypermethylated both in N2a/wt and N2a/APPswe cells at the fourth passage. This aberrant DNA methylation pattern of NEP gene at early passage suggests that it may be imprinted from primary N2a cells. Chen et al. have demonstrated that hypermethylation level of the same region on NEP promoter was associated with suppression of NEP expression. However, they also reported that 25 μM exogenous Aβ40 could induce changes of DNA methylation level of NEP in murine cerebral endothelial cells (15). N2a/APPswe cells are engineered to overexpress APP, the precursor for Aβ. We have examined expression of Aβ in these two cell lines by a sandwich ELISA and found that levels of the secreted Aβ40 and Aβ42 were 1.5 and 0.625 pM in N2a/wt cells, respectively. Compared with N2a/wt cells, levels of the secreted Aβ40 and Aβ42 were increased by 3.3- and 2.8-fold in N2a/APPswe cells, respectively (data not shown). These data indicate that the similar DNA methylation level of NEP in these two cell lines may be due to the far lower concentration of endogenous Aβ produced by N2a/APPswe cells that fails to play a role in NEP expression. Moreover, NEP is poorly expressed in N2a/wt cells (34), even if Aβ is at a baseline low level. Thus, it seems that Aβ level is not necessary for DNA methylation-associated changes of NEP in N2a cells.
Further, we found treatment of 5-aza-CdR, the well-known cytosine methylation inhibitor (29), resulted in remarkable increases of NEP expression in N2a/wt and N2a/APPswe cells, which is consistent with the upregulation of NEP induced by 5-aza-CdR in other age-associated diseases such as prostate cancer (16,35). These data indicate that DNA methylation of the NEP promoter is an important mechanism that mediates its transcriptional repression in the N2a cell lines at early passage in vitro. However, Belyaev et al. reported that 5-aza-CdR could not upregulate NEP in human neuroblastoma SH-SY5Y cells (36). We suppose this divergence may be due to the species-associated difference in NEP promoter or different approach of drug treatment.
Since epigenetic regulation is more amenable than irreversible genetic mutation (37), 5-aza-CdR has been extensively studied in cancer therapy. But its utility is limited due to its nonspecificity and undesirable side effects (29). Recent studies have identified CUR as a potential player in epigenetic regulation based on its inhibitory effect on DNMTs, histone deacetylases (HDACs), and histone acetyltransferases (HATs) (23). Using DNA methylation arrays, Link et al. reported that CUR-induced DNA demethylation occurred only in partially methylated genes (38), which suggests CUR may serve as a more valuable epigenetic inhibitor. However, no reports delineate the effect of CUR on the CpG methylation of potential genes in AD so far. We proceeded to test whether CUR could reactivate the expression of NEP gene in N2a/wt and N2a/APPswe cells through promoter DNA demethylation. Our findings showed that CUR demethylated CpG sites of NEP gene at 5 μM in these two cell lines, along with a significant upregulation of NEP level, which is as effective as 5-aza-CdR treatment. Similar effects of CUR on DNA demethylation have been shown in several genes such as Nrf2 and Neurog 1 (39,40). However, it is also reported that demethoxycurcumin and bisdemethoxycurcumin but not CUR could demethylate the promoter region (41). In the present study, we confirmed that CUR could restore the expression of NEP in a methylation-dependent manner in N2a/wt and N2a/APPswe cells, providing a new insight into the mechanism beyond the conventional effects of CUR in AD.
CUR has been demonstrated to exert its demethylation effect through covalent binding to the catalytic thiolate of C1226 of DNMT1, which was further validated by the inhibitory effect of CUR on M.Sss1 activity with an IC50 of 30 nM (24). Several studies have also reported that CUR could inhibit the activity or expression of DNMTs (40,42). Further, the effect of CUR and its analogues EF31 and UBS109 on inhibiting HSP-90 and NF-κB, the positive regulators of DNMT-1 transcription, has been suggested as a possible mechanism responsible for downregulation of DNMT-1 (25). Whether upregulation of NEP induced by CUR in these two cell lines was mediated by either or both of these mechanisms is needed to be further validated.
