

Abstract
A dual ligand (DL) system that combines high affinity streptavidin-biotin binding with lower affinity fibronectin-integrin ligand binding was developed to augment endothelial cell adhesion to polymers. In this study, we examined the utility of biotinylated fibronectin (bFN) as an enhancement to the previously developed DL approach. The goal was to make the system more amenable to clinical studies by eliminating xenogenic bovine serum albumin (bBSA). Fibronectin (FN) biotinylation was achieved with Sulfo-NHS-LC-Biotin. The affinity of conjugated biotin for wild-type streptavidin (WT-SA) and a mutant strain streptavidin (RGD-SA) was measured using surface plasmon resonance (SPR) spectroscopy. Enzyme-Linked ImmunoSorbent Assay (ELISA) absorbance values confirmed the accessibility of the cell binding domain on mildly biotinylated bFN when compared to unmodified native protein. SPR binding analysis confirmed similar binding behavior to bFN with WT-SA and RGD-SA. Kinetic analysis, however, showed no increase in affinity due to increased biotins per FN, an indication of the absence of positive cooperativity in the system. We verified the essential utility of bFN in affinity binding by SPR and confirmed the potential for integrin-FN linkages by ELISA. Finally, Vinculin immunostaining was used to determine focal adhesion formation using bFN in the DL system. Significantly greater focal adhesion density was achieved with the bFN in the DL system than with FN alone.
1.0. Introduction
Protein adsorption has been used in the field of vascular tissue engineering to enhance cell attachment, spreading, and formation of neointima in synthetic vascular grafts. The majority of studies have adsorbed films of single proteins like fibronectin, vitronectin, or laminin to bind cells to the material surface.1–4 Fibronectin, the most researched of the extracellular matrix proteins, is a 440 kDa glycoprotein composed of a dimer joined by a pair of disulfide bonds.5 These surface modifications, however, are often insufficient to achieve the firm cell attachment to vascular grafts necessary to resist thrombosis, intimal hyperplasia, and leukocyte adhesion.6,7 Insufficient adhesion may be attributed partially to the low binding affinity (KD ≈ 10−6 M) between transmembrane integrins and extracellular matrix proteins.5 Streptavidin is a tetrametric protein isolated from Streptomyces avidinii that is able to bind four submits of biotin with high affinity (KD in solution ≈ 10−15 M). A dual ligand (DL) strategy that uses high affinity streptavidin-biotin binding to augment lower affinity FN-integrin binding shows promise as a means to improve cell adhesion. Others have used the DL system to enhance chondrocyte adhesion to tissue culture polystyrene as an indication of future tissue engineering viability,8 hepatic cell binding and spreading on biodegradable, polymer-based, flat 2D surfaces, and in highly porous 3D scaffolds,9 and for drug delivery using avidin to immobilize biotinylated therapeutic agents.10
We have employed streptavidin-biotin to augment the binding of endothelial cells to vascular grafts.11 In our initial system, surfaces were co-adsorbed with fibronectin (FN) and biotinylated bovine serum albumin (bBSA) to augment the binding of endothelial cells decorated with biotin and subsequently incubated with streptavidin. The streptavidin-biotin binding stabilized the initial cell-substrate contact, bringing the cell membrane in close apposition to adsorbed fibronectin, thus facilitating the formation of intrinsic integrin-mediated focal contacts.12 DL augmented endothelial cell (EC) adhesion has been reported on Mylar and Teflon-AF that were employed as surrogates of Dacron and ePTFE vascular graft materials.13 The system was refined by employing an RGD-streptavidin (RGD-SA) mutant, isolated from E. coli cells transfected with a pET plasmid, which contained integrin-independent and integrin-dependent functionality in a single protein.14 Binding of the RGD-SA to integrin receptors on the EC membrane provided direct means of decorating the ECs with streptavidin, increasing the density of cell adhesion ligand, and also mitigating cohesive failure effects due to the nonspecificity of cell substrate biotinylation.15,16
The current study examined a second refinement that replaced co-adsorbed FN and bBSA with adsorbed biotinylated fibronectin (bFN) (Figure 1). Like the RGD-SA, bFN has dual functionality: biotin moieties that facilitate high affinity streptavidin-biotin binding and cell binding RGD domains that promote integrin-FN binding. This enhancement makes the system more amenable to future clinical studies by eliminating xenogenic effects due to bBSA. This study also examined whether the degree of biotinylation affected the streptavidin and integrin binding functions of biotinylated FN. Finally, this study examined whether a molecular laminate of bFN and RGD-SA promotes focal adhesion formation in a manner similar to native FN.
