

Abstract
Appropriate setting of dissolution specification of extended release (ER) formulations should include precise definition of a multidimensional space of complex definition and interpretation, including limits in dissolution parameters, lag time (t-lag), variability, and goodness of fit. This study aimed to set dissolution specifications of ER by developing drug-specific dissolution profile comparison tests (DPC tests) that are able to detect differences in release profiles between ER formulations that represent a lack of bioequivalence (BE). Dissolution profiles of test formulations were simulated using the Weibull and Hill models. Differential equations based in vivo–in vitro correlation (IVIVC) models were used to simulate plasma concentrations. BE trial simulations were employed to find the formulations likely to be declared bioequivalent and nonbioequivalent (BE space). Customization of DPC tests was made by adjusting the delta of a recently described tolerated difference test (TDT) or the limits of rejection of f2. Drug ka (especially if ka is small), formulation lag time (t-lag), the number of subjects included in the BE studies, and the number of sampled time points in the DPC test were the factors that affected the most these setups of dissolution specifications. Another recently described DPC test, permutation test (PT), showed excellent statistical power. All the formulations declared as similar with PT were also bioequivalent. Similar case-specific studies may support the biowaiving of ER drug formulations based on customized DPC tests.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-014-9615-6) contains supplementary material, which is available to authorized users.
KEY WORDS: bioequivalence, dissolution profile comparisons, f2 similarity factor, in vitro similarity, IVIVC
INTRODUCTION
After a drug formulation proves to be safe and effective in human trials, it is crucial to demonstrate that later formulations, following changes during drug development, post-approval stages, or production by generic manufacturers, possess the same efficacy and safety profile of the original formulation (1–3). Because human experiments to prove efficacy and safety are highly costly, time-consuming, and in some cases ethically questionable, the pharmaceutical industry is constantly searching for effective surrogates for judging biopharmaceutical equivalence of pharmaceutically equivalent drug products. One accepted procedure to ensure efficacy and safety of new formulations is the assessment of bioequivalence (BE) (4–9). On some cases, in vitro tests are sufficient to grant a waiver for an in vivo BE study (10–15). This regulatory acceptance of in vitro testing as a reliable surrogate for an in vivo BE is referred as “biowaiver” (14,16).
Biowaivers can be classified according to their requirements into three categories:
Dissolution-based biowaivers: For manufacturing site changes, scale-up changes, minor process changes, and lower strengths of a dosage forms, dissolution testing can be sufficient to waive BE requirements by demonstrating similarity in dissolution (10–13).
Biowaivers based on a biopharmaceutical classification system (BCS): For immediate release (IR), solid oral dosage forms already approved of (BCS) class I drugs (highly permeable and soluble), demonstration of ≥85% dissolution in three media (i.e., pH 1.2, 4.5, and 6.8) in 15 min is enough for conceding a biowaiving of BE studies. These biowaivers are applicable if the excipients of the formulation do not affect the absorption of the active pharmaceutical ingredient (API); the API is not a prodrug, does not have a narrow therapeutic index, and is not intended to be absorbed in the oral cavity (14,16).
In vivo–in vitro correlation (IVIVC)-based biowaiver: After an IVIVC is established, it can be applied as a surrogate for in vivo BE. Dissolution specifications should be set during the IVIVC development and the in vitro dissolution profiles of the compared formulations must be similar (12,13).
For dissolution-based and IVIVC-based biowaivers, the equivalence of drug formulations is assured by in vitro comparison of dissolution profiles. The term similarity has been employed to describe the lack of difference between dissolution profiles from two different sources (formulations), and it is normally established by using the f2 similarity factor (2,17–19). One objective of establishing BE for the range of the IVIVC is to set dissolution limits in such a way that products within the upper and lower dissolution limits should be biopharmaceutical equivalent to the appropriate reference formulation. However, these limits are difficult to be precisely set because the model fitting and variability of different formulations of the same type cannot be predicted beforehand; therefore, a similarity test like f2 to assure the similarity of both dissolution profiles is still required in the guidelines to assure consistency of quality of the new formulation.
Nevertheless, the limits of rejection of f2 (≤50) are not justified by any mechanistic or biopharmaceutical reasons. As it is an empirical and fixed limit, it is unlikely to exhibit the specific discriminatory power required in all scenarios in which it is currently used. Moreover, recent publications have stated that on the one hand, f2 may classify formulations that are nonbioequivalent as similar (20,21), while on the other hand, f2 can also be over-discriminative in some cases (22–24). Additionally, several publications have recognized some major statistical and conceptual limitations of f2, including uncertain level of confidence, low statistical power, lack of biopharmaceutical or statistical reasons, lack of flexibility to perform in different scenarios, and poor statistical consistency (20,21,24–26).
Advances in developing dissolution technologies which ensure clinical quality for each particular case are continuously being made (22,27,28), especially by developing more biorelevant dissolution methodologies (29–35), which has been identified as a major priority (27,36). Conversely, statistical tools to compare dissolution profiles have not been improved in the last years, despite the fact that a more rigorous application of statistics to understand and incorporate variability and uncertainty has also been identified as a priority in order to achieve a better integration of biopharmaceutics and quality for patient benefit (37,38).
We have recently proposed the theoretical background of a new strategy to perform case-by-case dissolution profile comparisons (26), including two new statistical tests for comparing drug dissolution profiles: the permutation test (PT), a very powerful and strict test to confer similarity, and the tolerated difference test (TDT), a flexible test in which the limits of rejection can be varied according to a desired level of tolerance without affecting its statistical properties. These two tests have the advantage of a better uncertainty quantification (type I and type II errors) and better statistical consistency of their estimators.
Using validated IVIVC models, plasma concentrations achieved by different formulations can be simulated from their dissolution profiles (39–41). It is further possible to simulate BE studies, incorporating proper variability in the formulation dissolution and in the subject pharmacokinetic (PK) parameters and to find which formulations are likely to produce differences in vivo large enough to be considered as nonbioequivalent (42). In this way, it is possible to explore the clinical relevance of the in vitro dissolution specifications set in the IVIVC development.
