

Abstract
Immunogenicity is a significant concern for biologic drugs as it can affect both safety and efficacy. To date, the descriptions of product immunogenicity have varied not only due to different degrees of understanding of product immunogenicity at the time of licensing but also due to an evolving lexicon that has generated some confusion in the field. In recent years, there has been growing consensus regarding the data needed to assess product immunogenicity. Harmonization of the strategy for the elucidation of product immunogenicity by drug developers, as well as the use of defined common terminology, can benefit medical practitioners, health regulatory agencies, and ultimately the patients. Clearly, understanding the incidence, kinetics and magnitude of anti-drug antibody (ADA), its neutralizing ability, cross-reactivity with endogenous molecules or other marketed biologic drugs, and related clinical impact may enhance clinical management of patients treated with biologic drugs. To that end, the authors present terms and definitions for describing and analyzing clinical immunogenicity data and suggest approaches to data presentation, emphasizing associations of ADA development with pharmacokinetics, efficacy, and safety that are necessary to assess the clinical relevance of immunogenicity.
KEY WORDS: anti-drug antibody, clinical relevance, harmonization
INTRODUCTION
Therapeutic proteins (also called biologics, biopharmaceuticals, biological products, or biological medicinal products) and peptides have the potential to induce immunogenicity (1,2). This drug class is referred together as “biologic drugs” in this article even though the US and European regulatory authorities currently distinguish peptides as recombinant (biologic) versus synthetic (chemical) entities with discrete Biologics License Application (BLA)/Marketing Authorisation Application (MAA) submission requirements. Peptides, which are not traditionally considered biologics or proteins, can have immunogenic properties similar to proteins.
The consequences of product immunogenicity vary from no evidence of clinical effect to severe, life-threatening responses (3–5). Anti-drug antibodies (ADA) have been implicated in infusion reactions and anaphylaxis (6,7) as well as immune complex-mediated diseases (8,9). ADA have also caused secondary treatment failures (loss of efficacy) (10–12) and, in rare occasions, more serious adverse events such as deficiency syndromes for instance thrombocytopenia and pure red cell aplasia (13,14). Therefore, ADA are a medical concern in terms of safety and long-term efficacy of the drug and it is critical to evaluate their development in all patients during clinical studies, not just in a symptom-driven manner (15). With a goal of guiding medical practice, the elucidation of ADA responses and their characteristics relative to clinical consequences is vital.
Despite the recent development of a variety of approaches to mitigate the immunogenicity potential of manufactured protein molecules, including the use of native human protein sequences, modification of known or predicted immunogenic epitopes, production in mammalian culture systems, advanced manufacturing practices, and analytical characterization techniques, the human immune system can perceive “foreignness/non-self” (16) or “danger signals/stressed self” (17) in the biologic drug product and launch specific immune responses against them. Indeed, most approved biologic drugs are immunogenic, and the incidence of ADA can reach over 90% (3–5,18). Importantly, the incidence of ADA and their clinical sequelae can vary greatly between same-class products and between patient populations hindering predictions of immunogenicity and necessitating clinical testing. Such differences may reflect disparate bioanalytical methods and interpretation approaches (19,20), as well as a plethora of product-specific and patient-specific factors (15,18,21,22). Further compounding this issue is a lack of standardization in the terminology and approaches used for the collection, analysis, and presentation of immunogenicity results.
Biologic drug package inserts or prescribing information documents describe clinical immunogenicity to varying degrees but frequently merely mention the overall incidence of ADA and/or neutralizing antibodies (NAb) in pivotal studies. Such limited information, although pertinent, is inadequate to inform physicians and patients of the true benefit/risk of the treatment in clinical practice. The lack of sufficient and consistent description of ADA-related events can cause confusion or erroneous patient management by clinicians. To provide optimal treatment plans for their patients, medical practitioners can benefit from having specific information on immunogenicity relating to the safety of initiating a treatment regimen, the maintenance of treatment efficacy with an acceptable safety profile, and clinical management options when ADA develop in their patients. Examples include risk of anaphylactic response with the initial administration or episodic re-administration of the biologic, dosing strategies tested to achieve a therapeutic effect despite the presence of ADA (“dosing through”), and situations requiring discontinuation of therapy, or consequences of switching to another same-class product or a product with a different mechanism of action. Therefore, it would be useful for labels to describe ADA incidence and magnitude, time of onset, duration, neutralizing ability, cross-reactivity with endogenous molecules or other marketed biologic drugs, and their clinically relevant thresholds to enable clinical management of disease with biologic drugs.
To foster a unified approach to assessing and describing product immunogenicity, the Therapeutic Protein Immunogenicity Focus Group of the non-profit professional organization American Association of Pharmaceutical Scientists (AAPS) convened a team of experts from industry and regulatory agencies and tasked them with producing a consensus document comprising best practices and recommendations. Accordingly, this article offers definitions for terminology commonly applied to the descriptions of product immunogenicity, as well as analytical and data presentation approaches, and guidance on the types of information necessary for clinicians to assess the clinical relevance of immunogenicity. It also provides specific recommendations on sampling schema for the assessment of ADA in clinical studies and on the interpretation and presentation of the corresponding results. However, because analyses and interpretations can vary for different products based on the drug development phase or pharmacovigilance objectives and specific assessments of risk, the authors’ recommendations are to be taken as a general approach to enhance the understanding of immunogenicity, but are not meant to be used in lieu of current regulatory guidance documents, consultations with health agencies, or sound scientific judgment.
TERMINOLOGY
Definitions for common terms applied in the descriptions of immunogenicity are offered below:
Biologic drug: This term includes biotechnology-derived therapeutic proteins (including mAbs) and peptides, some plasma-derived products (e.g., coagulation factor replacement products), and naturally derived proteins (e.g. therapeutic enzymes and toxins), but excludes oligonucleotides, cellular products, and vaccines. Peptides are included irrespective of the method of manufacture (synthetic or biological systems) or contemporary regulatory definitions.
Anti-drug antibody (ADA): Biologic drug-reactive antibody, including pre-existing host antibodies that are cross-reactive with the administered biologic drug (baseline ADA). It comprises neutralizing and non-neutralizing ADA. Other terms that have been used for ADA include anti-therapeutic antibody (ATA), anti-product antibody (APA), or anti-biologic antibody (ABA).
Binding ADA: All ADA are inherently “binding” antibodies because they bind the biologic drug molecule, as determined by an in vitro test method, regardless of their in vivo relevance (i.e., whether or not they produce clinical impact). A common misuse of this term is to apply it solely in reference to non-neutralizing antibodies, whereas neutralizing antibodies, in fact, are a subset of binding antibodies.
Neutralizing ADA (NAb): ADA that inhibits or reduces the pharmacological activity of the biologic drug molecule, as determined by an in vitro test or animal-based bioassay method, regardless of its in vivo clinical relevance (i.e., whether or not test method results relate to clinical impact in the subject).
Non-neutralizing ADA (non-neutralizing antibody, non-NAb): ADA that binds to the biologic drug molecule but does not inhibit its pharmacological activity in an in vitro test or animal-based bioassay method, regardless of its in vivo clinical relevance (i.e., whether or not test method results relate to clinical impact in the subject).
Drug-sustaining ADA response: An ADA immune response associated with a reduced clearance rate of the drug; the drug’s half-life is longer in its ADA-bound state than in its unbound state (inferred via statistical determination). The drug may (when bound by non-NAb) or may not (when bound by NAb) be pharmacologically active.
Clearing ADA response: when the presence of ADA (NAb or non-NAb) is associated with increased clearance of drug (inferred via statistical determination), the ADA immune response (not the ADA per se) is considered to be “clearing.” The impact of antibodies on drug clearance is a multifaceted mechanism involving ADA, circulating immune complex lattice, complement binding, Fc receptor binding, etc. Generally, ADA form immune complexes with the therapeutic that are cleared by the reticuloendothelial system; this can be in addition to the normal clearance of the drug. Because the size of an immune complex is dependent on the concentrations of antigen and antibody, in some instances, ADA may be clearing only above a certain titer threshold.
Human anti-murine antibody (HAMA): Human antibodies against epitopes of murine origin present in a murine or humanized mAb drug molecule. Taken literally, this term can be interpreted to mean that the ADA against one mAb drug can potentially cross-react with other murine sequence-based antibodies, raising concern over the administration of other mAb drugs containing murine sequences to ADA-positive subjects. When ADA cross-reactivity with other mouse antibodies is not confirmed, it is recommended to avoid the term HAMA to refer to ADA against murine mAb drugs.
Human anti-chimeric antibody (HACA): Human antibodies against non-human epitopes present in a chimeric (human + another species, usually mouse) mAb drug molecule. Taken literally, this term can be interpreted to mean that the ADA can potentially cross-react with other chimeric antibodies, raising concern over the administration of other chimeric mAb drugs to ADA-positive subjects. When cross-reactivity with other chimeric antibodies is not confirmed, it is recommended to avoid the term HACA to refer to ADA against chimeric mAb drugs.
Human anti-human antibody (HAHA): Human antibodies against human/humanized epitopes present in a humanized or fully human mAb drug molecule. Taken literally, this term can be interpreted to mean that the ADA can potentially cross-react with other human sequence-based antibodies, raising concern over the administration of other human mAb drugs to ADA-positive subjects. When cross-reactivity with other human sequence-based antibodies is not confirmed, it is recommended to avoid the term HAHA to refer to ADA against humanized or human mAb drugs.