Based on the amyloid cascade hypothesis, aggregation of Aβ could evoke inflammation stress. On the other hand, Aβ-induced neurotoxicity is exacerbated under conditions of inflammation dysregulation (43). Subsequent studies have demonstrated that the pro-inflammatory factor NF-κB activated by several molecules including AKT is involved in the inflammation progress of AD (7,8). Besides the property of Aβ clearance in AD brains, NEP has also been reported to have negative regulatory effect on AKT signaling (11). In this study, we confirmed the ability of CUR treatment to inhibit AKT/NF-κB signaling by suppressing the activation of AKT and NF-κB in N2a/APPswe cells in NEP-dependent way. We found CUR led to remarkable decreases of not only p-p65 levels but also the total amount of p65 proteins in nucleus, suggesting that downregulation of p-p65 by CUR is due to both dephosphorylation and downregulation of total p65 protein. The prevention of translocation of p-p65 to nucleus by CUR has also been demonstrated by immunofluorescence microscopy. However, when specific NEP inhibitor Thiorphan was added, the effects of CUR on this signaling were abolished. These above findings indicate that CUR-induced upregulation of NEP is indeed responsible for the inhibition of AKT/NF-κB signaling, whereas the ability of CUR to inhibit the activation of AKT/NF-κB signaling has been reported in many other diseases and is not involved in NEP gene (44,45). Thus, there may be more mechanisms contributing to the inhibitory effect of CUR on this signaling. In this study, we hypothesize the entire disruption of suppression of AKT/NF-κB signaling by CUR/Thiorphan combination treatment probably due to the higher effect of Thiorphan on activating this signaling because of its ability to exacerbate Aβ accumulation (46,47), thus masking CUR effect. Further, when we tested the proteins known to be downstream targets of NF-κB, we found that CUR reduced protein levels of COX-2 and iNOS in a time-dependent manner. These data suggest that disruption of AKT/NF-κB signaling may be at least one of the causes of modification of these proinflammatory genes by CUR in N2a/APPswe cells.
CONCLUSIONS
The present study was the first to show that CUR can elicit its neuroprotective effect, potentially at least in part, through DNA methylation modification of NEP gene with its subsequent induction of the NEP-mediated AKT/NF-κB pathway in the N2a cell line in vitro (Fig. 5). Our findings may help to fully appreciate the anti-inflammatory effects of CUR and provide a new basis for potential NEP-based epigenetic modifying therapeutic intervention in AD.
Fig. 5.
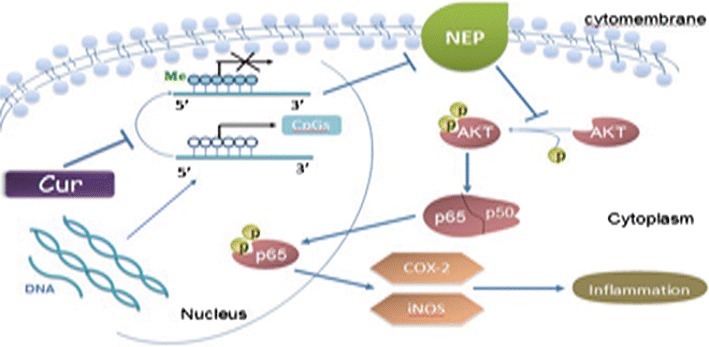
The possible mechanism of anti-inflammation induced by CUR in the N2a cell line. CUR induces CpGs demethylation of NEP promoter, leading to NEP restoration, then inhibiting the NEP-dependent activation of AKT/NF-κB signaling and its pro-inflammatory targets COX-2 and iNOS
Acknowledgments
We thank Dr. Huaxi Xu (Burnham Institute for Medical Research, La Jolla, USA) for generously providing N2a/wt and N2a/APPswe cell lines and Dr. Zhiqian Dong (Case Western Reserve University, Cleveland, USA) for meticulously going through the manuscript. This work was supported by the National Science Foundation of Chongqing Science and Technology Commission (CSTC, 2012JJA10044).