This study aimed, first, to investigate the effect of dissolution profile comparisons conditions, BE trial conditions, and drug/formulation in the determination of biorelevant dissolution specifications selecting three specific cases (ER formulations of metformin, diltiazem, and pramipexole) in which an IVIVC was previously established in order to extrapolate these findings to those drugs without an established IVIVC. Secondly, we aimed to explore a strategy to set dissolution specifications in an IVIVC by customizing the limit of rejections of dissolution profile comparison (DPC) tests.
METHODS
General Strategy
The strategy used to identify bioequivalent and nonbioequivalent formulations is illustrated in Fig. 1a, b, taking pramipexole as example. In vitro dissolution profiles were simulated for several formulations by modifying the Weibull model parameters, and PK profiles of the formulations were generated using IVIVC models. Through BE simulation studies, nonbioequivalent formulations were detected. The whole set of potential BE formulations is described as the BE space. Once the BE spaces were delimited, TDT, a DPC test, was customized to declare as nonsimilar, the formulations that were likely to be nonbioequivalent. The strategy used to investigate the effect of drug/formulation properties was similar (Fig. 1c). Starting with the same dissolution profile, different PK profiles can be generated by varying the IVIVC model input parameters. Investigation of the effect of such variation on the BE space for the theoretical drug is then possible.
Fig. 1.

Strategy to build the BE spaces and customize DPC tests. a Illustrative flow chart for pramipexole. b Reference formulation was modeled by = 0.076 h− 1, β = 0.732, formulation test 1: kd = 0.056 h − 1 (−26.45%), β = 0.082 (+39.68%), is bioequivalent to the reference formulation by AUC and Cmax. Formulation test 2: kd = 0.136 h− 1 (+79.36%), β = 1.032 (+40.98%), is bioequivalent to the reference formulation by AUC but not by Cmax. AUCtest/AUCReference ratios for test 1, test 2, and test 3 formulations were 1.037, 1.038, and 0.77, respectively; Cmax ratios were 1.017, 1.331, and 0.634. c Reference formulation was modeled as previously, test formulation by kd = 0.0756 h− 1 (+0%), β = 0.8784 (+20.00%). AUCtest/AUCReference ratios using the original and smaller ka were 1.0429 and 0.9256, respectively; Cmax ratios were 1.061 and 0.716
IVIVC and Dissolution Models
Two published differential equation-based IVIVC models were used for analysis. For diltiazem and metformin (Fig. 2a), a one compartment pharmacokinetic model with a first order rate elimination was employed for describing plasma concentrations in which the rate of in vivo input is connected to the rate of in vitro dissolution through a functional dependency that allows inclusion of time scaling, time shifting, and absorption window (43).
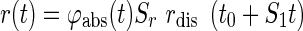 |
1 |
Fig. 2.

Schematic representation of the biopharmaceutic/pharmacokinetic models for the IVIVC of dialtiazem and metformin (a) and pramipexole (b). Parameters used are listed in Table I
Where S1 is the time-scaling factor, Sr is the scaling factor, rdis is the dissolution rate, and t0 is an input parameter that depends on the lag time (t-lag) of the formulation. To account for the variability of the in vivo absorption as the drug moves along the gastrointestinal tract including an absorption window, a function φabs(t) is introduced including a truncated absorption at time tcut:
 |
2 |
The dissolution rate (rdis) was described by the Hill function (44) in which:
 |
3 |
Where fdis(t) is the fraction (%) of drug released at time t, t50 is the time at which 50% of the drug is released from the formulation, and n is a shape parameter.
Resulting in the differential expression for rdis:
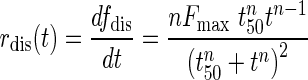 |
4 |
Data were generated to reproduce the data of Gillespie (45) for diltiazem and the data of Balan and co-workers (46) for metformin.
For pramipexole (Fig. 2b), a two compartment model with first order absorption and elimination was used for describing plasma concentrations (47) in which the dissolution rate was described by the Weibull function (48–50):
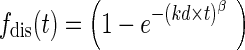 |
5 |
Leading to:
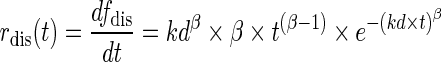 |
6 |
Where fdis(t) is the fraction (%) of drug released at time t, kd is the dissolution constant, and β is the shape parameter. The relationship between the kdin vivo and kdin vitro was modeled by kdin vivo = kdin vitro + θSCL where θSCL is a scale factor representing the increment in the in vivo dissolution. θSCL of 0.0581 (13.75% CV) was used in all simulations.
For the purpose of this study, only differential equation-based IVIVC methods were included because of the more mechanistic nature of these models (36,51,52). All parameters employed are listed in Table I.
Table I.
Population Pharmacokinetic Models Parameters Used in the IVIVC Models
| BCS | Pramipexole I | Diltiazem | Metformin |
|---|---|---|---|
| I | I | III | |
| k el (h−1) | 0.087 (13) | 0.138 (10) | 0.23 (10) |
| tlag (h) | 0.22 (66.3) | 0.57 (10) | 0.86 (10) |
| tcut (h) | NA | 6.36 (20) | 4.77 (20) |
| k a (h−1) | 5.26 (91.8) | NA | NA |
| V1 (L) | 351 (14.1) | NA | NA |
| V2 (L) | 60.9 (10) | NA | NA |
| CLD (L/h) | 33.2 (10) | NA | NA |
Parameters are listed with the IIV in parenthesis
k el elimination constant, t lag lag time, t cut absorption window, k a absorption constant, V 1 , V 2 volumes of distribution in the central and peripheral compartments, respectively, C LD apparent distribution clearance, NA parameter not used in that model, BCS biopharmaceutical classification system
Test and Reference Formulations
Reference formulations were modeled as follows:
Metformin: t50 = 1.77 h; n = 2.6 (Hill Model , equation 3)
Diltiazem: t50 = 1.61 h; n = 1.85 (Hill Model , equation 3)
Pramipexole: kd = 0.076 h−1; β = 0.732 (Weibull Model, equation 5)
Test formulations were generated by varying simultaneously the two dissolution model parameters (t50 and n or kd and β) from −95 to 200% around those of the reference formulation. Variability (CV 10%) was included at all dissolution points to mimic experimental data. Approximately, 1,000 test formulations were compared to a reference in every experiment.