Rheumatoid factor (RF): an endogenous immunoglobulin that typically binds the Fc portion of IgG. RF is often found in the serum of patients with autoimmune diseases like rheumatoid arthritis. Rheumatoid factor can sometimes also be found in serum of patients with other diseases or even healthy individuals and can interfere with ADA detection methods.
Pre-existing ADA: refers to antibodies reactive with the biologic drug that are present in subjects before treatment (or before initiation of the clinical study). This term, analogous with “baseline ADA,” is used strictly on the basis of detecting drug-reactive antibodies prior to treatment administration, irrespective of the etiology of this reactivity (i.e., whether or not a patient received the same drug in the past, had cross-reactive antibodies from exposure to other antigens, etc.).
Treatment-induced ADA: ADA developed de novo (seroconversion) following biologic drug administration (i.e., formation of ADA any time after the initial drug administration in a subject without pre-existing ADA).
Treatment-boosted ADA: Pre-existing ADA that were boosted to a higher level following biologic drug administration (i.e., any time after the initial drug administration the ADA titer is greater than the baseline titer by a scientifically reasonable margin such as fourfold or ninefold as explained in “Bioanalytical Considerations”).
ADA prevalence: The proportion of all individuals having drug-reactive antibodies (including pre-existing antibodies) at any point in time. This term is distinct from ADA incidence (see below).
ADA incidence: The proportion of the study population found to have seroconverted or boosted their pre-existing ADA during the study period. Synonymous with “treatment-emergent ADA”, ADA incidence is the sum of both treatment-induced and treatment-boosted ADA-positive subjects as a proportion of the evaluable subject population. The term “rate of ADA” should not be used to mean ADA incidence because “rate” usually implies a measured unit over time, whereas “incidence” relates the measured unit to the total population of units. This term is distinct from ADA prevalence (see above).
Titer: a quasi-quantitative expression of the level of ADA in a sample. By employing a serial dilution-based test method, titer is defined as the reciprocal of the highest dilution of the sample (including MRD) that yields a positive result (e.g., dilution of 1/100 = titer of 100), i.e., a result above a predetermined “cut point” value. It may be presented after logarithmic conversion if preferred. Alternatively, titer can be derived at the cut point value by interpolating from the dilution curve.
ANALYSIS AND REPORTING OF CLINICAL IMMUNOGENICITY
An integrated immunogenicity analysis strategy and tactical plan that is relevant to the intended clinical treatment strategy (dosage, tested in pivotal trials) is critical for elucidating the clinical relevance of immunogenicity data.
ADA should be tested using sensitive and valid methods and employing an appropriate strategy for elucidating immunogenicity (15,19,20,23,24). Detection of ADA is typically followed by assessments of the magnitude (titer) of the ADA response and the in vitro neutralizing ability of ADA, especially in late-stage clinical studies. Additional characterization of ADA such as immunoglobulin subclass or isotype determinations, domain-mapping, relative binding affinity, cross-reactivity with endogenous proteins, or complement-activating ability of the ADA may be driven by product-specific, indication-specific, or risk assessment-based objectives (19,20,25). These attributes of ADA can be delineated further based on their kinetic characteristics, that is, how early after drug exposure these antibodies develop (“onset” of ADA) and how long they last (“duration” of ADA). Any of these attributes and/or kinetic characteristics of ADA could potentially correlate with clinical consequences. Thus, it is useful to present ADA results from clinical studies as (a) characteristics of the ADA immune response, (b) relationship of ADA with pharmacokinetics (PK) and, when relevant, pharmacodynamic (PD) biomarkers, and (c) relationship of ADA with clinical safety and efficacy, as described below.
Clinical consequences of ADA can range from no apparent clinical effect to lack of efficacy (primary treatment failure), loss of efficacy (secondary treatment failure) or heightened effect due to altered exposure to the biologic drug, adverse drug reactions (administration-related systemic or site reactions), and severe adverse drug reactions (anaphylaxis and unique clinical problems associated with cross-reactivity and neutralization of endogenous molecules). Thus, it becomes important to examine any associations between ADA or any of its attributes with the various clinical sequelae. Whereas a clearing ADA immune response (effect evident on PK) and non-clearing but neutralizing ADA response (such as a low-titer NAb whose effect may be seen on PD) can negatively impact clinical efficacy, the presence of ADA may not preclude the administration of drug to ADA-positive patients because the outcome is dependent upon the magnitude of the impact of ADA on PK and PD. Hence, the relationship of ADA with PK/PD is an important additional consideration, but does not necessarily result in a clinically impactful consequence per se.
Determining the Characteristics of the ADA Immune Response
A fundamental metric that informs clinical immunogenicity interpretation is the incidence of ADA in a study or across comparable studies. Validated ADA test methods enable characterization of samples into ADA-positive versus ADA-negative. Additionally, when test methods are applied that are susceptible to interference by drug, it is recommended that samples containing interfering levels of drug be separated into a third category—ADA-inconclusive (26). A prior publication (20) had originally advised that such samples be “reported as negative with possible drug interference” with the intent of conveying that the ADA status of such samples may be unresolved or inaccurate. However, newer bioanalytical technologies and sample pre-treatment steps (e.g., acid dissociation), when carefully optimized and validated to produce accurate results, may lead to fewer ADA-inconclusive samples. Although bioanalytical strategies and technical details are outside the scope of this manuscript, a brief discussion of methodological concerns that can impact immunogenicity results are presented in “Bioanalytical Considerations.”
To classify the ADA status of a subject using data from an in vitro test method, it is recommended that each sample from the subject be categorized (the “sample ADA status”) based on the following definitions:
ADA-positive sample: When ADA is detected in a sample, the sample is considered positive.
ADA-negative sample: A sample is considered negative when ADA is not detected, and drug is absent in the same sample or present at a level that has been demonstrated to not interfere in the ADA detection method.
ADA-inconclusive sample: When ADA is not detected in a sample but drug is present in the same sample at a level that can interfere in the ADA detection method then the negative ADA result cannot be incontrovertibly confirmed and it is better that the sample be classified as ADA-inconclusive.
Unevaluable sample: When a sample could not be tested for ADA status (“un-assayed sample”) due to inadequate sample volume, mishandling, or errors in sample collection, processing, storage, etc.
The above definitions warrant the following clarifications. The term “detected” implies that drug-specific ADA was confirmed (24). Also, the “drug tolerance” of an assay (highest drug concentration that does not interfere in the ADA detection method) is not an absolute value and differs between individuals due to the varying avidities of ADA immune responses. Although it is understood that immune responses in humans may vary between subjects and performance of the ADA-positive control in the ADA assay cannot be extrapolated to clinical samples, a contemporary practical approach has been to use one or more ADA-positive controls to develop methods that can tolerate drug levels that are higher than the concentrations expected at the sampling time points. Thus, study sponsors might consider taking a conservative approach such as applying a method with drug tolerance level that is twofold higher than the anticipated peak drug concentration in the ADA samples. When feasible, combining this with a sampling strategy of collecting samples at times when the least drug concentration is anticipated (trough concentrations) or eliminated (drug “washed out”) can increase the likelihood of accurate ADA detection.
Of note, the term “borderline positive” to describe a sample (with confirmed drug-specific ADA) that produced laboratory results just above the assay cut point is inappropriate; these are positive samples whose titer equals at least the minimum required dilution (MRD) of the assay method.
Next, using the sample ADA status, it is recommended that the treatment-emergent immunogenicity status of each subject (“subject ADA status”) be determined using the following definitions:
Evaluable subject: A subject with at least one sample taken after drug administration during the treatment or follow-up observation period that is appropriate for ADA testing (with reportable result). Only evaluable subjects are considered for computing treatment-induced ADA incidence. It is advised that sample(s) be taken at time points appropriate for the detection of antibodies, as described in “Sampling.”
Unevaluable subject: A subject without a single sample taken (or without a reportable result) after drug administration during the treatment or follow-up observation period. However, whereas such a subject is excluded from treatment-emergent immunogenicity analyses, this subject ought to be included in the reporting of pre-existing ADA if the baseline sample had a reportable result. On the other hand, an unevaluable subject with all unevaluable samples cannot be evaluated at all, so therefore, is excluded from all immunogenicity analyses.
ADA-positive subject: A subject with at least one treatment-induced or treatment-boosted ADA-positive sample at any time during the treatment or follow-up observation period.
ADA-negative subject: Subject without a treatment-induced or treatment-boosted ADA-positive sample during the treatment or follow-up observation period.
- ADA-inconclusive subject: A subject who cannot irrefutably be classified as ADA-negative. To recommend a single definition for this category is not feasible because there can be any of several possible grounds warranting this category for different product classes and disparate circumstances. Thus, sound scientific rationale incorporating the drug’s immunogenicity risk profile, prior experience with the drug, label information or publications on same-class drugs, and/or discussions with regulatory authorities ought to be considered in developing a fit-for-purpose definition of the ADA-inconclusive subject category. For example:
-
aDespite observing some ADA-negative samples during a subject’s treatment (including follow-up observation period) with a higher risk drug, multiple other samples were found to be inconclusive precluding a definitive conclusion on the ADA status of the subject.
-
bDespite all ADA-negative samples during a subject’s treatment (including follow-up observation period) with a lower risk drug, the last evaluable sample was found to be inconclusive, lending to a conservative ADA-inconclusive subject status.
-
a
Nevertheless, if the ADA-inconclusive status is not utilized a sound scientific rationale ought to be developed as a justification.