References
- 1.Abbott A. Dementia: a problem for our age. Nature. 2011;475:S2–4. doi: 10.1038/475S2a. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Inflammation and therapeutic vaccination in CNS diseases. Nature. 2002;420:879–84. doi: 10.1038/nature01325. [DOI] [PubMed] [Google Scholar]
- 3.Revesz T. Inflammation in Alzheimer’s disease: insights from immunotherapy. Brain. 2013;136:2654–6. doi: 10.1093/brain/awt231. [DOI] [PubMed] [Google Scholar]
- 4.Manning BD. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delhase M, Li N, Karin M. Kinase regulation in inflammatory response. Nature. 2000;406:367–8. doi: 10.1038/35019154. [DOI] [PubMed] [Google Scholar]
- 6.Robertson M. Alzheimer’s disease and amyloid. Nature. 1992;356:103. doi: 10.1038/356103a0. [DOI] [PubMed] [Google Scholar]
- 7.Ferrer I, Martí E, López E, Tortosa A. NF-κB immunoreactivity is observed in association with beta A4 diffuse plaques in patients with Alzheimer’s disease. Neuropathol Appl Neurobiol. 1998;24:271–7. doi: 10.1046/j.1365-2990.1998.00116.x. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalo MR, Martin B, Sanz-Anquela JM, Arevalo-Serrano J, Gonzalo-Ruiz A. Oligomers of beta-amyloid protein (Abeta1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp Neurol. 2012;236:215–27. doi: 10.1016/j.expneurol.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Hama E, Shirotani K, Iwata N, Saido TC. Effects of neprilysin chimeric proteins targeted to subcellular compartments on amyloid beta peptide clearance in primary neurons. J Biol Chem. 2004;279:30259–64. doi: 10.1074/jbc.M401891200. [DOI] [PubMed] [Google Scholar]
- 10.Marr RA, Guan H, Rockenstein E, Kindy M, Gage FH, Verma I, et al. Neprilysin regulates amyloid beta peptide levels. J Mol Neurosci. 2004;22:5–11. doi: 10.1385/JMN:22:1-2:5. [DOI] [PubMed] [Google Scholar]
- 11.Dai J, Mikhail M, Navarro D, Taneja SS, Lee P, Christos P, et al. Loss of neutral endopeptidase and activation of protein kinase B (Akt) is associated with prostate cancer progression. Cancer. 2006;107:2628–36. doi: 10.1002/cncr.22312. [DOI] [PubMed] [Google Scholar]
- 12.Ando H, Nagasaka T, Shibata D, Harata T, Shimomura Y, Goto M, et al. Neutral endopeptidase expressed by decidualized stromal cells suppresses akt phosphorylation and deoxyribonucleic acid synthesis induced by endothelin-1 in human endometrium. Endocrinology. 2006;147:5153–9. doi: 10.1210/en.2006-0172. [DOI] [PubMed] [Google Scholar]
- 13.Russo R, Borghi R, Markesbery W, Tabaton M, Piccini A. Neprylisin decreases uniformly in Alzheimer’s disease and in normal aging. FEBS Lett. 2005;579:6027–30. doi: 10.1016/j.febslet.2005.09.054. [DOI] [PubMed] [Google Scholar]
- 14.Martin C, Zhang Y. Mechanisms of epigenetic inheritance. Curr Opin Cell Biol. 2007;19:266–72. doi: 10.1016/j.ceb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Chen KL, Wang SS, Yang YY, Yuan RY, Chen RM, Hu CJ. The epigenetic effects of amyloid-beta (1–40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem Biophys Res Commun. 2009;378:57–61. doi: 10.1016/j.bbrc.2008.10.173. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi M, Yoshida M, Igarashi M, Nakae D. Methylation of neutral endopeptidase 24.11 promoter in rat hepatocellular carcinoma. Cancer Sci. 2006;97:611–7. doi: 10.1111/j.1349-7006.2006.00250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aggarwal BB, Sung B. Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci. 2009;30:85–94. doi: 10.1016/j.tips.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Shehzad A, Rehman G, Lee YS. Curcumin in inflammatory diseases. Biofactors. 2013;39:69–77. doi: 10.1002/biof.1066. [DOI] [PubMed] [Google Scholar]