Simulations and Bioequivalent Studies
Simulations of plasma concentration were conducted in the R software environment (version 2.14.2.). Interindividual variability (IIV) was included for each parameter (Table I) to fit the reported experimental variability (43,47) including an overall CV of area under the curve (AUC)0-∞ and Cmax of 15% in order to mimic the previously published data (43,47). In total, 1,000 BE crossover-simulated studies per scenario were conducted. In each study, 12 subjects (minimum number of subjects accepted by FDA and EMEA BE guidelines) were generated. Each subject received an oral dose of the test and reference formulation with a washout period between the administrations. AUC0-∞ and maximum plasma concentration (Cmax) were calculated from the generated plasma concentrations. BE between formulations was determined by calculating 90% confidence intervals (90% CI) of the ratio between test and reference means after log transformation of AUC0-∞ and Cmax (equivalent to a two one-sided test procedure). The formulations were considered bioequivalent if the 90% CI of AUC0-∞ and Cmax ratios were contained within the acceptance interval of 80.00–125.00% (R scripts included in the supplemental data).
DPC Tests
Dissolution profiles from the reference and test formulations were compared using the f2 similarity factor (Eq. 7) and two recently described tests, PT and TDT (26). In all cases, 12 units of the reference formulation and 12 units of the test formulation were simulated. The f2 similarity factor was calculated according to:
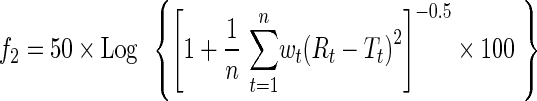 |
7 |
Where Rt is the mean of the dissolved drug from the reference batch at time t, Tt is the mean of the drug dissolved from the test batch at time t, n is the number of time points, and wt is a weight factor that can be used to enhance the influence of particular time points. If the calculation yielded f2 ≥ 50, similarity of R and T was declared. Theoretically, if the difference in drug dissolution between R and T is exactly 10% at every time point, the value of f2 is 50.
PT and TDT were calculated as previously described (26). Briefly, in PT, the information of each tablet is considered as vector of length defined by the number of time points sampled in the dissolution profile. A mathematical distance D0 between the reference and test profiles is first calculated by the sum of the squared differences of the means at each time point. The same distance Di is calculated for each of the 5,000 permutations of the vectors of the original data and 5,000 values of Di are obtained. The D0 is compared to the distribution of the 5,000 calculated Di. According to a predetermined type I error (typically, alpha = 0.05), a rejection value that is greater than the 1-alpha percent of all Di values in this empirical distribution is calculated. If the profiles are similar, D0 is expected to be below this rejection value; D0 above the rejection value indicates lack of similarity between the two profiles.
TDT is based on a tolerated difference (δ) in dissolution between two tablets at each time point. This test attempts to statistically prove whether the differences between the reference and test samples exceed the predetermined tolerated difference or not.
Having at any time point the dissolved drug for m tablets from the reference and m from the test
 |
Differences between all Rti and Tti are evaluated, and the number of events for which this difference is greater than the established tolerated difference (δ) is counted.
 |
8 |
Where Di is the sum of differences greater than δ at the ith time point. p value is calculated by comparing the calculated Dd statistic from the samples with the distribution of Dd under the null hypothesis |μRt – μTt| ≤ δ at every time point (rejection values in supplemental data S1).
Statistical Power Explorations
Sets of 12 reference formulation tablets and 12 test formulation tablets were generated under conditions of nonsimilarity when different model parameters were employed for the reference and test formulations. Each pair of batches was compared using f2, TDT, and PT. At every condition, 5,000 sets of reference test were generated, and the percentage of rejections (% detections of no similarity) was evaluated. More powerful tests are expected to detect smaller differences in the parameters used. In all cases, equidistant sample points were sampled from which the last sample point was the smaller t85 (time to reach 85% release) of the two formulations.
RESULTS
Illustrative Example
In Fig. 1b, results from pramipexole are outlined as an example. Test 1 is bioequivalent to the reference formulation by AUC and Cmax (PK profiles). Formulation test 2 is bioequivalent to the reference formulation by AUC but not by Cmax, and formulation test 3 is not bioequivalent to the reference formulation by either AUC or Cmax. The BE space delimits the bioequivalent formulations from the nonbioequivalent. TDT was customized to declare as nonsimilar, formulations that are likely to be nonbioequivalent (BE formulations). To investigate the effect of drug/formulation properties, the same dissolution profile (test 1) was employed. Different PK profiles can be generated by varying the IVIVC model input parameters. In Fig. 1c, using the same test formulation (equal kd and β), two different PK profiles are generated by using different ka values (PK profiles). The Cmax BE space for the theoretical drug with a smaller ka was reduced (green BE space) and the test formulation is no longer bioequivalent to the reference formulation by Cmax (BE formulations). When the larger ka is used, the same test formulation is bioequivalent to the reference formulation (inside the red BE space).
Bioequivalent and Similarity Spaces
For each drug, test formulations were compared to the reference formulation in their dissolution profiles and plasma levels. Figure 3 illustrates the effect of dissolution parameters on BE and similarity. The X- and Y-axes show the difference in each one of the dissolution parameters of the test formulation compared to the reference formulation. When the difference in the two parameters is 0 (coordinates 0,0 in the contour plots), the test and reference formulations are the same. Response surfaces for AUC and Cmax display the test formulations (combination of dissolution parameters) with probability ≥80% of being nonbioequivalent by AUC or Cmax, respectively. Density contour plots displaying probability of rejection from 0 to 100% for AUC and Cmax for the three ER formulations are included in the supplemental data (S2–S7).
Fig. 3.

BE spaces of pramipexole when population variability is or is not included in the BE space delimitation. Solid line no variability included, dashed line population variability included in the subjects of the BE studies
When the BE space of the formulations is calculated considering population variability to mimic experimental variation, the BE space is considerably smaller compare to the BE space calculated without considering variability (Fig. 3). The same effect was observed for the other two drugs (S8 and S9).