Classifying samples as being apparently negative for ADA yet having detectable drug on board (or at a level above what is defined as the drug tolerance limit of the ADA assay) as “ADA-inconclusive” can be a contentious issue. A commonly held opinion is that the results of ADA assays ought to be reported “as is” (i.e., only as positive or negative) and those results placed into perspective based upon results from other tests (such as PK and PD) and that if there is further evidence (based on the other tests) that affects the interpretation of an ADA-negative result, then an explanation would be warranted. However, this approach assumes that other (and appropriate) tests are performed, that they are tolerant to the presence of ADA, and that they are sensitive and specific enough to demonstrate the effect of ADA. Another debatable issue that can arise is whether the ADA-inconclusive subjects ought to be included or excluded in the sum total of subjects (the “denominator”) bearing ADA results (proportions of which are reported as ADA-positive or negative). One point of view is that ADA-inconclusive subjects should not be included in the denominator because a “drug-intolerant” ADA detection method could report false-negative data (26). On the other hand, including them may be acceptable if ADA-inconclusive subjects comprised a minor proportion of the evaluated subjects, immunogenicity risk was considered low (15,19,20,27), and/or because “drug tolerance” of an assay could not be applied with confidence as it is not an exact limit (24). For example, including ADA-inconclusive subjects in the denominator may be justifiable in certain oncology studies where typically the dosage is high (leading to high trough serum concentration levels of the drug) and drug washout periods are frequently impossible to achieve. However, if this were the case, the caveats of the interpretation ought to be made clear in the label. A decision to proceed on either of these paths is enabled by a good-working knowledge of the ADA detection method, an understanding of the drug tolerance range using multiple ADA-positive controls, supportive data from an orthogonal approach or technology, and/or consultations with the relevant health regulatory agency. Lastly, but notably, an important consideration in clinical study designs is to incorporate a sampling strategy of collecting samples at times when the least drug concentration is anticipated (trough concentrations) or eliminated (drug washed out) to increase the likelihood of accurate ADA detection and reporting.
After classifying subjects and their samples into the above “ADA status” categories, it is recommended that the combined data set be examined from various angles, and sample size permitting, also broken down by each relevant variable such as dose, dose frequency, route of administration, number of drug administrations, number of exposure days (days on each of which one or more infusions of biologic drug were administered), concomitant medications (particularly immunomodulators), etc. The first step for evaluating the clinical relevance of ADA-positive samples is to visualize the data in different ways. The extent of analysis will depend upon the phase of drug development, sample size, and ADA incidence (the more ADA-positive subjects, the more likely statistical correlations or observations of “trends” might prove worthwhile); thus, the type and extent of analyses ought to be driven by sound scientific judgment and/or consultations with the relevant health regulatory agency. The following types of analyses of ADA attributes can be useful:
- Pre-existing ADA, titer, and boosting:
- Baseline ADA-positive subjects as a percentage of the total number of subjects whose baseline samples were tested (with reportable results) for ADA.
- Titer range (median and interquartile range (IQR)) of the baseline ADA-positive samples
- Percentage of baseline ADA-positive subjects with significant increases in ADA titer after biologic drug administration (i.e., when any one sample taken after the initial drug administration has an ADA titer that is greater than the baseline titer by a scientifically reasonable margin, such as fourfold or ninefold as explained in “Bioanalytical Considerations”).
- ADA incidence and titer:
- Overall ADA incidence: combined results of treatment-boosted ADA-positive subjects and treatment-induced ADA-positive subjects. Compute as a percentage of the total number of evaluable subjects, excluding baseline positive subjects without any samples available after drug administration.
- Treatment-induced ADA incidence computed as a percentage of the total number of evaluable subjects that were ADA-negative at baseline. Also, report peak positive titer and range (median, IQR) for this group of subjects.
- Treatment-boosted ADA computed as a percentage of the total number of evaluable subjects that were ADA-positive at baseline. Also, compute the fold increase in titer (ratio of peak post-administration titer to baseline titer) and range of titer increases (median, IQR).
Neutralizing ADA: When applicable, report pre-existing NAb, boosting, and incidence as described above. If all ADA are neutralizing in all subjects, a separate analysis is obviously redundant.
Kinetics of ADA: The timing of ADA development and its duration can be useful information for a clinician to monitor treatment progress. The persistence of antibodies has been found to associate with clinical impact in several cases (28–30). To the drug developer, knowledge of ADA kinetics can help optimize the sampling schedule in subsequent studies of the same biologic drug, and in instances of ADA monitoring post-marketing as part of pharmacovigilance plans, the surveillance schedule, and the design of risk management and mitigation strategy can be optimized by understanding ADA kinetics.
Graphical representations of ADA kinetics are most useful. For example, a bivariate plot of ADA onset and ADA duration is illustrated in Fig. 1, and a transient versus persistent ADA frequency plot depicted in Fig. 2. These types of graphs are most informative when there are higher numbers of ADA-positive subjects (e.g., ≥20) and the study duration is long enough to discern persistent antibodies after their development (e.g., ≥1 year). In addition to such graphics, a supplementary statistical description of the results is also advised when the sample size is adequate. This objective approach prevents misinterpretation of the results due to subjective biases. Of note however, sample size adequacy ought to be decided on a case-by-case basis, depending upon the design of the clinical study. The following computational approaches are advised:
Onset of ADA: refers to the time period between the initial administration of the biologic drug (in a study) and the first instance of treatment-induced ADA. The use of real-elapsed days is ideal for the calculations, although the use of nominal study time points can be adequate. Compute the “median time to ADA development” and the quartiles Q1 and Q3, which can enable an understanding of onset of ADA in one half, 25%, and 75% of the ADA-positive subjects, respectively. Other analyses relevant to the onset of ADA can be “number of drug doses to first incidence of ADA” or “number of exposure days to first incidence of ADA.”
-
Duration of ADA: refers to the longevity of treatment-induced ADA. Computing and reporting the median duration of an induced ADA response and IQR are the most objective approaches and useful for correlation with clinical consequences, yet colloquial expectations to classify ADA as transient versus persistent predominate. While it is unnecessary to categorize using such terms, when they are applied it becomes important to utilize harmonized definitions. Because natural (endogenous) human IgG1, IgG2, and IgG4 have approximate half-lives in the range 21–25 days, five half-lives are approximately equal to 16 weeks. If an ADA were induced and never re-stimulated or boosted (a “transient” antibody) so that it would be subject to its natural clearance mechanisms, then the ADA is expected to be eliminated at the end of five half-lives (actually, only a negligible 3% remains). It is reasonable, therefore, to apply this phenomenon to differentiate transient (“sero-reverting”) versus persistent ADA and suggest the following approaches to evaluate ADA duration:
- Transient ADA response:
- Treatment-induced ADA detected only at one sampling time point during the treatment or follow-up observation period (excluding the last sampling time point, which ought to be considered persistent unless shown to be undetectable at a later time) or
- Treatment-induced ADA detected at two or more sampling time points during the treatment (including follow-up period if any), where the first and last ADA-positive samples (irrespective of any negative samples in between) are separated by a period less than 16 weeks, and the subject’s last sampling time point is ADA-negative.
- Persistent ADA response:
- Treatment-induced ADA detected at two or more sampling time points during the treatment (including follow-up period if any), where the first and last ADA-positive samples (irrespective of any negative samples in between) are separated by a period of 16 weeks or longer or
- Treatment-induced ADA incidence only in the last sampling time point of the treatment study period or at a sampling time point with less than 16 weeks before an ADA-negative last sample.
Although rare, if IgG3 or IgA ADA are known to predominate in a study population, a 5-week period ought to be applied to modify the definitions of transient and persistent ADA (instead of the 16 weeks). This is because IgG3 and IgA have shorter half-lives than other IgGs (IgG3, 7 days; IgM and IgA, 5 days).
Note that treatment-boosted ADA are excluded from the analyses of ADA kinetics because this type of immune response differs mechanistically. In instances where pre-existing ADA are significantly prevalent, it may be useful to separately delineate the kinetics of boosting.
Instead of categorizing ADA responses into transient versus persistent, computation of the median (the “median duration of ADA”) and quartiles (Q1 and Q3) enables an understanding of the duration of ADA in half, 25%, and 75% of the ADA-positive subjects respectively. The quartile approach may enable better elucidation of the relationship of ADA duration with clinical impact, if any.
Lastly, it is inappropriate to define transient antibodies as those that disappear before the end of the study versus persistent antibodies as those that remain through the last study time point. This is because the definitions of transient and persistent ADA would then become dependent upon the length of the study rather than the actual duration of the ADA; longer studies would bias the results in favor of “transient antibodies” if such a definition was applied.- NAb incidence and kinetics: When study results indicate distinct NAb-containing versus non-NAb-containing subject groups, it can be useful to examine NAb incidence and kinetics separately for each group in the same manner as described above for ADA.
- Cross-reactivity: When a biologic drug molecule is identical or nearly identical to an endogenous protein (whole or in part), it is important that the cross-reactivity of the ADA with the endogenous protein be evaluated because of heightened concern that such ADA might cause an autoimmune-like syndrome characterized by depletion of the endogenous protein. Comparing the kinetics and titers of the cross-reactive ADA and the ADA against the whole drug molecule can help evaluate worsening of disease manifestations.
Fig. 1.