Figure 1.
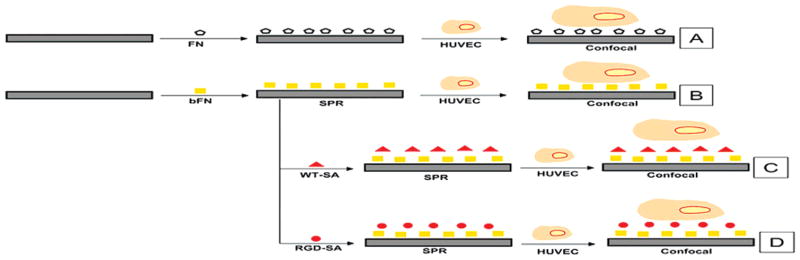
Experimental flowchart of the different formulations examined in the current studies and their method of analysis. Surface plasmon resonance (SPR) spectroscopy and confocal microscopy were the primary analysis tools used for the different formulations.
2.0. Methods
2.1. Biotinylation of Fibronectin
Fibronectin was biotinylated with Sulfo NHS-LC-Biotin (Pierce, Rockford, IL) with a spacer arm of 22 Å. A solution of FN (1 mg/mL, Chemicon, Temecula, CA) was mixed with 10 mM Sulfo NHS-LC-Biotin dissolved in ultrapure water for 30 min at room temperature. Excess biotin was removed by dialysis in Dulbecco’s phosphate buffered saline (dPBS) (Invitrogen, Carlsbad, California) for 24 h at 4 °C with a buffer change after 12 h. Multiple biotin conjugation levels were achieved by stoichiometrically varying the moles of biotin reagent per FN. The amount of biotin incorporation was measured using EZ-Link-NHS-chromogenic-Biotin (Pierce) with a polyethylene glycol spacer arm of 41 Å and a bis-aryl hydrazone group with a characteristic absorbance at 354 nm. Absorbance values were indicative of number of biotins per fibronectin.
2.2. Surface Plasmon Resonance Spectroscopy (SPR)
Glass slides were cleaned as previously detailed.13 Chromium (4 nm) and gold (20 nm) were evaporated onto the slides under vacuum to yield transparent gold films17 and stored under argon until used. Self-assembled monolayer (SAM) formation was achieved by immersing gold-coated chips in a solution of 2 mM 16-mercaptohexadecanoic acid (HDA) in 100% ethanol for ≥24 h. Afterward, chips were rinsed and dried under N2 gas.
The SPR experiments were carried out on a BIACOREX (Pharmacia Biosensor) microfluidic system as described previously.17 After chip insertion, the surface with HDA SAM was either activated by injection of 35 μL of an equal mix of 0.1 M N-hydroxysuccinimide (NHS) and 0.4 M 1-ethyl-3-(3-(dimethyl-amino)propyl)carbodiimide (EDC) in PBS at a flow rate of 5 μL/min or left unmodified for direct ligand adsorption to the SAM layer. After rinsing and baseline equilibration, 20 μg/mL bFN with different molar ratios was immobilized onto the chip surface at a flow rate of 2 μL/min. Subsequently, either 50 μg/mL WT-SA (Pierce, Rockford, IL) or RGD-SA was flowed over the protein at 2 μL/min and response units (RU) were measured based on differences in refractive index. These units were eventually correlated to protein surface density based on the relationship: 1000 RU = 1 ng/mm2 protein. The binding kinetics and amount immobilized were calculated with BIAevalution software (Biacore AB, Uppsala, Sweden).