Response surfaces for f2 and PT (Fig. 4) delimit the combination of dissolution parameters at which the probability of declaring nonsimilarity using the corresponding test is ≥80%. This set of in vitro similar formulations is described as the similarity space (Sim space).
Fig. 4.

BE spaces of the three formulations. Contour lines for AUC and Cmax display the test formulations (combination of dissolution parameters) with probability ≥80% of being declaring nonbioequivalent by AUC or Cmax, respectively
PT was the most powerful test to declare as non-similar, test formulations with changes in the dissolution model parameters compared to those of the Reference formulation. Consequently, formulations declared as similar with PT were always also bioequivalent and differences in Cmax and AUC from the test and reference formulations were less than 1.5%. For all three drugs, f2 showed less power than PT, and several formulations declared as similar with f2 were not bioequivalent.
Some contour lines may appear wavy in the BE spaces due to limited number of points in the response surface of the contour plots (≈1,000). Smoother contour lines can be drawn by increasing this number by 50-fold (as in Fig. 3 compared to Fig. 4). However, the computation time is also increased by 65-fold (from 9 h to 24.5 days per line in every contour plot).
Effect of Drug/Formulation Properties on Bioequivalence
The effect of changes in ka, kel (CL), t-lag, V1, V2, and CLD (model II in Fig. 2) on the BE space of the pramipexole formulation was studied. Changes in V1, V2, and CLD of 10-fold had no effect on the BE space (S10). Changes in ka of 10-fold had no apparent effect on the BE space of AUC or Cmax. However, reductions in ka of 50-fold or bigger showed an increasing reduction in the BE space of Cmax (Fig. 5a). Changes in t-lag had a direct impact in the BE space of both AUC and Cmax (Fig. 5b, c); when t-lag was not considered (t-lag = 0), the BE space was increased. For a t-lag of 2 h, the BE space was reduced to 25% of the original area. Changes in kel had no effect on the BE space of AUC. A slight effect in the BE space of Cmax by changes in kel was observed; however, there was no change in the total area of the BE space (less than 5%), but a small modification in the shape regardless if the kel was increased or decreased. The same effect was observed for metformin and diltiazem (supplemental data S11 and S12). Studied effects are summarized in Table II.
Fig. 5.

Effect of drug/formulation parameters in the determination of BE space. a Effect of ka on BE spaces. b Effect of tlag on AUC BE space. c Effect of tlag on Cmax BE space
Table II.
Effect of Volunteers Sample Size and Drug Parameters on the BE Space
| Pramipexole | Diltiazem | Metformin | ||||
|---|---|---|---|---|---|---|
| AUC | Cmax | AUC | Cmax | AUC | Cmax | |
| Increase sample size (volunteers) | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ |
| Change k el | ↔ | ↑ ↓ | ↔ | ↑ ↓ | ↔ | ↑ ↓ |
| Increase t-lag | ↓ | ↓ | NA | NA | NA | NA |
| Reduce k a | ↔ | ↓ | NA | NA | NA | NA |
| Change V1 | ↔ | ↔ | NA | NA | NA | NA |
| Change V2 | ↔ | ↔ | NA | NA | NA | NA |
| Change CLD | ↔ | ↔ | NA | NA | NA | NA |
↑ Increase BE space, ↓ reduce BE Space, ↑↓ change BE space and some zones are augmented and some are reduced but no total increase or decrease, ↔ no apparent effect, NA parameter not used in that model, AUC area under the curve
Effect of BE Trials Conditions and DPC Tests Conditions on BE space and Sim Space
BE trial simulations were performed with different numbers of subjects to study the effect of sample size in the BE space. Figure 6a shows the increment in the BE space, in both AUC and Cmax, at increasing numbers of subjects. Nevertheless, the increment in the area from 12 to 18 subjects was less than 3%, and the reduction in the BE space from 12 to 6 subjects was less than 5%.
Fig. 6.

Effect of BE trials conditions and DPC tests conditions on BE space and Sim space. a Effect of number of subjects included in BE trials on BE spaces. b Effect dissolution sampled time points on Sim space
The Sim space was sensitive to the number of points used in the dissolution profile comparison; a reduction in Sim space (increasing statistical power) at higher number of time points was detected (Fig. 6b). These effects of number of subjects and of time points were also observed for the metformin and diltiazem formulations (supplemental data S13 and S14).
Customization of TDT and f2
Dissolution profiles of the generated formulations were compared using TDT at different values of δ or different limits for f2. Figure 7 shows the different similarity space (Sim space) of TDT at increasing values of δ for the pramipexole formulation. A δ of 3.45 is the maximum at which all the formulations declared as similar with this test are also bioequivalent. A δ of 3.6 for metformin and 5.95 for diltiazem were found following the same procedure (S15 and S16). This value represents the average tolerated difference (in %) between two formulations at any time point to produce bioequivalent formulations under both criteria, AUC and Cmax. Customized limits of f2 were 71 for pramipexole, 60 for metformin, and 60 for diltiazem. The customized Sim spaces of TDT and f2 were very similar in size and shape.
Fig. 7.

Customization of TDT and f2. a Sim spaces of TDT at different values of δ. b f2 Sim spaces at different values of rejection for pramipexole formulations
BE space Compared to MDT and MRT
Mean dissolution time (MDT) and mean residence time (MRT) were calculated for all the reference formulations and compared to the BE space. Surface responses of MDT and MRT are depicted in Fig. 8 for pramipexole formulations. There is no overlap between the MDT or MRT response surfaces with the BE space. Test formulations with the same MRT or MDT of the reference formulation could be nonbioequivalent. Likewise, formulations with very different values of MDT and MRT are included in the BE space. Similarly, BE space did not match with the surface spaces of MDT or MRT for metformin or diltiazem (S17 and S18).
Fig. 8.

Comparison of BE spaces with a MDT and b MRT
DISCUSSION
In this study, we compared the BE space of three ER formulations against the Sim spaces calculated with three different DPC tests trying to identify which DPC test could be more suitable to set dissolution specifications (best overlap between BE and Sim spaces) and what factors could influence this setting of dissolution limits and its clinical relevance.