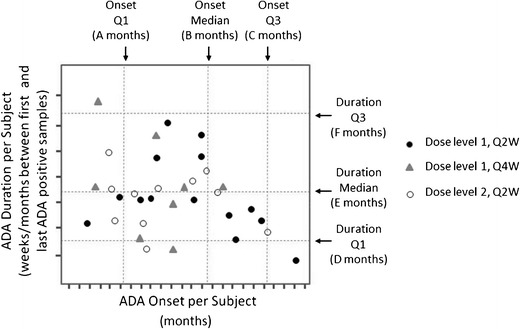
Treatment-induced ADA kinetics: onset and duration. A plot of the duration of ADA-positive results versus the ADA onset time, with vertical and horizontal grid lines drawn at the quartiles of the distributions. This helps to determine whether the persistent or transient nature of the ADA is related to the time when the ADA levels are observed in the patients. For a clear assessment, only those patients for whom the ADA onset time is at least 16 weeks before the last visit or those that were ADA-negative at or before the last visit should be included in this graph. When interpreting this graph, it should be kept in mind that the maximal duration at later onset times will be proportionately less. Symbols in the plot could indicate a variable of choice (dosage in this example), clinical impact such as effects on efficacy (e.g., yes, no, and study discontinuation), adverse reactions (e.g., yes, no, and study discontinuation), etc.
Fig. 2.
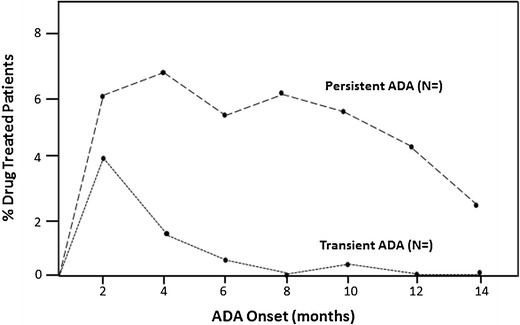
Treatment-induced ADA incidence kinetics: The development of transient and persistent ADA immune responses in an example study is illustrated. Each point indicates the percentage of biologic drug-treated patients who developed ADA at indicated onset times and whose duration may have been transient or persistent. In this example, 10 % of treated subjects had a 2-month ADA development time, with 4 % having a transient ADA response and 6 % having a persistent ADA response. Likewise, 6 % of treated subjects had a 6-month ADA development time, with 0.5 % having a transient ADA response and 5.5 % having a persistent ADA response. Data can be plotted by dosage
Alternate approaches that describe these ADA attributes may be used. However, subjective terminology ought to be avoided because they can mistakenly be interpreted as implying association with a certain level of clinical impact. For example, titers among the ADA-positive population can be reported as median and interquartile range (IQR), but not using terms such as “high” or “low” because one might incorrectly assume that high-titer antibodies are clinically relevant (i.e., cause adverse events) whereas low-titer antibodies were not (i.e., benign). Similarly, instead of describing the onset of ADA after drug exposure as early versus late antibodies, it is objective to report median time to ADA development (or median number of doses to ADA) and the interquartile range. Characterizing ADA responses as transient or persistent could be useful for understanding their duration once they develop in subjects, yet such demarcation also must not imply that clinical consequences are associated with persistent antibodies alone. This is another instance where simple descriptive statistics (median duration of ADA and the interquartile range) can enable better associations with clinical outcomes. Figure 3 illustrates a useful way to visualize titer distribution over time in a study.
Fig. 3.
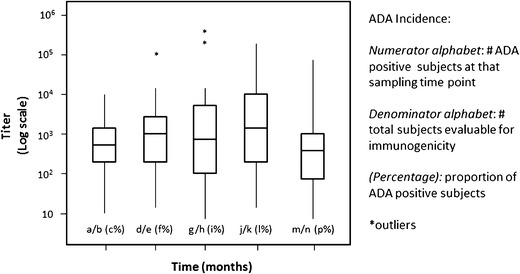
ADA titer kinetics. This plot of titers over time in a study is useful in determining whether the ADA levels tend to change over time during the treatment. Each box plot represents the titer range, Q1, Median (Q2), Q3, excluding outliers (asterisks)
Results may be presented in tabular, text, or graphical form as appropriate. Additionally, providing raw data in a tabular format can allow regulatory agencies to be able to conduct their own analyses in order to validate the results in the packages submitted. When sample results are provided in a table, it is useful to include information such as patient identification number, clinical site identification (name or number), planned study visit or drug administration visit (nominal time point), dose level/frequency grouping, date sample was taken (actual time point), measured serum drug concentration, sample ADA status and titer, neutralizing ability status, etc. However, studies resulting in very low numbers of ADA-positive subjects may limit some analyses.
Determining the Relationship of ADA Results with PK and PD
ADA bind the biologic drug in circulation to form immune complexes which, depending upon their size, may be cleared faster from the body than unbound drug. Alternatively, for some products, the formation of immune complexes leads to recirculation and prolonged half-life. Regardless of their ability to bind to the active site of the drug molecule, these clearing or drug-sustaining ADA responses can affect the PK profile such that drug clearance rates are increased or decreased respectively leading to altered drug exposure (15,31). Thus, it is important to examine the effects of ADA response on PK. The simplest way to illustrate this form of assessment is through graphical plots of relevant PK parameters against ADA status. A plot of median trough serum drug concentrations over time in ADA-positive versus ADA-negative groups of drug-treated patients (and, when appropriate, statistical significance of differences shown by p values) can be very informative (Fig. 4a). If high variability of serum concentrations precludes meaningful information from median plots, it may be useful to plot individual profiles (trough concentrations over time) within a cohort for ADA-positive versus ADA-negative subjects. Plots showing the effect of ADA response on drug clearance rate (Fig. 4b) or relevant PD parameter can further unravel the effects of ADA on PK. These PK parameters can also be plotted against ADA attributes such as titer (quartiles), onset (quartiles), and duration (quartiles or transient/persistent categories). Similarly, when efficacy surrogate PD markers are included in clinical studies, it can be useful to examine the effect of ADA development on PD. In addition to correlations with titer, onset, and duration of ADA as described above, presenting the effects of NAbs on PD is important.
Fig. 4.
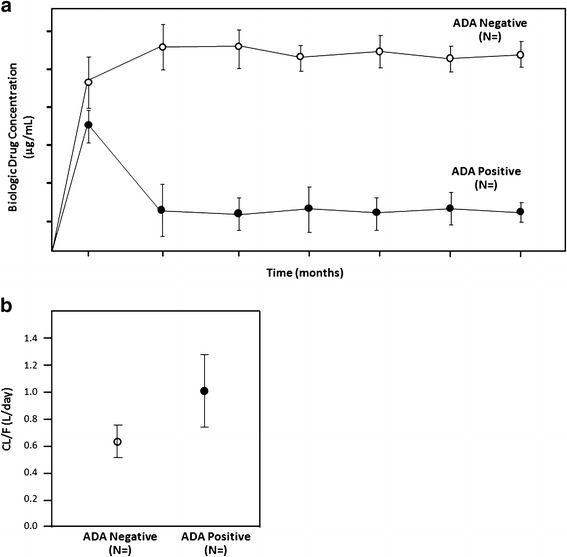
a Influence of ADA on PK (trough serum concentrations). This plot of serum concentrations of the biologic drug in ADA-positive subjects (closed symbols) and ADA-negative subjects (open symbols) over time provides a useful visual assessment of the effect of ADA on circulating drug levels. The ADA status (positive or negative) is considered in a cumulative manner at each time point (i.e., if a subject had a positive sample at any prior time before an efficacy assessment visit and that subject would be counted as positive through that time point). If differences are not easily apparent between the ADA-positive and ADA-negative subjects, it can be informative to present the ADA-positive subjects in groups of titers (quartiles) versus the ADA-negative subjects. Alternatively, if a clinically relevant threshold of ADA titer was already known, serum concentration values from ADA-positive patients with titers above the threshold, those below the threshold, and ADA-negative patients can each be plotted. At each time point, the geometric mean drug concentration and 90 or 95% confidence intervals are shown. Additional plots may be added for each dose level. When ADA-inconclusive subjects comprise a significant proportion, it should also be included in this plot. Similarly, such a graphical plot may be useful to illustrate the effect of transient versus persistent immune responses on serum concentrations of drug. b Influence of ADA PK (clearance rate). This plot of apparent clearance rate (CL/F) of the biologic drug in ADA-positive subjects (closed symbols) and ADA-negative subjects (open symbols) provides a useful visual assessment of the effect of ADA on drug elimination. The ADA status (positive or negative) is considered in a cumulative manner at each time point (i.e., if a subject had a positive sample at any prior time before an efficacy assessment visit and that subject would be counted as positive through that time point). If differences are not easily apparent between the ADA-positive and ADA-negative subjects, it can be informative to present the ADA-positive subjects in groups of titers (quartiles) versus the ADA-negative subjects. Alternatively, if a clinically relevant threshold of an ADA attribute was already known, drug clearance values from ADA-positive patients with results above the threshold, those below the threshold, and ADA-negative patients can each be plotted. When ADA-inconclusive subjects comprise a significant proportion, it should also be included in this plot. Similarly, such a graphical plot may be useful to illustrate the effect of transient versus persistent immune responses on clearance rate. Alternative visualizations as box-plots with individual points overlaid may also be used. Data may also be grouped by dosing period, demographics, etc.
Determining the Relationship of ADA Results with Clinical Safety and Efficacy
The primary goal of ADA assessments is to determine the clinical relevance of ADA. The following analyses in tabular or graphical depictions could shed light on whether ADA affect clinical efficacy and safety:
- Impact of ADA on clinical efficacy:
- Primary non-response: While infrequent, patients with pre-existing ADA may not respond to drug treatment from the outset. Hence, examining the lack of response or inadequate response in baseline ADA-positive patients is important.