2.3. Enzyme-Linked ImmunoSorbent Assay (ELISA)
Ninety six well plates were coated with Teflon-AF and dried under vacuum overnight at room temperature according to the procedure of Koenig et al.18 bFN with five biotins per FN or native FN in dPBS at a concentration of 20 μg/mL was adsorbed to 96 well plates for 1 h at room temperature. Wells were blocked with 1% BSA in dPBS for 3 h and rinsed 2× with dPBS. The wells were incubated with 1:100 mouse anti-human FN cell binding peptide antibody (Chemicon, Temecula, CA) in dPBS for 2 h at room temperature and rinsed 2× with dPBS. These wells were then incubated with 1:200 sheep anti-mouse horseradish peroxidase (HRP) secondary antibody in 0.5% BSA for 20 min and rinsed 2× with dPBS. Wells were then incubated with 100 μL of TMB/E developing solution in the dark for 20 min at room temperature. The reaction was halted with 50 μL of 2 N sulfuric acid, and the plate was read within 5 min at 450 nm with a reference wavelength of 540 nm to correct for background absorbance due to the plate.
2.4. Cell Culture
All cell culture reagents were obtained from Cambrex (Walkersville, MD) unless otherwise specified. Human umbilical vein endothelial cells (HUVEC) were grown to confluence in gelatin coated T25 polystyrene flasks (Corning Inc., Corning, NY) with endothelial basal media (EBM) supplemented with 0.5 mL of 10 mg/mL hEGF (human recombinant Epidermal Growth Factor), 0.5 mL of 1.0 mg/mL Hydrocortisone, 0.5 mL of 50 mg/mL Gentamicin and 50 mg/mL Amphotericin-B mix, 3 mg/mL bovine brain extract (BBE), and 10 mL of Fetal Bovine Serum (FBS). Cells were cultured in an incubator with 95% air/5% CO2 at 37 °C. HUVECs from passage 3–5 were used for all experiments. For all experiments, confluent p25 flasks were trypsinized with 0.025% Trypsin/EDTA for 4 min at 37 °C, neutralized with trypsin neutralizing solution (TNS), and centrifuged at 2200 rpm for 5 min. The cells were resuspended in fresh serum-free media or dPBS prior to experimentation.
2.4.1. Immunofluorescent Stain for Focal Adhesions
Glass coverslips were cleaned as described above13 and inserted into sterile 12 well plates. Slides were incubated with 20 μg/mL of FN or bFN with nine biotins for 1 h at 37 °C, rinsed 3× with dPBS, and blocked with 2 mg/mL BSA for 30 min at 37 °C. Coverslips were rinsed 3× with dPBS then incubated with 50 μg/mL of either wild-type streptavidin (WT-SA) or mutant strain streptavidin (RGD-SA) for 40 min in an incubator. Coverslips were rinsed and incubated with 40,000 cells in 1 mL of serum-free media in the incubator. After a 1 h attachment, cells were fixed with 3.7% fresh paraformaldehyde for 20 min at room temperature, permeabilized for 5 min with 0.2% Triton X-100 (Sigma), and washed 3× for 2 min intervals on a shaker with dPBS. Cells were blocked with 15% goat serum for 30 min in the incubator to prevent nonspecific binding, then incubated with 1:100 mouse anti-human vinculin antibody in 1% goat serum for 1 h in incubator. Cells were washed 3× for 10 min cycles on shaker using PBS with 0.05% Tween-20 at room temperature. Cells were then incubated with 1:700 goat anti-mouse Alexa-Fluor 488 secondary antibody for 45 min in the incubator, washed, and mounted onto glass slides using FluorSave (Calbiochem). Images were captured the next day using a confocal laser scanning microscope (Carl Zeiss Inc.). The LSM 510 image processing tool was used to quantify the size of focal contacts, and the number of focal adhesions was quantified using ImageJ.
2.5. Statistical Analysis
StatView 5.0 was used to statistically compare data and measure variability. One-way ANOVA plus Tukey-Kramer post hoc analysis were conducted to determine p values. All data reported as the mean ± SEM.
3.0. Results
3.1. Fibronectin Biotinylation Characterizations
bFN was generated by reacting from 10 to 500 molar excesses of Sulfo-NHS-LC-Biotin with FN. Figure 2 shows the increase in the total number of biotins conjugated to FN expressed as biotins per FN molecule. Biotinylation yielded 2–21 biotins per FN across the range of molar excesses. The number of total biotins per fibronectin in solution was determined using the absorbance of the EZ-Link-NHS-chromogenic-Biotin. The number of biotins conjugated per FN molecule increased linearly until ~20 biotins at a molar excess of 300, after which the degree of biotinylation appeared to saturate. Note that there was no significant difference between the values at 300, 400, and 500 molar excess biotin.
Figure 2.