The calculated BE space of the formulations is considerably reduced if population variability is considered (Fig. 3). This should be analyzed in conjunction with the observed increment in the BE spaces when the subject sample size is incremented in the BE studies (Fig. 6). Because of interindividual variability, it is more likely to declare BE of two formulations when more subjects are included in a BE trial, and a BE space calculated with a large sample of subjects is expected to be closer to the BE space when variability is not considered. In principle, the BE space when variability is not considered should be more aligned with results from large trials. However, the fact that there is a difference between the two BE spaces (with or without considering variability) manifest that the smaller BE space could be more relevant concerning individual safety and efficacy.
Likewise, the Sim space of TDT and PT was reduced (increased statistical power) when the number of time points sampled in the dissolutions profiles was increased (Fig. 6). Sim space of f2 was erratically increased and decreased by the increased number of sample time points manifesting the lack of consistency of this estimator and the lack of proper statistical basis of the f2 test (S19). These results manifest that conclusions about BE or similarity can be affected by the specific setup of each particular comparison. Precise limits must be fixed in order to standardize comparisons and rejections criteria. Based on these results, we propose that a number between 6 and 10 equidistant time points must be sampled in the dissolution comparisons, as a good predictor of BE studies with 12 subjects.
It has been stated that ER formulations can produce flip flop-like kinetics when the apparent ka < kel (kel/ka ratio >1) and could lead to miscalculations of the PK parameters (53). For pramipexole, we observed reductions in the Cmax BE space for values of ka ≤0.4 h−1 (kel/ka ratio = 0.2). Calculating the BE space for theoretical drugs with different kel and ka, the reduction in the Cmax BE space due to reduction in ka was produced only at kel/ka ratios of 0.2 or higher (S20–S22). This seems to be the limit at which the ka is small enough in comparison with the kel to reduce the Cmax in the PK profile. This aspect should be accounted for in the design of ER formulations since a slow enough release can have, in practical terms, the same effect as reducing the ka of the drug. We suggest that this flip flop-like phenomenon could be present at kel/ka ratios smaller than 1 (i.e., 0.2).
Absorption is recognized as the rate-limiting step for BCS class III drug formulations (54,55); however, for the metformin formulation, test formulations with faster release (Fig. 4, smaller t50 and larger n) than the reference formulation generated plasma profiles with higher Cmax and AUC, resulting in declaration of nonbioequivalence. Formulations with slower release (larger t50 and smaller n) were also declared as nonbioequivalent, generating plasma profiles with smaller Cmax and AUC. These results suggest that in the case of ER products, dissolution may also play a substantial role in the in vivo performance of BCS class III formulations.
AUC and Cmax from formulations with larger t-lag were smaller than those of formulations with smaller t-lag (Fig. 5, S23); therefore, changes in the dissolution parameters of the reference formulations have a bigger relative impact on the BE space in comparison to BE spaces of formulations with smaller t-lag, explaining the reduced BE space of formulations with larger t-lag. This may have a considerable impact on the setting of dissolution specifications of the IVIVC because the definitive BE space should be define as a multidimensional space of dissolution parameters, t-lag, and variability.
The differences in dissolution necessary to produce nonbioequivalent formulations vary from one drug formulation to another, depending on the drug and formulation. When the original limits of similarity are used (f2 ≥ 50), f2 failed to associate the in vitro similarity of two formulations with their comparative in vivo performance in the three cases investigated. Moreover, f2 declared as similar not only formulations that are likely to be bioequivalent in vivo but also formulations that are likely to be nonbioequivalent in vivo (Fig. 4). This observation is in agreement with one of the criticisms of f2 (21,22) of lack of flexibility to perform in a wider range of cases.
We aimed to set the set dissolution limits of each formulation by customizing cases, specifically the limits of rejection of DPC tests. We were able to associate f2 (with customized limits different than 50) and TDT limits of rejection with an in vivo property (BE) of the tested formulations. Proper setting of dissolution specifications in an IVIVC should define a complex multidimensional space with clear limits in dissolution parameters, t-lag, variability, and goodness of fit; therefore, setting dissolution limits through customized DPC tests offer a simpler solution with clear rejection criteria. The customized Sim spaces of TDT and f2 were very similar in size and shape, and test formulations declared as similar to the reference formulation using these customized DPC tests are expected to be also bioequivalent and consequently to retain the same efficacy and safety profile. However, we think that a customized TDT should be preferred due to its superior statistical properties (known uncertainty and consistency).
PT was the most restrictive test to confer similarity of dissolution profiles; typically, formulations declared as similar with this test did not differ in AUC and Cmax more than 1.5% assuming the same patient with no intra-occasion variability. Formulations declared as similar with PT are expected with high accuracy to be bioequivalent, even formulations with different release mechanism or from a different manufacturer, situations for which an IVIVC is not recommended by the current guidelines to waive BE studies. From the regulatory point of view, an established DPC test of an innovator formulation represents a fast, cheap, and reliable protocol with simple acceptance criteria to test new generic formulations, reducing the costs and time of these submissions and potentially avoiding unnecessary in vivo BE studies. PT should also be a more suitable test for monitoring similarity as a critical quality attribute (CQA) (56,57) because it would be more sensitive to detect CQA changes.
Ideally, all the bioequivalent formulations should be declared as similar with the customized DPC tests; however, a total overlap of the BE space and the Sim space was not achieved with the investigated tests. Nevertheless, according to risk management principles (37,58), the reduction of risk caused to subjects by declaring as similar formulations that are not bioequivalent must be considered as a higher priority than reducing manufacturer risk of declaring as non-similar formulations that are bioequivalent.
Setting of dissolution specifications through customized DPC tests is only possible when differential equations based IVIVC are employed because it allows a complete and detailed exploration of the multidimensional BE space (S24), which is not possible with conventional convolution/deconvolution IVIVC.
MDT and MRT (40,54) were not useful to discriminate between nonbioequivalent and bioequivalent formulations nor to set dissolution specifications (Fig. 8 and S17, S18, and S25) highlighting the complex composition of the multidimensional BE space.