- Secondary non-response (loss of response): Patients who develop ADA de novo and patients with benign baseline ADA (i.e., patients who initially responded to the drug despite pre-existing ADA) that were boosted after biologic drug treatment may lose efficacy after a period of time. Thus, examining the potential impact of ADA on maintenance of efficacy in subjects with primary response to drug is important. Assessing levels of efficacy in ADA-positive versus ADA-negative patients can be useful (example shown in Fig. 5). Similar evaluations of ADA attributes such as titer (quartiles), onset (quartiles), and duration (quartiles or transient versus persistent ADA response) could also be useful.
-
Impact of ADA on clinical safety:
Safety issues may not necessarily correlate with any effects on PK or efficacy; thus, it is important to independently evaluate the relationship of ADA with the anticipated or relevant adverse events (AEs) driven by product-specific immunogenicity risk assessment and risk management plan. Generally, the following types of AEs can result from ADA development in human subjects:- Acute AEs: ADA could cause devastating hypersensitivity reactions during or within hours after exposure to the biologic drug. Thus, it is important to examine the relationship between ADA and the following types of acute AEs.
- Type-I hypersensitivity (anaphylaxis): ADA-positive patients could experience anaphylactic reactions (27,32). In some settings, the term anaphylaxis is conventionally restricted to IgE-mediated reactions such as hypotension, bronchospasm, laryngeal or pharyngeal edema, wheezing, and/or urticaria (15). However, including all cases with such acute clinical signs under anaphylaxis, irrespective of the pathophysiology, may be needed for drug development purposes (27). In such cases, the investigation of drug-specific IgE may be warranted, particularly if such AEs were not rare. Depending on the time of the event relative to the initiation of treatment, the role of pre-existing antibodies may require investigation. Such acute reactions can have implications for the need to develop a drug-specific companion diagnostic test to screen prospective patients for pre-existing antibodies and to implement precautionary measures at the time of treatment (including exclusion from treatment) or to develop post-dose safety monitoring strategies. It is advised that diagnosis and management of anaphylaxis during clinical trials be discussed with regulatory authorities because they may have different approaches to managing anaphylaxis.
- Other drug administration-related acute reactions: Biologic drug exposure can elicit a range of acute effects other than anaphylaxis from symptomatic discomfort and local (administration site) inflammatory reactions to sudden, fatal reactions. Infusion reactions include a range of symptoms such as headache, nausea, fever or chills, dizziness, flush, pruritis, and chest or back pain (15). It can be challenging to distinguish between such reactions and anaphylaxis or cytokine release syndrome (15,27); therefore, it may be best to examine associations of ADA with specific signs and symptoms of acute clinical AEs (27).
- Non-acute AEs: Delayed hypersensitivity and inflammatory responses secondary to immune complex and complement-mediated reactions could occur hours to days after exposure to the biologic drug. Hence, it is important to examine the relationship between ADA and non-acute AEs such as fever, rash, arthralgia, myalgia, hematuria, proteinuria, serositis, central nervous system complications, and hemolytic anemia.
- Type III hypersensitivity: ADA-positive patients could experience immune complex-mediated inflammatory reactions (27,32) after the initial dose if pre-existing antibodies are present or later when treatment-induced ADA develop. Type III hypersensitivity symptoms may be acute or delayed in relation to the initial exposure to the biological drug. Relationships between such adverse reactions and ADA and its various attributes ought to be examined. However, these reactions typically have a subacute presentation (27), and as a result, such relationships may be difficult to establish.
- Type IV hypersensitivity: These reactions are mediated by the activation of different types of lymphocytes leading to delayed reactions after exposure to the biologic drug. These reactions are mechanism-dependent and usually not within the scope of ADA-mediated reactions.
- Worsening of disease: In addition to loss of efficacy, ADA-positive patients could experience worsening of underlying disease or disease manifestations due to the depletion of the biologic drug molecule’s endogenous counterpart or potentially a novel morbidity, as a result of cross-reactive ADA to another endogenous entity. Such investigations usually depend on the clinical risk of immunogenicity. Thus, relationships between ADA (incidence and other attributes) versus clinical symptoms of worsened disease and other adverse events are important.
- Increased drug toxicity: ADA-positive patients could experience clinical symptoms of drug toxicity due to biologic drug overexposure. In instances where an integrated analysis of PK and ADA indicates prolonged exposure to the drug (due to a “drug-sustaining” ADA response), signs of increased drug toxicity ought to be examined.
-
Impact of ADA on other clinically relevant outcomes:
Other associations of clinical outcomes may be useful to examine by ADA status. For example, the relationship between ADA and study discontinuation (33) and the probability of drug maintenance by ADA status over time (34) can be useful supplementary information.
Fig. 5.
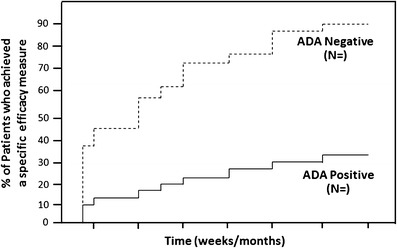
Influence of ADA on clinical efficacy. This plot of an efficacy measure in ADA-positive subjects and ADA-negative subjects over time provides a useful visual assessment of the effect of ADA on drug efficacy. The ADA status (positive or negative) is considered in a cumulative manner at each time point (i.e., if a subject had a positive sample at any prior time before an efficacy assessment visit then that subject would be counted as positive through that time point). If differences are not easily apparent between the ADA-positive and ADA-negative subjects, it can be informative to present the ADA-positive subjects in groups of titers (quartiles) versus the ADA-negative subjects. Similarly, such a graphical plot may be useful to illustrate the effect of transient versus persistent immune responses on efficacy. Alternatively, if a clinically relevant threshold of an ADA attribute was already known, efficacy data from ADA-positive patients with results above the threshold, those below the threshold, and ADA-negative patients can each be plotted. Sample sizes are indicated
DETERMINATION OF CLINICALLY RELEVANT THRESHOLDS OF ADA
The type and degree of clinical consequence is probably related to differences in the quality (attributes) or quantity (magnitude) of ADA. We refer to this clinically discriminating characteristic of ADA as a clinically relevant threshold of ADA. The final goal of immunogenicity assessments and analyses is to derive a clinically relevant threshold of any of the various attributes of ADA for categories of harmful clinical consequences observed during drug development.
In its simplest form, an analysis includes a listing of relevant clinical results in order of increasing severity alongside the continuous or discrete ADA data (incidence or the various attributes) per subject. A visual examination of “no observed effects” or benign effects versus significant adverse effects could provide an approximate indication of a clinically relevant threshold. This approach can be laborious, error-prone, and subject to bias. Thus, generally, clinically relevant thresholds can be decisively established only when statistical approaches are applied. It is not possible to present all, or the best, statistical approaches for the determination of clinically relevant thresholds of ADA; however, for statistical analysis, sufficiency of sample size is important. Hence, we caution that subjective approaches or those involving statistically insufficient sample size be avoided or minimally applied only under exceptional circumstances in consultation with relevant health regulatory agencies. For example, the overall risk-benefit profile of a biologic drug may warrant the application of an empirical ADA threshold in order to ensure patient safety despite the lack of statistically supported evidence of clinical relevance. Complex study designs with multiple arms and satellite studies may not permit statistical assessments of immunogenicity. Hence, we recommend that while most descriptive characteristics be reported by dosage, attempts ought to be made to combine data from the whole study or similar dosage arms in order to enable statistical analyses, particularly for correlations with safety issues.
The association of ADA level with clinical endpoints is best evaluated by classifying the ADA characteristic/attribute values into groups (“Determining the characteristics of the ADA Immune Response”). It may be tempting to group them as low, mid, and high based on experience with other products. However, such arbitrary distinction is suboptimal for associating with clinical efficacy, PK, or safety endpoints. On the other hand, grouping by quartiles (or higher percentiles if appropriate) is a more objective approach. Any statistical analysis, however, will be dependent upon the availability of an adequate sample size. Extremely low numbers of ADA-positive subjects may preclude the application of statistics and limit the determination to subjective inference. However, when sufficient data are available, clinically relevant ADA thresholds can be determined using a partition analysis or a multivariate approach. It is useful to apply an approach that can uncover nonlinear relationships between the predictor and response. For example, if the decline in efficacy is negligible up to a certain titer level and then progressively worsens for patients that have higher titers.
For evaluating the association of ADA attributes with binary clinical endpoints (such as adverse reaction/no adverse reaction, or worsened/not worsened disease status), receiver operating characteristic (ROC) curves can be used. ROC analysis is frequently used in assessments of diagnostic test performance and is a graphical plot which illustrates the performance of a binary classifier system as its discrimination threshold is varied (35). Positive versus negative clinical outcomes are assessed above and below each discrete value of an ADA attribute in a 2 × 2 tabular format. Then, the true-positive rate (sensitivity) and true-negative rate (specificity) can be calculated and based on the latter; the false-positive rate is also calculated (1-specificity). Similarly, these rates are calculated for all the other titer values. Then, the true-positive rate (y-axis) is plotted against the false-positive rate (x-axis), and the resulting curve informs two important aspects: (a) if the area under the curve is greater than 0.5, the test attribute is associated with clinical outcome and (b) the value of this attribute at which the maximal sensitivity and minimal false-positive rate occur is considered the optimal discriminator of positive versus negative clinical outcomes or the clinically relevant threshold. ROC analysis has been used to determine clinically relevant thresholds of ADA and drug levels recently (36) and can be a reliable tool; however, this approach enables the association of only one ADA attribute at a time.