Number of biotins attached per fibronectin in solution after dialysis measured as a function of biotin solution molar excess. This number was determined by absorbance of EZ-Link-NHS-chromogenic-Biotin at 354 nm (n = 3).
3.2. SPR Spectroscopy
SPR spectroscopy was used to determine the binding characteristics of WT-SA and the RGD-SA mutant to immobilized bFN as a function of biotins per FN molecule. Panels a and b of Figure 3 compare traces of passively adsorbed and immobilized bFN. Table 1 shows the surface densities of bFN and RGD-SA before and after buffer flushing steps. The amount of bFN immobilized by EDC/NHS was not significantly different when compared to passive adsorption, but the amount of RGD-SA bound to immobilized bFN was significantly higher than with passive adsorption. RGD-SA desorption was negligible on surfaces functionalized with EDC/NHS.
Figure 3.

Sample SPR scan of WT-SA or RGD-SA bound to immobilized bFN. (a) A representative scan using EDC/NHS functionalization. (b) A scan with bFN passively adsorbed to HDA SAM.
Table 1.
Comparison of bFN(9) Immobilized via Passive Adsorption Versus EDC/NHS Activation and Subsequent Binding and Desorption of RGD-SAa
| surface | bFN(9) | desorbed | RGD-SA | desorbed |
|---|---|---|---|---|
| SAM | 1.76 ± 0.04 | 0 | 0.78 ± 0.03 | 0.37± 0.01 |
| SAM + EDC/NHS | 1.87 ± 0.23 | 0 | 1.54 ± 0.17b | 0.12 ± 0.03c |
Values are reported as average SEM for n = 3. Column statistics was performed using ANOVA and Tukey-Kramer post hoc tests.
Significantly higher binding compared to passive adsorption (p < 0.05).
Significantly lower desorption compared to passive adsorption (p < 0.01).
Figure 4a shows the mass of bFN immobilized on the SPR chip and the mass of WT-SA bound to the immobilized bFN. Figure 4b similarly shows the mass of bFN immobilized on the SPR chip and the mass of RGD-SA bound to the immobilized bFN. Results indicated that below 10 biotins per FN the mass of bFN immobilized was relatively constant at a value of 2ng/mm2 (note boxed region). Above 10 biotins per FN the amount of immobilized FN decreased. The values of bound WT-SA and RGD-SA increased linearly and appeared to plateau at a value of 2 ng/mm2 after 10 biotins per FN. These data suggest well-behaved binding for bFN with <10 biotins per molecule. In contrast, higher levels of biotinylation appeared to introduce steric effects which reduced binding.
Figure 4.

Amount of bound (a) WT-SA and (b) RGD-SA bound per immobilized bFN. All these values were measured as a function of biotins per FN (n = 4).
Figure 5 contains the molar binding ratio of WT-SA and RGD-SA per mole of immobilized bFN as a function of biotins per bFN molecule. There was remarkable similarity when these data were examined on a molar basis. The amount of bound WT-SA and RGD-SA were statistically indistinguishable (p < 0.01) at each biotinylation level and increased linearly with slopes of 0.83 ± 0.033 and 0.75 ± 0.026, respectively.
Figure 5.
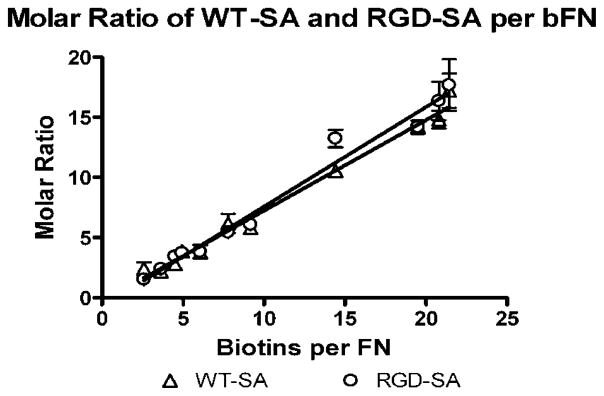
Molar ratio of WT-SA and RGD-SA per mole of bFN. Both are measured as a function of biotins per FN (n = 3).