Several questions remain open as to which is the most suitable procedure to declare BE (38,55,59–62) and which other mathematical expressions could model drug release more mechanistically (63,64). If these customized DPC tests are attempted to be used for generic drugs, serious harmonization efforts should be made to share these IVIVC models between agencies and manufacturers.
The strategy presented in this study of setting dissolution specifications for IVIVC using customized DPC tests can be refined, improved, and applied to other drug formulations when more IVIVC mechanistic models become available. For BCS class III drugs, further studies are required before analyzing the possibility of biowaiving ER formulations of these drugs (65) or generic ER formulations of BCS class I drugs. However, we propose that under a deep case-specific analysis, biowaiving of ER of BCS class I drug formulations may be possible through customized DPC tests or by PT.
CONCLUSIONS
In the present study, we have set the dissolution specifications of three IVIVC according to the probabilities of the theoretical reference formulations of being bioequivalent or nonbioequivalent. Because the full BE space could be a multidimensional space of difficult analysis, we proposed an easy to implement and interpret customization of DPC tests to set dissolution specifications for three different ER formulations. According to these simulations, formulations that can prove to be similar with the reference formulation under the established conditions are likely to be bioequivalent. Established conditions were TDT with δ of 3.6, 5.95, and 3.45 and f2 with limits of 60, 55, and 71 for metformin, diltiazem, and pramipexole, respectively, and sampling at least six time points in the release profiles. Once a specific test is developed for a particular formulation, this DPC test can be used to explore post-approval changes by the manufacturer or to evaluate BE between products from different manufacturers. Drug ka (especially if ka is small), formulation t-lag, the number of subjects included in the BE studies, and the number of sampled time points in the DPC tests were the factors that affected the most this setups of dissolution specifications. PT was the most powerful DPC test, and differences in Cmax and AUC produced by formulations declared as similar with PT were less than 1.5% in all cases.
Electronic supplementary material
(DOCX 8296 kb)
(R 11 kb)
(R 14 kb)
(R 3 kb)
(RDATA 2 kb)
Acknowledgments
JDGM thanks the Deutscher Akademischer Austausschdienst (DAAD) and Colciencias (Colombia) for the financial support.
Footnotes
Vicente Germán Casabó passed away during the preparation of this study.
REFERENCES
- 1.CDER. Guidance for industry, immediate release solid oral dosage forms, scale-up and postapproval changes. Rockville, MD 208551995.
- 2.FDA. Guidance for industry, SUPAC-MR: modified release solid oral dosage forms scale-up and postapproval changes: chemistry, manufacturing, and controls; in vitro dissolution testing and in vivo bioequivalence documentation. CMC 8. Rockville, MD 20857: Center for Drug Evaluation and Research (CDER); 1997.
- 3.EMEA. Guidance on pharmaceutical Develo. London, E14 4HB, UK; 2009.
- 4.FDA. Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products—general considerations. In: FDA, editor. Rockville, MD; 2003.
- 5.Midha KK, McKay G. Bioequivalence; its history, practice, and future. AAPS J. 2009;11(4):664–670. doi: 10.1208/s12248-009-9142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.EMEA. Guideline on the investigation of bioequivalence. London, E14 4HB, UK; 2010.
- 7.NIHS. Guideline for bioequivalence studies of generic products. In: Bureau PaFS, editor. Japan; 2012.
- 8.WHO. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability. In: Fortieth report of the WHO Expert Committee on Specifications for Pharmaceutical Preparations, World Health Organization. WHO Technical Report Series, No. 937, Annex 7. Geneva; 2006. [PubMed]
- 9.Karalis V, Macheras P. Current regulatory approaches of bioequivalence testing. Expert Opin Drug Metab Toxicol. 2012;8(8):929–942. doi: 10.1517/17425255.2012.690394. [DOI] [PubMed] [Google Scholar]
- 10.FDA. Guidance for industry. SUPAC-IR, immediate release solid oral dosage forms. Scale-up and post-approval changes. 1995.
- 11.CDER/FDA. Guidance for industry. Dissolution testing of immediate release solid oral dosage forms. Rockville, MD; 1997.
- 12.CDER/FDA. Guidance for industry. SUPAC-MR: modified release solid oral dosage forms Scale-up and postapproval changes: chemistry, manufacturing, and controls; in vitro dissolution testing and in vivo bioequivalence documentation. CMC 8. Rockville, MD 20857: Center for Drug Evaluation and Research (CDER); 1997.
- 13.CDER/FDA. Guidance for industry. Extended release oral dosage forms: development, evaluation, and, application of in vitro/in vivo correlations. Rockville, MD 208571997. [DOI] [PubMed]
- 14.CDER/FDA. Guidance for industry. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. Rockville, MD 208572000.
- 15.CDER/FDA. Guidance for industry. Bioavailability and bioequivalence studies for orally administered drug products -general considerations. In: FDA, editor. Rockville, MD; 2003.
- 16.Gupta E, Barends DM, Yamashita E, Lentz KA, Harmsze AM, Shah VP, et al. Review of global regulations concerning biowaivers for immediate release solid oral dosage forms. Eur J Pharm Sci Off J Eur Fed Pharm Sci. 2006;29(3–4):315–324. doi: 10.1016/j.ejps.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Moore JW, Flanner HH. Mathematical comparison of dissolution profiles. Pharm Technol. 1996;20(6):64–74. [Google Scholar]
- 18.FDA. Guidance for industry, dissolution testing of immediate release solid oral dosage forms. Rockville, MD; 1997.