More powerful approaches explore the associations of ADA attributes with clinical outcomes in a multivariate framework. One such approach is the Classification and Regression Tree (CART) model, which provides decision rules that are visualized in the form of a tree for predicting a categorical clinical outcome (classification tree) or a continuous clinical outcome (regression tree). The CART model, generated by repeated partitioning of the dataset (Fig. 6), can provide useful insights on the ADA characteristics that impact the efficacy, safety, and PK endpoints. Hence, this approach is also referred to as “recursive partitioning” or “decision tree” analysis. Among all the ADA attributes (listed in “Determining the characteristics of the ADA Immune Response”, such as ADA incidence, titer, neutralizing ability, kinetics, etc.), the one that best separates (with statistical significance) the data into two clinically distinct groups is first chosen, along with the “split” point on the predictor that provided this optimal separation. This process is repeated separately for each of the two groups, and the process continues to grow the tree until there is no statistical significance in splitting (typically the robust “logworth” value is used to represent statistical significance in CART; however, logworth can be translated into the more familiar p value. For example, a logworth of 2 is the p value 0.01 and a logworth of 3 is the p value 0.001). This type of analysis requires large datasets, at least 30 (but preferably >50) ADA-positive patients are preferred, with a fairly wide distribution of titer levels, ADA duration, onset times, or other ADA characteristics of interest. In contrast with ROC, the CART approach is useful also for assessing interactions (dependencies) between predictors, for example, if the titer levels have a stronger impact on efficacy decline when the ADA duration is longer or when the ADA onset time is earlier. CART models can be performed with user-friendly programs such as JMP® (SAS Institute Inc., Cary, NC, USA), but more rigorous and powerful versions of tree-based models are available in R and other specialized statistical programs. Collaboration with a statistician is highly recommended when carrying out these analyses. Additional modeling methods may be considered depending on the nature of the data.
Fig. 6.

An example CART model for the association to safety (adverse event) incidence. Among the 89 patients found to be ADA-positive through 2 years of treatment, ∼18% had a specific type of adverse event (AE). The association of this binary safety endpoint (1 = yes AE; 0 = no AE) with the ADA attributes titer, onset, and duration was evaluated. The model picked ADA onset time as the most important predictor; 26% of the 43 patients with onset time before 174 days had the AE, whereas only 11% of the 46 patients with later onset time had AE. Among 43 patients with ADA onset earlier than 174 days, the model found titer levels to be a useful predictor; 31% of the 36 patients with titer level >20 had the AE, where none of the seven patients that had titer levels <20 had any AE. Using this model, it could be inferred that biologic drug-treated patients were 2.4-fold more likely to develop ADA in the initial 25-week period than later. Furthermore, at least among those who developed ADA in the initial 25 weeks, titers less than 20 were not associated with the AE
SAMPLING
For the registration (BLA/MAA) of chronic treatments (or repeat drug administration), regulatory authorities typically expect immunogenicity data at least through the first year of treatment (27), although shorter terms may be justifiable on a case-by-case basis. It is recommended that ADA be evaluated in all study patients and not in a symptom-driven manner (15) and the sampling schedule for each product be determined on a case-by-case basis, taking into account the risks associated with immunogenicity (15), risk of extensive blood collection on the health of the patient, dose frequency, trough concentration time points, and previous experience with the drug’s immunogenicity profile. Sampling frequency during treatment ought to be designed to maximize the opportunity of detecting treatment-induced ADA and, when applicable, to understand the kinetics of this immune response. Ideally, samples are collected at baseline (prior to receiving treatment) and depending on the length of the clinical study at approximately the following time points following initial drug administration: 2 weeks (optional); 1, 2, 3, 6, 9, 12, 18, and 24 months; and every year thereafter during treatment, including an end-of-study sample. When feasible, attempts ought to be made to collect an end-of-study sample at a time point where the drug has washed out, i.e., after at least five half-lives of the drug since its last dose (when only a negligible 3% of the drug remains). This sampling scheme enables the differentiation of transient versus persistent ADA response optimally during the first year of treatment, but less so in longer studies. Hence, for some high-risk products, one might consider continued frequent sampling in year 2 and thereafter (the pattern of sampling used in the first year is suggested). If alternate time points are applied, it is advised that at least four appropriately spaced samples are collected in studies ≤1 year in length. A risk-based approach (19,20,37,38) may also justify collecting the samples at the suggested time points, but only testing a limited set of samples during clinical trials while storing the remainder for contingent testing. However, strategies for contingent testing may require discussion with regulatory authorities.
Additionally, some high-risk products may require post-study monitoring (follow-up of ADA-positive subjects) to determine the off-treatment persistence of ADA. Such decisions are based on considerations of the drug’s immunogenicity risk profile, prior experience with the drug and/or label information or publications on same-class drugs. If a subject is found to be ADA-positive at the end-of-study, it may be important to assess the persistence of the ADA. To assess this, at least one more sample ought to be taken at least 4 months later, that is, a period of time where any transient ADA would be expected to have cleared. ADA detected after the above period should be considered persistent off-treatment. Regulatory agencies may also advise that patients with persistent antibodies be followed up until their ADA become negative.
BIOANALYTICAL CONSIDERATIONS
Recommendations for ADA assay development, method validation, and testing strategies have been published by the Ligand-Binding Assay Bioanalytical Focus Group (LBABFG) of AAPS (20,24,39–41). Additionally, scientific publications on risk-based approaches to immunogenicity assessments (19,20,37,38,42) and regulatory documents from the US Food and Drug Administration (FDA) and the European Medicine Agency (EMA) are also available (15,23,27,43). Together, these documents provide ample guidance for the application of appropriate ADA detection methods in clinical studies. Beyond these efforts to harmonize bioanalytical approaches, however, some caveats remain that can impact the interpretation of immunogenicity data, as described below. Due to such caveats, the US Food and Drug Administration (FDA) requires that biological product package inserts explicitly state that cross-product comparisons of immunogenicity can be misleading due to methodological differences and are therefore inappropriate.
ADA detection is limited by the assay method used. For example, lower affinity ADA can be detected by surface plasmon resonance, bio-layer interferometry, or other platforms that minimize washing steps, albeit with lower sensitivity compared with enzyme-linked immunosorbent assays (ELISA) and electrochemiluminescence immunoassays (ECLIA) (44). In contrast, the latter types of immunoassays involving multiple wash steps are typically able to detect higher affinity ADA (39), yet because immunoassays offer higher sensitivity and throughput, they remain the major platform of choice for ADA detection. Whether lower or higher affinity ADA is clinically relevant cannot be generalized, but such relevance can only be reliably assessed if a method was capable of detecting a reasonable range of affinities of ADA. Similarly, ADA assay sensitivity is dependent on the affinity of the positive controls (usually generated from hyperimmunized animals) and thereby ought to be interpreted with this caveat in mind. Antibodies of different affinities produce different assay sensitivity results in the same assay, and the same antibody produces different assay sensitivity results in different assay platforms or sometimes even below detection (45). Hence, when possible, it is recommended that more than one positive control be used during assay development and validation. It is also possible that an ADA assay optimized using a high-affinity positive control may inherently be biased toward overestimating the method sensitivity and missing the detection of lower affinity ADA unless the detection of lower affinity positive controls is also demonstrated with reasonable sensitivity. While optimal, it is understandable that such a variety of positive controls may not be available during assay development and validation.
Rheumatoid factor (RF) is often found in the serum of patients with autoimmune diseases like rheumatoid arthritis and infrequently in healthy individuals. It binds the Fc portion of IgG and therefore could be detected inaccurately as “ADA” in the case of monoclonal drug products. Hence, efforts are needed during the development of ADA methods for certain disease populations to minimize RF interference. Additionally, interference by other factors, such as soluble receptors, also need to be evaluated as indicated based on the disease population.
The biologic drug itself typically interferes with ADA detection in the test methods to produce inaccurate results; this can lead to false-negative results or underestimated titer values for positive samples (39). Details on assay interference are absent in drug labels and in most publications, so ADA results ought to be accepted with caution. Characterization of this drug tolerance of the assay (i.e., sensitivity of ADA detection in the presence of drug) has been stressed in recent years (24), but it is likely that some methods, particularly the older methods used for established products, have not addressed the impact of drug on reported ADA results (26). Nevertheless, because the sensitivity and drug tolerance of an ADA method are dependent upon the positive control used in the assay and vary when positive controls are changed, a risk-based approach can be applied in determining the circumstances when it may be appropriate to apply this value in decision-making. For example, whether or not to apply the “drug tolerance limit” in inferring an apparently ADA-negative sample with drug on board as truly negative, instead of ADA-inconclusive, or whether or not to wait for a drug washout period before sampling (or at least one final sample) in clinical studies. Interference by soluble drug target is increasingly also being recognized as another confounding factor recently, in particular, due to the development of “drug-tolerant” ADA detection methods employing “acid dissociation.” When low pH conditions are validated to leave the ADA structure unaffected but break apart the ADA-drug immune complex into individual components, it becomes possible to detect ADA in samples that would otherwise have produced false-negative data. However, in such methods, acid pre-treatment of test samples can break apart not only the drug-ADA immune complex but also the drug-target complex (“bound target”) (46). Depending upon the abundance of epitopes in its structure, released target can interfere in ADA detection methods and produce either false-negative or false-positive results. Because binding of drug to its target is required for efficacy, bound target in patients can accumulate to supraphysiological concentrations when in complex with long-acting biologics such as mAbs or pegylated drugs. Thus, a majority of subjects’ samples may contain drug-bound target, which if released during laboratory analyses, can produce significantly inaccurate ADA results.