Figure 6a and b show the molar equilibrium dissociation constants for WT-SA and RGD-SA bound to immobilized bFN over the range of biotin molar excesses. The equilibrium dissociation constants (KD) for all experimental groups ranged from 1.1 × 10−8 to 1.6 × 10−11 M for WT-SA and 1.1 × 10−7 to 5.3 × 10−11 M for RGD-SA bound to bFN. These dissociation constants were much higher than reported values for SA-biotin interactions in solution.19 However, dissociation constants of 10−7–10−11 M are consistent with literature values observed with biotin-functionalized macromolecules bound to immobilized streptavidin.20,21
Figure 6.

Equilibrium dissociation kinetic constant of WT-SA and RGD-SA bound to immobilized bFN (n = 4).
3.3. ELISA
ELISA with an antibody to the cell binding domain of FN was used to determine whether biotinylation affected accessibility of fibronectin’s cell-binding domain for bFN with <10 biotins per molecule. The binding of the antibody to the cell-binding domain of FN and bFN was statistically indistinguishable (data not shown). These results indicated that relatively mild biotinylation did not affect the accessibility of the cell binding domain when compared to the native unmodified protein.
3.4. Vinculin Localization
The biotinylation level of nine biotins per FN and 50 μg/mL RGD-SA ratio used for the immunofluorescence studies were based in Figure 4. Immunostaining for vinculin was used to examine two phenomena: (1) the similarity in formation of focal adhesions between native FN and bFN and (2) the ability to modulate the formation of focal adhesions using either WT-SA or RGD-SA. Figure 7 contains images of focal adhesions for the four conditions shown in Figure 1: (A) HUVEC bound to FN; (B) HUVEC bound to bFN; (C) HUVEC bound to RGD-SA-bFN; (D) HUVEC bound to WT-SA-bFN. Table 2 summarizes the percentage of HUVECs that exhibited focal contacts, the number of focal contacts per cell, and the average size of the focal contacts. These values were averaged over 30 confocal images of adherent cells with 10 images analyzed per experiment.
Figure 7.

Focal adhesion formation at 100× with HUVEC bound to FN (A); HUVEC bound to bFN (B); HUVEC bound to RGD-SA-bFN (C); HUVEC bound to WT-SA-bFN (D) (n = 3).
Table 2.
Quantitative Analysis of Focal Adhesion for HUVEC Seeded onto Glass Cover Slips for 1 h with Surface Treatments A–Da
| surface treatment | percent of cells with focal adhesions | av no. of focal adhesions per cell | av size of focal adhesions (μm2) |
|---|---|---|---|
| FN (A) | 100 | 308.3 ± 30.7 | 1.73 ± 0.19 |
| bFN (B) | 100 | 309.0 ± 18.0 | 1.57 ±0.10 |
| bFN + WT-SA | 62.4 ± 23.1b | 203.2 ± 12.2 | 1.22 ± 0.15 |
| bFN + RGD-SA | 100 | 430.0 ± 19.9c | 1.84 ± 0.17 |
Values are reported as average SEM for n = 3. Column statistics was performed using ANOVA and Tukey-Kramer post hoc tests.
Significantly different compared to all other treatment groups (p < 0.01).
Significantly different compared to all other treatment groups (p < 0.05).
All of the cells on FN, bFN, and bFN+RGD-SA exhibited focal contacts, while only 62% of cells showed focal contacts when bFN was partially blocked with WT-SA. Cells on FN and bFN exhibited nearly identical focal adhesion density per cell. Cells on bFN+RGD-SA had 40% more focal adhesions per cell when compared to FN and bFN alone; whereas bFN+WT-SA had 44% fewer focal adhesions per cell compared to FN and bFN. There were no differences in the size of focal adhesions between any treatment groups at p < 0.01, but differences were observed between the FN and bFN+RGD-SA and the other treatment groups at p < 0.05.