- 19.Korakianiti E, Rekkas D. Statistical thinking and knowledge management for quality-driven design and manufacturing in pharmaceuticals. Pharm Res. 2011;28(7):1465–1479. doi: 10.1007/s11095-010-0315-3. [DOI] [PubMed] [Google Scholar]
- 20.Duan JZ, Riviere K, Marroum P. In vivo bioequivalence and in vitro similarity factor (f2) for dissolution profile comparisons of extended release formulations: how and when do they match? Pharm Res. 2011;28(5):1144–1156. doi: 10.1007/s11095-011-0377-x. [DOI] [PubMed] [Google Scholar]
- 21.Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–133. doi: 10.1016/S0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 22.Dickinson PA, Lee WW, Stott PW, Townsend AI, Smart JP, Ghahramani P, et al. Clinical relevance of dissolution testing in quality by design. AAPS J. 2008;10(2):380–390. doi: 10.1208/s12248-008-9034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kocic I, Homsek I, Dacevic M, Parojcic J, Miljkovic B. An investigation into the influence of experimental conditions on in vitro drug release from immediate-release tablets of levothyroxine sodium and its relation to oral bioavailability. AAPS PharmSciTech. 2011;12(3):938–948. doi: 10.1208/s12249-011-9660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polli JE, Rekhi GS, Augsburger LL, Shah VP. Methods to compare dissolution profiles and a rationale for wide dissolution specifications for metoprolol tartrate tablets. J Pharm Sci. 1997;86(6):690–700. doi: 10.1021/js960473x. [DOI] [PubMed] [Google Scholar]
- 25.O'Hara T, Dunne A, Kinahan A, Cunningham S, Stark P, Devane J. Review of methodologies for the comparison of dissolution profile data. Adv Exp Med Biol. 1997;423:167–171. doi: 10.1007/978-1-4684-6036-0_14. [DOI] [PubMed] [Google Scholar]
- 26.Gomez-Mantilla JD, Casabo VG, Schaefer UF, Lehr CM. Permutation test (PT) and tolerated difference test (TDT): two new, robust and powerful nonparametric tests for statistical comparison of dissolution profiles. Int J Pharm. 2013;441(1–2):458–467. doi: 10.1016/j.ijpharm.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Polli JE, Cook JA, Davit BM, Dickinson PA, Argenti D, Barbour N, et al. Summary workshop report: facilitating oral product development and reducing regulatory burden through novel approaches to assess bioavailability/bioequivalence. AAPS J. 2012;14(3):627–638. doi: 10.1208/s12248-012-9376-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu Y. In vitro-in vivo correlations: fundamentals, development considerations, and applications. . In: Qiu Y. CHen Y ZG, editor. Developing solid oral dosage forms: pharmaceutical theory & practice. Academic; 2009. p. 379–408.
- 29.Dressman JB, Vertzoni M, Goumas K, Reppas C. Estimating drug solubility in the gastrointestinal tract. Adv Drug Deliv Rev. 2007;59(7):591–602. doi: 10.1016/j.addr.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 30.Pluvinage B, Chitayat S, Ficko-Blean E, Abbott DW, Kunjachen JM, Grondin J, et al. Conformational analysis of StrH, the surface-attached exo-beta-d-N-acetylglucosaminidase from Streptococcus pneumoniae. J Mol Biol. 2013;425(2):334–349. doi: 10.1016/j.jmb.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Juenemann D, Bohets H, Ozdemir M, de Maesschalck R, Vanhoutte K, Peeters K, et al. Online monitoring of dissolution tests using dedicated potentiometric sensors in biorelevant media. Eur J Pharm Biopharm. 2011;78(1):158–165. doi: 10.1016/j.ejpb.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 32.Garbacz G, Klein S. Dissolution testing of oral modified-release dosage forms. J Pharm Pharmacol. 2012;64(7):944–968. doi: 10.1111/j.2042-7158.2012.01477.x. [DOI] [PubMed] [Google Scholar]
- 33.Jantratid E, De Maio V, Ronda E, Mattavelli V, Vertzoni M, Dressman JB. Application of biorelevant dissolution tests to the prediction of in vivo performance of diclofenac sodium from an oral modified-release pellet dosage form. Eur J Pharm Sci Off J Eur Fed Pharm Sci. 2009;37(3–4):434–441. doi: 10.1016/j.ejps.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 34.Lobenberg R, Kramer J, Shah VP, Amidon GL, Dressman JB. Dissolution testing as a prognostic tool for oral drug absorption: dissolution behavior of glibenclamide. Pharm Res. 2000;17(4):439–444. doi: 10.1023/A:1007529020774. [DOI] [PubMed] [Google Scholar]
- 35.Jantratid E, Janssen N, Reppas C, Dressman JB. Dissolution media simulating conditions in the proximal human gastrointestinal tract: an update. Pharm Res. 2008;25(7):1663–1676. doi: 10.1007/s11095-008-9569-4. [DOI] [PubMed] [Google Scholar]
- 36.Karalis V, Magklara E, Shah VP, Macheras P. From drug delivery systems to drug release, dissolution, IVIVC, BCS, BDDCS, bioequivalence and biowaivers. Pharm Res. 2010;27(9):2018–2029. doi: 10.1007/s11095-010-0220-9. [DOI] [PubMed] [Google Scholar]
- 37.Selen A, Cruanes MT, Mullertz A, Dickinson PA, Cook JA, Polli JE, et al. Meeting report: applied biopharmaceutics and quality by design for dissolution/release specification setting: product quality for patient benefit. AAPS J. 2010;12(3):465–472. doi: 10.1208/s12248-010-9206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang W, Kim S, Zhang X, Lionberger RA, Davit BM, Conner DP, et al. The role of predictive biopharmaceutical modeling and simulation in drug development and regulatory evaluation. Int J Pharm. 2011;418(2):151–160. doi: 10.1016/j.ijpharm.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 39.Chilukuri DM, Sunkara G, Young D. Pharmaceutical product development : in vitro-in vivo correlation. New York: Informa Healthcare; 2007. [Google Scholar]
- 40.FDA. Guidance for industry extended release oral dosage forms: development, evaluation, and application of in vitro/in vivo correlations. Rockville, MD 208571997. [DOI] [PubMed]