In addition to possibly reduced titer values caused by drug interference in the assay, the determination of changes in ADA titers over time can be significantly affected by the type of serial dilution scheme applied in titration assays. For example, if a tenfold dilution scheme is applied, a sample’s titer value could be 10, 100, 1,000, or a higher multiple of ten. In this case, ADA levels with titer values between 10 and 100 would be judged as 10, values between 100 and 1,000 would be judged as 100, and so on. An ADA-positive subject with two samples—an initial one with a titer value of 10 and a subsequent one with an obviously higher titer value of 80—would both appear to possess a titer of 10, leading to an inaccurate conclusion that the ADA levels remained unchanged over time. Whether such changes in ADA levels are clinically meaningful cannot be generalized, but such relevance can be reliably assessed only if the titration method was capable of discriminating close ADA titers. Hence, after the determination of the MRD of the assay, a low serial dilution scheme such as twofold or threefold is recommended; higher dilution factors (e.g., tenfold) are not recommended as they can be imprecise. Alternatively, it is also suitable to define the serial dilution scheme based on the precision of the titration assay. Due to the format of titration assays, close titer values ought to be compared cautiously based on a reasonable margin of the imprecision of the titration method. Because a single dilution difference in titration experiments is often within the expected imprecision of the method, a difference in titer values between two samples representing twice the dilution level is generally considered significant (i.e., a fourfold difference in titer values may be the least significant change when a twofold serial dilution is applied for titration or ninefold difference in titer values may be the least significant change when a threefold serial dilution is applied for titration).
Neutralizing antibody test methods—particularly cell-based bioassays—are often limited by sensitivity compared to ADA detection immunoassays; a lack of neutralizing activity in these assays does not necessarily confirm that the ADA is non-neutralizing (19). Statements regarding non-neutralizing antibodies in labels and publications ought to be taken cautiously, preferably only after examining the relevant clinical (pharmacodynamic, efficacy, and safety) correlates supporting the absence of drug neutralization and possibly also including a statement of relative sensitivity differences between the screening and neutralizing antibody methods.
Last but not least, a seldom considered factor that can impact the interpretation of the influence of ADA on PK is the type of bioanalytical method employed for drug concentration determination. Typically, PK immunoassays detect “free” drug because its levels are critical to clinical efficacy, and such methods are relatively easier to develop. Alternatively, some assay formats can detect “total” drug, which comprises free drug, target-bound drug, and possibly ADA-bound drug. Like ADA methods, PK immunoassays cannot be taken for granted; the format of PK immunoassays can vary widely in their susceptibility to interference by ADA (47). For example, the accuracy of PK immunoassays based on a format of capture and detection of a mAb drug using the drug target or anti-idiotypic antibodies is not expected to be affected by the presence of non-neutralizing ADA (non-NAb), whereas NAb can produce significantly undervalued drug concentration results. On the other hand, PK immunoassays employing an anti-Fc antibody-based capture or detection scheme may be interfered by the presence of non-NAb. A PK immunoassay may undervalue drug concentration when a fraction of the drug may have existed in vivo in an immune complex with non-NAb (therefore remaining bioactive). The same sample may have shown evidence of ADA if the ADA method was drug-tolerant. In this instance, one might erroneously conclude that PK was unaffected by ADA. Additionally, it would be difficult to reconcile the paradox that drug was efficacious at a lower concentration in ADA-positive subjects whereas the same concentration in ADA-negative subjects precluded efficacy. Thus, it is important to closely examine whether apparent changes in drug concentrations were caused by changes in drug clearance (due to immune complex formation with ADA), interference of ADA in the PK assay, or both, and this etiology is independent from other modes of drug elimination such as target-mediated drug disposition, renal filtration, or degradation. Lack of such knowledge could hinder a clear understanding of the impact of ADA on pharmacokinetics and the relationship with efficacy.
CONCLUSION
To date, the description of the immunogenicity of most biologic drug products has been of limited utility in informing physicians and patients of the true benefit/risk of the treatment and to guide post-marketing clinical practice. Clearly, a better understanding of ADA incidence and magnitude, time of onset, duration, neutralizing ability, cross-reactivity with endogenous molecules or other marketed biologic drugs, and their clinically relevant thresholds would enable enhanced clinical management of disease with biologic drugs.
We have defined terms for describing and analyzing immunogenicity data. We also presented analytical and data presentation approaches and our perspective on the types of information necessary to assess the clinical relevance of immunogenicity. The recommended descriptive and statistical analyses may not be equally relevant in all phases of drug development nor for all drugs; one ought to develop an analytical plan that is suitable for the intended purpose. Describing the characteristics of ADA development and associations of PK, efficacy and safety are meaningful when reporting individual clinical study findings, albeit limited when ADA incidence is too low or when study size is small. ADA characteristics and associations with clinical outcomes may vary among studies; hence, determinations of clinically relevant ADA thresholds may be unnecessary until after the completion of the pivotal studies supporting registration. At that time, if feasible, attempts ought to be made to combine data from all the studies with comparable dosage in order to enable a more robust assessment of the clinically relevant ADA threshold.
Acknowledgments
This work was sponsored by The Therapeutic Protein Immunogenicity Focus Group (TPIFG) of the BIOTEC Section, American Association of Pharmaceutical Scientists (AAPS). A global physician survey was conducted by TPIFG in 2010 and 2011 to assess the viewpoints and needs of medical practitioners relative to immunogenicity. Survey results and a call for the harmonization of terminology and the analysis and reporting of clinical immunogenicity were presented at the European Immunogenicity Platform (EIP) Symposium in December 2010 (Gent, Belgium) and at an Open Forum of the AAPS National Biotechnology Conference (NBC) in May 2011 (San Francisco, USA). Draft definitions and work-in-progress data presentation tactics were presented at the EIP symposia in February 2012 (Copenhagen, Denmark) and February 2013 (Munich, Germany) and at AAPS-NBC conventions in May 2012 and May 2013 (San Diego, USA). Finally, a draft manuscript was posted on the AAPS website for public feedback. The authors thank all those who provided feedback, which was considered during the finalization of this manuscript. During the preparation of this manuscript, our recommended terminology, definitions, and approaches for ADA characterization were shared with the ABIRISK (Anti-Biopharmaceutical Immunization: prediction and analysis of clinical relevance to minimize the RISK) consortium, which agreed to adopt them. ABIRISK is a project of the Innovative Medicines Initiative (IMI), a public-private partnership between the European Union and the European Federation of Pharmaceutical Industries and Associations (EFPIA), and aims to conduct immunogenicity studies of several biologic drugs, which will be used to develop a database comprising their evaluations of factors underlying immunogenicity and to generate tools for determining how individual patients are likely to respond.
Disclaimer
The contents of this article reflect the personal opinions of the authors and may not represent the official positions or perspectives of their affiliated organizations.
Footnotes
The contents of this article reflect the consensus perspective of the authors and may not represent the official positions or expectations of their affiliated organizations. This article is not a substitute or equivalent of a guidance document from a health regulatory agency.
References
- 1.Schellekens H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov. 2002;1(6):457–462. doi: 10.1038/nrd818. [DOI] [PubMed] [Google Scholar]
- 2.Schnabel CA, Fineberg SE, Kim DD. Immunogenicity of xenopeptide hormone therapies. Peptides. 2006;27(7):1902–1910. doi: 10.1016/j.peptides.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 3.Kuus-Reichel K, Grauer LS, Karavodin LM, Knott C, Krusemeier M, Kay NE. Will immunogenicity limit the use, efficacy, and future development of therapeutic monoclonal antibodies? Clin Diagn Lab Immunol. 1994;1(4):365–372. doi: 10.1128/cdli.1.4.365-372.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koren E, Zuckerman LA, Mire-Sluis AR. Immune responses to therapeutic proteins in humans—clinical significance, assessment and prediction. Curr Pharm Biotechnol. 2002;3(4):349–360. doi: 10.2174/1389201023378175. [DOI] [PubMed] [Google Scholar]
- 5.Schellekens H, Casadevall N. Immunogenicity of recombinant human proteins: causes and consequences. J Neurol. 2004;251(Suppl 2):II4–II9. doi: 10.1007/s00415-004-1202-9. [DOI] [PubMed] [Google Scholar]
- 6.Mayer L, Young Y. Infusion reactions and their management. Gastroenterol Clin N Am. 2006;35(4):857–866. doi: 10.1016/j.gtc.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Scheinfeld N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. J Dermatol Treat. 2004;15(5):280–294. doi: 10.1080/09546630410017275. [DOI] [PubMed] [Google Scholar]
- 8.D’Arcy CA, Mannik M. Serum sickness secondary to treatment with the murine-human chimeric antibody IDEC-C2B8 (rituximab) Arthritis Rheum. 2001;44(7):1717–1718. doi: 10.1002/1529-0131(200107)44:7<1717::AID-ART299>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 9.Korswagen LA, Bartelds GM, Krieckaert CL, Turkstra F, Nurmohamed MT, van Schaardenburg D, et al. Venous and arterial thromboembolic events in adalimumab-treated patients with antiadalimumab antibodies: a case series and cohort study. Arthritis Rheum. 2011;63(4):877–883. doi: 10.1002/art.30209. [DOI] [PubMed] [Google Scholar]
- 10.Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis. 2007;66(7):921–926. doi: 10.1136/ard.2006.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolbink GJ, Aarden LA, Dijkmans BA. Dealing with immunogenicity of biologicals: assessment and clinical relevance. Curr Opin Rheumatol. 2009;21(3):211–215. doi: 10.1097/BOR.0b013e328329ed8b. [DOI] [PubMed] [Google Scholar]
- 12.Yanai H, Hanauer SB. Assessing response and loss of response to biological therapies in IBD. Am J Gastroenterol. 2011;106(4):685–698. doi: 10.1038/ajg.2011.103. [DOI] [PubMed] [Google Scholar]
- 13.Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469–475. doi: 10.1056/NEJMoa011931. [DOI] [PubMed] [Google Scholar]
- 14.Macdougall IC. Antibody-mediated pure red cell aplasia (PRCA): epidemiology, immunogenicity and risks. Nephrol Dial Transplant. 2005;20(Suppl 4):iv9–iv15. doi: 10.1093/ndt/gfh1087. [DOI] [PubMed] [Google Scholar]
- 15.Guideline On Immunogenicity Assessment Of Biotechnology-Derived Therapeutic Proteins, EMEA/CHMP/BMWP/14327/2006. http://www.tga.gov.au/pdf/euguide/bmwp1432706en.pdf. Accessed 18 Mar 2014.