- 19.Thangapazham RL, Sharma A, Maheshwari RK. Multiplemolecular targets in cancer chemoprevention by curcumin. AAPS J. 2006;8:E443–9. doi: 10.1208/aapsj080352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagaraju GP, Aliya S, Zafar SF, Basha R, Diaz R, El-Rayes BF. The impact of curcumin on breast cancer. Integr Biol (Camb) 2012;4:996–1007. doi: 10.1039/c2ib20088k. [DOI] [PubMed] [Google Scholar]
- 21.Hatcher H, Planalp R, Cho J, Torti FM, Torti SV. Curcumin: from ancient medicine to current clinical trials. Cell Mol Life Sci. 2008;65:1631–52. doi: 10.1007/s00018-008-7452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhillon N, Aggarwal BB, Newman RA, Wolff RA, Kunnumakkara AB, Abbruzzese JL, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14:4491–9. doi: 10.1158/1078-0432.CCR-08-0024. [DOI] [PubMed] [Google Scholar]
- 23.Fu S, Kurzrock R. Development of curcumin as an epigenetic agent. Cancer. 2010;116:4670–6. doi: 10.1002/cncr.25414. [DOI] [PubMed] [Google Scholar]
- 24.Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu J, et al. Curcumin is a potent DNA hypomethylation agent. Bioorg Med Chem Lett. 2009;19:706–9. doi: 10.1016/j.bmcl.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 25.Nagaraju GP, Zhu S, Wen J, Farris AB, Adsay VN, Diaz R, et al. Novel synthetic curcumin analogues EF31 and UBS109 are potent DNA hypomethylating agents in pancreatic cancer. Cancer Lett. 2013;341:195–203. doi: 10.1016/j.canlet.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 26.Nagaraju GP, Zhu S, Ko JE, Shoji M, El-Rayes B. Anti-angiogenic effects of curcumin and its novel analogs EF-31 and UBS-109 in colorectal cancer. FASEB J. 2013;27(Meeting Abstract Supplement):lb574. [Google Scholar]
- 27.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 28.Kim HS, Lim JY, Sul D, Hwang BY, Won TJ, Hwang KW, et al. Neuroprotective effects of the new diterpene, CBNU06 against beta-amyloid-induced toxicity through the inhibition of NF-kappaB signaling pathway in PC12 cells. Eur J Pharmacol. 2009;622:25–31. doi: 10.1016/j.ejphar.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 29.Lyko F, Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J Natl Cancer Inst. 2005;97:1498–506. doi: 10.1093/jnci/dji311. [DOI] [PubMed] [Google Scholar]
- 30.Wang ZF, Li HL, Li XC, Zhang Q, Tian Q, Wang Q, et al. Effects of endogenous β-amyloid overproduction on tau phosphorylation in cell culture. J Neurochem. 2006;98:1167–75. doi: 10.1111/j.1471-4159.2006.03956.x. [DOI] [PubMed] [Google Scholar]
- 31.Ding GJ, Fischer PA, Boltz RC, Schmidt JA, Colaianne JJ, Gough A, et al. Characterization and quantitation of NF-kappaB nuclear translocation induced by interleukin-1 and tumor necrosis factor-alpha. Development and use of a high capacity fluorescence cytometric system. J Biol Chem. 1998;273:28897–905. doi: 10.1074/jbc.273.44.28897. [DOI] [PubMed] [Google Scholar]
- 32.Adwan L, Zawia NH. Epigenetics: a novel therapeutic approach for the treatment of Alzheimer’s disease. Pharmacol Ther. 2013;139:41–50. doi: 10.1016/j.pharmthera.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miners JS, Van Helmond Z, Chalmers K, Wilcock G, Love S, Kehoe PG. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2006;65:1012–21. doi: 10.1097/01.jnen.0000240463.87886.9a. [DOI] [PubMed] [Google Scholar]