4.0. Discussion
Rapid cell attachment and spreading is essential in many biomedical applications. Most efforts to improve cell adhesion in vitro have used immobilized ECM proteins, but this approach has had limited success.22–24 Any strategy to enhance cell binding and retention must bind cells in a manner that brings trans-membrane integrins in close apposition to immobilized proteins and in turn promotes the formation of mature focal contacts. Streptavidin-biotin binding is being explored as an adjuvant to the natural cell adhesion formulation because of its high binding affinity with a solution phase KD of ~10−15 M. Our group introduced a DL protocol that takes advantage of the high affinity streptavidin-biotin interaction to buttress the integrin dependent cell adhesion interaction system. The current DL system employs two bifunctional protein constructs, RGD-SA and bFN, that both possess integrin-independent and integrin-dependent binding properties. This construct is a shift from the previous use of RGD-SA with bBSA as the biotin source in the system.14 The use of human FN leaves open the potential for future clinical applications. Other groups have used the avidin-biotin system to bind biotinylated chondrocytes to Avidin-coated tissue culture polystyrene plates (TCPS),8 and more recently, Kojima et al. demonstrated the effectiveness of binding biotinylated hepatic cells to poly(L-lactic acid) (PLLA) surfaces.9,25 Others have examined the binding affinity of streptavidin to surface-immobilized biotin with varying densities and showed that the dissociation rate is a function of surface biotin density.25,26 Single biotin-functionalized FN has also been used but primarily as a separation or extraction ligand.27,28
The essential benefit of the DL system comes from interaction between biotin and streptavidin. SPR was used in this study to monitor the binding of WT-SA and RGD-SA to biotin-functionalized FN. SPR binding efficiency and high kinetic binding affinity was best using EDC/NHS functionalization. The amount of bFN immobilized was relatively constant up to 10 biotins per FN at which point there was a decline in amount bound. The decrease in bFN immobilization can be attributed to depletion of available amine groups by biotinylation, thus limiting accessibility of amine groups to EDC/NHS coupling. This drop, however, did not affect the proportionate increase in binding of WT-SA or RGD-SA to immobilized bFN. This observation was also evident in the molar binding ratio of WT-SA or RGD-SA per bFN. The nearly 1:1 molar correlation with the number of biotins per FN indicates that only one SA binding site was occupied during each interaction.
The 1:1 correspondence was unexpected because there should be two accessible binding sites on each streptavidin. The reasons for the lack of bivalent binding may be due to surface-induced conformational shifts in the binding sites or the mass transfer limitations at the surface of the substrate.29 More accurate control of surface biotin coverage has been shown by other groups to affect the SA binding site occupation.25 It is clear from our results that the random orientation of the bound WT-SA or RGD-SA does not allow for structured binding where all SA binding sites are accessible to surface biotins.
SPR was used to confirm that the affinity of the SA-biotin interaction was conserved when biotin was attached to FN. Different levels of biotin incorporation were examined under the assumption that increased biotinylation would lead to increased binding affinity. This assumption was ultimately proven to be incorrect by SPR kinetic models. The binding kinetic analysis of WT-SA or RGD-SA for immobilized bFN was measured with the aid of a Langmuir model. The KD in this system was 1.1 × 10−8–1.6 × 10−11 M for WT-SA and 1.1 × 10−7–5.3 × 10−11 M for RGD-SA. These values were a few orders of magnitude higher than that for SA-biotin binding observed in solution but were consistent with literature values for binding biotin-functionalized molecules to SA.20,21 Though Huang et al. worked with polystyrene beads with micrometer-size diameters, there was evidence from their results to suggest that increased ligand size increased dissociation constants. This could explain the increase in dissociation kinetics on the 2-dimensional FN surface.
Monovalent SA-biotin binding appears to have dominated over the range of biotins per FN. This indicates that though biotin density is increasing, there was no cooperative effect from the proximal biotins, i.e., binding occurs independently between one binding pocket of SA and biotin conjugated to FN. This noncooperative effect was also observed previously.30 Flow experiment results were consistent with the SPR studies with no positive correlation between percent retention and biotinylation levels. Cell retention ranged from 80% to 91% with an average of 86.26% ± 3.5% across all biotinylation levels (data not shown).
Although our model ultimately showed no difference in affinity for the wild-type compared to the RGD-SA, the binding curves showed a slight desorption using the mutant strain (not shown). The overall results were promising because the affinity of biotin for SA was not diminished by coupling to fibronectin. The consistency of binding affinity and concentration levels between WT-SA and RGD-SA was further evidence that conjugation to these high-molecular-weight glycoproteins did not negatively affect the high-affinity interaction. The most important information gained from this SPR analysis is that the binding affinity of WT-SA and RGD-SA for immobilized bFN was the same.
Finally, biotin functionalization had no apparent effect on the availability of cell binding domains, as seen by ELISA and focal adhesion formation. Quantification of the focal adhesions confirmed that the biotinylation of fibronectin had no adverse effect on its utility in forming focal contacts after short seeding times. This was essential given that the effectiveness of the system would be diminished if the cell-binding domain was blocked.