- 16.Janeway C. Immunogenicity signals 1,2,3 … and 0. Immunol Today. 1989;10(9):283–286. doi: 10.1016/0167-5699(89)90081-9. [DOI] [PubMed] [Google Scholar]
- 17.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13(1):114–119. doi: 10.1016/S0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 18.Schellekens H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clin Ther. 2002;24(11):1720–1740. doi: 10.1016/S0149-2918(02)80075-3. [DOI] [PubMed] [Google Scholar]
- 19.Shankar G, Pendley C, Stein KE. A risk-based bioanalytical strategy for the assessment of antibody immune responses against biological drugs. Nat Biotechnol. 2007;25(5):555–561. doi: 10.1038/nbt1303. [DOI] [PubMed] [Google Scholar]
- 20.Koren E, Smith HW, Shores E, Shankar G, Finco-Kent D, Rup B, et al. Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J Immunol Methods. 2008;333(1–2):1–9. doi: 10.1016/j.jim.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Ponce R, Abad L, Amaravadi L, Gelzleichter T, Gore E, Green J, et al. Immunogenicity of biologically-derived therapeutics: assessment and interpretation of nonclinical safety studies. Regul Toxicol Pharmacol. 2009;54(2):164–182. doi: 10.1016/j.yrtph.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Jahn EM, Schneider CK. How to systematically evaluate immunogenicity of therapeutic proteins—regulatory considerations. New Biotechnol. 2009;25(5):280–286. doi: 10.1016/j.nbt.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 23.Guidance for Industry: Assay Development for Immunogenicity Testing of Therapeutic Proteins. U.S. Department of Health and Human Services (DHHS), Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). http://www.fda.gov/downloads/Drugs/…/Guidances/UCM192750.pdf. Accessed 18 Mar 2014.
- 24.Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–1281. doi: 10.1016/j.jpba.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 25.Buttel IC, Chamberlain P, Chowers Y, Ehmann F, Greinacher A, Jefferis R, et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals. 2011;39(2):100–109. doi: 10.1016/j.biologicals.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 26.Wang YM, Fang L, Zhou L, Wang J, Ahn HY. A survey of applications of biological products for drug interference of immunogenicity assays. Pharm Res. 2012;29(12):3384–3392. doi: 10.1007/s11095-012-0833-2. [DOI] [PubMed] [Google Scholar]
- 27.Guidance for industry: immunogenicity assessment for therapeutic protein products. In: U.S. Department of Health and Human Services (DHHS), Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM338856.pdf2013. Accessed 18 Mar 2014.
- 28.Naserke HE, Bonifacio E, Ziegler AG. Prevalence, characteristics and diabetes risk associated with transient maternally acquired islet antibodies and persistent islet antibodies in offspring of parents with type 1 diabetes. J Clin Endocrinol Metab. 2001;86(10):4826–4833. doi: 10.1210/jcem.86.10.7931. [DOI] [PubMed] [Google Scholar]
- 29.Male C, Foulon D, Hoogendoorn H, Vegh P, Silverman E, David M, et al. Predictive value of persistent versus transient antiphospholipid antibody subtypes for the risk of thrombotic events in pediatric patients with systemic lupus erythematosus. Blood. 2005;106(13):4152–4158. doi: 10.1182/blood-2005-05-2048. [DOI] [PubMed] [Google Scholar]
- 30.Calabresi PA, Giovannoni G, Confavreux C, Galetta SL, Havrdova E, Hutchinson M, et al. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69(14):1391–1403. doi: 10.1212/01.wnl.0000277457.17420.b5. [DOI] [PubMed] [Google Scholar]
- 31.Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012;14(2):296–302. doi: 10.1208/s12248-012-9340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guidance for industry: immunotoxicology evaluation of investigational new drugs. U.S. Department of Health and Human Services (DHHS), Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079239.pdf2002. Accessed 18 Mar 2014.
- 33.Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–1468. doi: 10.1001/jama.2011.406. [DOI] [PubMed] [Google Scholar]
- 34.Ducourau E, Mulleman D, Paintaud G, Miow Lin DC, Lauferon F, Ternant D, et al. Antibodies toward infliximab are associated with low infliximab concentration at treatment initiation and poor infliximab maintenance in rheumatic diseases. Arthritis Res Ther. 2011;13(3):R105. doi: 10.1186/ar3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beck JR, Shultz EK. The use of relative operating characteristic (ROC) curves in test performance evaluation. Arch Pathol Lab Med. 1986;110(1):13–20. [PubMed] [Google Scholar]
- 36.Steenholdt C, Bendtzen K, Brynskov J, Thomsen OO, Ainsworth MA. Cut-off levels and diagnostic accuracy of infliximab trough levels and anti-infliximab antibodies in Crohn’s disease. Scand J Gastroenterol. 2011;46(3):310–318. doi: 10.3109/00365521.2010.536254. [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg AS, Worobec A. A risk-based approach to immunogenicity concerns of therapeutic protein products—Part 1–considering consequences of the immune response to a protein. Biopharm Int. 2004;17:22–26. [Google Scholar]
- 38.Rosenberg AS, Worobec A. A risk-based approach to immunogenicity concerns of therapeutic protein products—Part 2–considering host-specific and product-specific factors impacting immunogenicity. Biopharm Int. 2004;17:34–42. [Google Scholar]
- 39.Mire-Sluis AR, Barrett YC, Devanarayan V, Koren E, Liu H, Maia M, et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods. 2004;289(1–2):1–16. doi: 10.1016/j.jim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 40.Gupta S, Indelicato SR, Jethwa V, Kawabata T, Kelley M, Mire-Sluis AR, et al. Recommendations for the design, optimization, and qualification of cell-based assays used for the detection of neutralizing antibody responses elicited to biological therapeutics. J Immunol Methods. 2007;321(1–2):1–18. doi: 10.1016/j.jim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Gupta S, Devanarayan V, Finco D, Gunn GR, 3rd, Kirshner S, Richards S, et al. Recommendations for the validation of cell-based assays used for the detection of neutralizing antibody immune responses elicited against biological therapeutics. J Pharm Biomed Anal. 2011;55(5):878–888. doi: 10.1016/j.jpba.2011.03.038. [DOI] [PubMed] [Google Scholar]
- 42.Rosenberg AS, Worobec A. A risk-based approach to immunogenicity concerns of therapeutic protein products—Part 3–effects of manufacturing changes in immunogenicity and the utility of animal immunogenicity studies. Biopharm Int. 2005;18:32–36. [Google Scholar]
- 43.Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. EMA/CHMP/BMWP/86289/2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128688.pdf. Accessed 18 Mar 2014.
- 44.Lofgren JA, Dhandapani S, Pennucci JJ, Abbott CM, Mytych DT, Kaliyaperumal A, et al. Comparing ELISA and surface plasmon resonance for assessing clinical immunogenicity of panitumumab. J Immunol. 2007;178(11):7467–7472. doi: 10.4049/jimmunol.178.11.7467. [DOI] [PubMed] [Google Scholar]
- 45.Li J, Schantz A, Schwegler M, Shankar G. Detection of low-affinity anti-drug antibodies and improved drug tolerance in immunogenicity testing by Octet((R)) biolayer interferometry. J Pharm Biomed Anal. 2011;54(2):286–294. doi: 10.1016/j.jpba.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 46.Dai S, Schantz A, Clements-Egan A, Cannon M, Shankar G. Development of a method that eliminates false positive results due to nerve growth factor interference in the assessment of fulranumab immunogenicity. AAPS J. 2014; doi:10.1208/s12248-014-9581-z. [DOI] [PMC free article] [PubMed]
- 47.Kelley M, Ahene AB, Gorovits B, Kamerud J, King LE, McIntosh T, et al. Theoretical considerations and practical approaches to address the effect of anti-drug antibody (ADA) on quantification of biotherapeutics in circulation. AAPS J. 2013;15(3):646–658. doi: 10.1208/s12248-013-9468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]