- 34.Lentzen H, Monden I, Linke J, Palenker J. No evidence for enkephalinase A on neuronal cells. Life Sci. 1983;33:105–8. doi: 10.1016/0024-3205(83)90455-1. [DOI] [PubMed] [Google Scholar]
- 35.Hong Y, Beckett C, Belyaev ND, Turner AJ. The impact of amyloid precursor protein signalling and histone deacetylase inhibition on neprilysin expression in human prostate cells. Int J Cancer. 2012;130:775–86. doi: 10.1002/ijc.26028. [DOI] [PubMed] [Google Scholar]
- 36.Belyaev ND, Nalivaeva NN, Makova NZ, Turner AJ. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Rep. 2009;10:94–100. doi: 10.1038/embor.2008.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wright J. Epigenetics: reversible tags. Nature. 2013;498:S10–1. doi: 10.1038/498S10a. [DOI] [PubMed] [Google Scholar]
- 38.Link A, Balaguer F, Shen Y, Lozano JJ, Leung HC, Boland CR, et al. Curcumin modulates DNA methylation in colorectal cancer cells. PLoS ONE. 2013;8:e57709. doi: 10.1371/journal.pone.0057709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khor TO, Huang Y, Wu TY, Shu L, Lee J, Kong AN. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs emethylation. Biochem Pharmacol. 2011;82:1073–8. doi: 10.1016/j.bcp.2011.07.065. [DOI] [PubMed] [Google Scholar]
- 40.Shu L, Khor TO, Lee JH, Boyanapalli SS, Huang Y, Wu TY, et al. Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. AAPS J. 2011;13:606–14. doi: 10.1208/s12248-011-9300-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu YL, Yang HP, Gong L, Tang CL, Wang HJ. Hypomethylation effects of curcumin, demethoxycurcumin and bisdemethoxycurcumin on WIF-1 promoter in non-small cell lung cancer cell lines. Mol Med Rep. 2011;4:675–9. doi: 10.3892/mmr.2011.435. [DOI] [PubMed] [Google Scholar]
- 42.Yu J, Peng Y, Wu LC, Xie Z, Deng Y, Hughes T, et al. Curcumin down-regulates DNA methyltransferase 1 and plays an anti-leukemic role in acute myeloid leukemia. PLoS ONE. 2013;8:e55934. doi: 10.1371/journal.pone.0055934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 44.Shakibaei M, John T, Schulze TG, Lehmann I, Mobasheri A. Suppression of NF-κB activation by curcumin leads to inhibition of expression of cyclo-oxygenase-2 and matrix metalloproteinase-9 in human articular chondrocytes: implications for the treatment of osteoarthritis. Biochem Pharmacol. 2007;73:1434–45. doi: 10.1016/j.bcp.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 45.Moriyuki K, Sekiguchi F, Matsubara K, Nishikawa H, Kawabata A. Curcumin inhibits the proteinase-activated receptor-2-triggered prostaglandin E2 production by suppressing cyclooxygenase-2 upregulation and Akt-dependent activation of nuclear factor-κ B in human lung epithelial cells. J Pharmacol Sci. 2010;114:225–9. doi: 10.1254/jphs.10126SC. [DOI] [PubMed] [Google Scholar]
- 46.Mosca M, Lucciarini R, Perfumi MC, Santoni G. Thiorphan-induced survival and proliferation of rat thymocytes by activation of Akt/survivin pathway and inhibition of caspase-3 activity. J Pharmacol Exp Ther. 2008;327:215–25. doi: 10.1124/jpet.108.138719. [DOI] [PubMed] [Google Scholar]
- 47.Li W, Wu Y, Min F, Li Z, Huang J, Huang R. A nonhuman primate model of Alzheimer’s disease generated by intracranial injection of amyloid-beta42 and thiorphan. Metab Brain Dis. 2010;25:277–84. doi: 10.1007/s11011-010-9207-9. [DOI] [PubMed] [Google Scholar]