The prevalence, number, and size of focal adhesions were not different between native FN and bFN, indicating that biotin functionalization did not have an adverse effect on the ability to form focal adhesions. Additionally, the DL system showed enhanced ability over both FN and bFN in promoting statistically higher focal adhesion density, as well as larger focal contacts (the latter was an indication of the maturity of the focal adhesion31). This effect was enhanced in the DL because of the availability of additional RGD motifs that promoted greater formation of focal adhesions. Conversely, surfaces coated with bFN then incubated with WT-SA showed no enhanced effect and even showed decreased prevalence, number, and size of focal adhesions when compared to the other treatment groups. The large SA molecule on top of the bFN layer may quench the cell adhesion effect by blocking essential domains necessary to promote formation of focal adhesions.
5.0. Conclusion
This manuscript examined the utility of biotinylated FN in the DL cell adhesion system. Effective biotinylation of fibronectin was achieved without masking accessibility to the cell-binding domain for integrins at <10 biotins per FN. Kinetic analysis by SPR verified that affinity of bound biotin was the same for WT-SA, as well as RGD-SA. Additionally, no enhanced kinetic effect was observed with higher biotins per FN, underscoring the lack of positive cooperativity in the system. Biotinylated FN behaved similar to FN in promoting focal adhesion formation but had an enhanced effect in density of focal adhesion when used in the DL system. Further studies need to be conducted with this system to determine effect on improving cell adhesion to synthetic vascular graft surrogates.
Acknowledgments
This project was supported by NIH Grant No. HL44972 and an NIH Graduate Development Fellowship. The authors kindly acknowledge Dr. Pat Stayton of the University of Washington for the RGD-SA plasmid, Dr. Mort Friedman for the use of the plate reader, Dr. Ashutosh Chilkoti for the use of the Biacore instrument, and Mr. Charles S. Wallace for assistance with confocal studies.
References
- 1.Danen EH, Yamada KM. J Cell Physiol. 2001;189 (1):1–13. doi: 10.1002/jcp.1137. [DOI] [PubMed] [Google Scholar]
- 2.Grainger DW, Pavon-Djavid G, Migonney V, Josefowicz M. J Biomater Sci Polym Ed. 2003;14 (9):973–88. doi: 10.1163/156856203322381456. [DOI] [PubMed] [Google Scholar]
- 3.Wilson CJ, Clegg RE, Leavesley DI, Pearcy MJ. Tissue Eng. 2005;11(1–2):1–18. doi: 10.1089/ten.2005.11.1. [DOI] [PubMed] [Google Scholar]
- 4.Boateng SY, Lateef SS, Mosley W, Hartman TJ, Hanley L, Russell B. Am J Physiol Cell Physiol. 2005;288 (1):C30–8. doi: 10.1152/ajpcell.00199.2004. [DOI] [PubMed] [Google Scholar]
- 5.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson J. Molecular Biology of the Cell. 2. Garland Publishing; New York: 1989. [Google Scholar]
- 6.Poole-Warren LA, Schindhelm K, Graham AR, Slowiaczek PR, Noble KR. J Biomed Mater Res. 1996;30 (2):221–29. doi: 10.1002/(SICI)1097-4636(199602)30:2<221::AID-JBM12>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 7.Hagerty RD, Salzmann DL, Kleinert LB, Williams SK. J Biomed Mater Res. 2000;49 (4):489–97. doi: 10.1002/(sici)1097-4636(20000315)49:4<489::aid-jbm7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 8.Tsai WB, Wang MC. Biomaterials. 2005;26 (16):3141–51. doi: 10.1016/j.biomaterials.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Kojima N, Matsuo T, Sakai Y. Biomaterials. 2006;27 (28):4904–10. doi: 10.1016/j.biomaterials.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 10.Hoya K, Guterman LR, Miskolczi L, Hopkins LN. Drug Delivery. 2001;8 (4):215–22. doi: 10.1080/107175401317245895. [DOI] [PubMed] [Google Scholar]