- 41.Fotaki N, Aivaliotis A, Butler J, Dressman J, Fischbach M, Hempenstall J, et al. A comparative study of different release apparatus in generating in vitro-in vivo correlations for extended release formulations. Eur J Pharm Biopharm. 2009;73(1):115–120. doi: 10.1016/j.ejpb.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 42.Polli JE. In vitro studies are sometimes better than conventional human pharmacokinetic in vivo studies in assessing bioequivalence of immediate-release solid oral dosage forms. AAPS J. 2008;10(2):289–299. doi: 10.1208/s12248-008-9027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchwald P. Direct, differential-equation-based in-vitro-in-vivo correlation (IVIVC) method. J Pharm Pharmacol. 2003;55(4):495–504. doi: 10.1211/002235702847. [DOI] [PubMed] [Google Scholar]
- 44.Eddington ND, Marroum P, Uppoor R, Hussain A, Augsburger L. Development and internal validation of an in vitro-in vivo correlation for a hydrophilic metoprolol tartrate extended release tablet formulation. Pharm Res. 1998;15(3):466–473. doi: 10.1023/A:1011988601696. [DOI] [PubMed] [Google Scholar]
- 45.Gillespie WR. Convolution-based approaches for in vivo-in vitro correlation modeling. Adv Exp Med Biol. 1997;423:53–65. doi: 10.1007/978-1-4684-6036-0_5. [DOI] [PubMed] [Google Scholar]
- 46.Balan G, Timmins P, Greene DS, Marathe PH. In vitro-in vivo correlation (IVIVC) models for metformin after administration of modified-release (MR) oral dosage forms to healthy human volunteers. J Pharm Sci. 2001;90(8):1176–1185. doi: 10.1002/jps.1071. [DOI] [PubMed] [Google Scholar]
- 47.Soto E, Haertter S, Koenen-Bergmann M, Staab A, Troconiz IF. Population in vitro-in vivo correlation model for pramipexole slow-release oral formulations. Pharm Res. 2010;27(2):340–349. doi: 10.1007/s11095-009-0027-8. [DOI] [PubMed] [Google Scholar]
- 48.Langenbucher F. Linearization of dissolution rate curves by the Weibull distribution. J Pharm Pharmacol. 1972;24(12):979–981. doi: 10.1111/j.2042-7158.1972.tb08930.x. [DOI] [PubMed] [Google Scholar]
- 49.Papadopoulou V, Kosmidis K, Vlachou M, Macheras P. On the use of the Weibull function for the discernment of drug release mechanisms. Int J Pharm. 2006;309(1–2):44–50. doi: 10.1016/j.ijpharm.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 50.Dokoumetzidis A, Papadopoulou V, Macheras P. Analysis of dissolution data using modified versions of Noyes-Whitney equation and the Weibull function. Pharm Res. 2006;23(2):256–261. doi: 10.1007/s11095-006-9093-3. [DOI] [PubMed] [Google Scholar]
- 51.Dunne A. Approaches to developing in vitro–in vivo correlation models. In: Murthy CD SG, Young D, editors. Pharmaceutical product development: in vitro-in vivo correlation (drugs and the pharmaceutical sciences) New York: Informa Healthcare; 2007. pp. 47–70. [Google Scholar]
- 52.Farrel CHS. IVIVC for oral drug delivery: immediate release and extended release dosage forms in pharmaceutical product development. In: Chilukuri DM SG, Young D, editors. Pharmaceutical product development: 165 (drugs and the pharmaceutical sciences) New York: Informa Healthcare; 2007. pp. 125–140. [Google Scholar]
- 53.Proctor WR, Bourdet DL, Thakker DR. Mechanisms underlying saturable intestinal absorption of metformin. Drug Metab Dispos. 2008;36(8):1650–1658. doi: 10.1124/dmd.107.020180. [DOI] [PubMed] [Google Scholar]
- 54.Drewe J, Guitard P. In vitro-in vivo correlation for modified-release formulations. J Pharm Sci. 1993;82(2):132–137. doi: 10.1002/jps.2600820204. [DOI] [PubMed] [Google Scholar]
- 55.Endrenyi L, Tothfalusi L. Metrics for the evaluation of bioequivalence of modified-release formulations. AAPS J. 2012;14(4):813–819. doi: 10.1208/s12248-012-9396-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.CDER/FDA. Guidance for industry. Q8(R2) Pharmaceutical development. 2009.
- 57.Lionberger RA, Lee SL, Lee L, Raw A, Yu LX. Quality by design: concepts for ANDAs. AAPS J. 2008;10(2):268–276. doi: 10.1208/s12248-008-9026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.CDER/FDA. Guidance for industry. Q9 Quality Risk Management. Rockville, MD 208572006.
- 59.Garcia-Arieta A, Gordon J. Bioequivalence requirements in the European Union: critical discussion. AAPS J. 2012;14(4):738–748. doi: 10.1208/s12248-012-9382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karalis V, Macheras P, Van Peer A, Shah VP. Bioavailability and bioequivalence: focus on physiological factors and variability. Pharm Res. 2008;25(8):1956–1962. doi: 10.1007/s11095-008-9645-9. [DOI] [PubMed] [Google Scholar]
- 61.Karalis V, Macheras P. An insight into the properties of a two-stage design in bioequivalence studies. Pharm Res. 2013;30(7):1824–1835. doi: 10.1007/s11095-013-1026-3. [DOI] [PubMed] [Google Scholar]
- 62.Karalis V, Symillides M, Macheras P. Novel methods to assess bioequivalence. Expert Opin Drug Metab Toxicol. 2011;7(1):79–88. doi: 10.1517/17425255.2011.539202. [DOI] [PubMed] [Google Scholar]
- 63.Siepmann J, Siepmann F. Mathematical modeling of drug delivery. Int J Pharm. 2008;364(2):328–343. doi: 10.1016/j.ijpharm.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 64.Siepmann J, Siepmann F. Mathematical modeling of drug dissolution. Int J Pharm. 2013;453(1):12–24. doi: 10.1016/j.ijpharm.2013.04.044. [DOI] [PubMed] [Google Scholar]
- 65.Polli JE, Abrahamsson BS, Yu LX, Amidon GL, Baldoni JM, Cook JA, et al. Summary workshop report: bioequivalence, biopharmaceutics classification system, and beyond. AAPS J. 2008;10(2):373–379. doi: 10.1208/s12248-008-9040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 8296 kb)
(R 11 kb)
(R 14 kb)
(R 3 kb)
(RDATA 2 kb)