- 11.Bhat VD, Klitzman B, Koger K, Truskey GA, Reichert WM. J Biomater Sci Polym Ed. 1998;9:1117–35. doi: 10.1163/156856298x00686. [DOI] [PubMed] [Google Scholar]
- 12.Bhat VD, Truskey GA, Reichert WM. J Biomed Mater Res. 1998;40:57–65. doi: 10.1002/(sici)1097-4636(199804)40:1<57::aid-jbm7>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 13.Anamelechi CC, Truskey GA, Reichert WM. Biomaterials. 2005;26 (34):6887–96. doi: 10.1016/j.biomaterials.2005.04.027. [DOI] [PubMed] [Google Scholar]
- 14.Chan BP, Reichert WM, Truskey GA. Biotechnol Prog. 2004:566–575. doi: 10.1021/bp034215z. [DOI] [PubMed] [Google Scholar]
- 15.Chan BP, Chilkoti A, Reichert WM, Truskey GA. Biomaterials. 2003;24:559–570. doi: 10.1016/s0142-9612(02)00367-8. [DOI] [PubMed] [Google Scholar]
- 16.McDevitt TC, Nelson KE, Stayton PS. Biotechnol Prog. 1999;15 (3):391–6. doi: 10.1021/bp990043n. [DOI] [PubMed] [Google Scholar]
- 17.Smith JT, Tomfohr JK, Wells MC, Beebe TP, Jr, Kepler TB, Reichert WM. Langmuir. 2004:8279–8286. doi: 10.1021/la0489763. [DOI] [PubMed] [Google Scholar]
- 18.Koenig AL, Gambillara V, Grainger DW. J Biomed Mater Res, Part A. 2003;64 (1):20–37. doi: 10.1002/jbm.a.10316. [DOI] [PubMed] [Google Scholar]
- 19.Savage MD, Mattson G, Desai S, Nielander GW, Morgensen S, Conklin EJ. Avidin-Biotin Chemistry: A Handbook. 2. Pierce Chemical Company; Rockford, IL: 1992. [Google Scholar]
- 20.Huang SC, Stump MD, Weiss R, Caldwell KD. Anal Biochem. 1996;237:115–122. doi: 10.1006/abio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 21.Yang N, Su X, Tjong V, Knoll W. Biosens Bioelectron. 2007;22:2700–2706. doi: 10.1016/j.bios.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 22.Lambert AW, Fox AD, Williams DJ, Horrocks M, Budd JS. Cardiovascular Surgery. 1999;7 (5):491–4. doi: 10.1016/s0967-2109(99)00012-5. [DOI] [PubMed] [Google Scholar]
- 23.Becquemin JP, Riff Y, Kovarsky S, Ardaillou N, Benhaien-Sigaux N. J Cardiovascular Surgery. 1997;38:7–14. [PubMed] [Google Scholar]
- 24.Shimada T, Nishibe T, Miura H, Hazama K, Kato H, Kudo F, Murashita T, Okuda Y. Surgery Today. 2004;34 (12):1025–30. doi: 10.1007/s00595-004-2862-x. [DOI] [PubMed] [Google Scholar]
- 25.Jung LS, Nelson KE, Stayton PS, Campbell CT. Langmuir. 2000;16:9421–9432. [Google Scholar]
- 26.Nelson KE, Gamble L, Jung LS, Boeckl MS, Naeemi E, Golledge SL, Sasaki T, Castner DG, Campbell CT, Stayton PS. Langmuir. 2001;17:2807–2816. [Google Scholar]
- 27.Wolz C, Pohlmann-Dietze P, Steinhuber A, Chien YT, Manna A, van Wamel W, Cheung A. Mol Microbiol. 2000;36 (1):230–43. doi: 10.1046/j.1365-2958.2000.01853.x. [DOI] [PubMed] [Google Scholar]
- 28.Talamas-Rohana P, Rosales-Encina JL, Gutierrez MC, Hernandez VI. Archives Med Res. 1992;23 (2):119–23. [PubMed] [Google Scholar]
- 29.Fujita K, Silver J. Biotechniques. 1993;14 (4):608–17. [PubMed] [Google Scholar]
- 30.Jones ML, Kurzban GP. Biochemistry. 1995;34 (37):11750–6. doi: 10.1021/bi00037a012. [DOI] [PubMed] [Google Scholar]
- 31.Romer LH, Birukov KG, Garcia JG. Circ Res. 2006;98 (5):606–16. doi: 10.1161/01.RES.0000207408.31270.db. [DOI] [PubMed] [Google Scholar]