

Abstract
The Shb adapter protein is a signaling intermediate that operates downstream of vascular endothelial growth factor receptor‐2 (VEGFR‐2) in endothelial cells. The Shb knockout mouse displays a dysfunctional microvasculature and impaired growth of subcutaneously implanted tumor cells. We decided to investigate tumor growth and angiogenesis in the absence of Shb in an inheritable tumor model, the RIP‐Tag2 mouse, which produces insulinomas in a manner highly dependent on de novo angiogenesis. We observed a reduced tumor incidence and burden in both RIP‐Tag2 Shb−/− and RIP‐Tag2 Shb+/− mice. This correlated with a reduced microvascular density, measured as a percentage of insulinoma area positive for CD31 staining, and altered vascular morphology. However, treatment with a VEGF‐A blocking antibody was without effect on the Shb mutant tumor volume whereas it significantly inhibited tumor volume in the wild‐type mice, suggesting that in mice with reduced Shb expression tumor angiogenesis was primarily sustained by VEGF‐A independent pathway(s). This notion was further substantiated by gene expression analysis of angiogenic markers showing reduced VEGF‐A expression in Shb‐deficient tumors. Considerable heterogeneity with respect to the gene expression profiles of other angiogenic markers and the signal‐transduction characteristics was observed between different tumors, suggesting that multiple “rescue” pathways could be operating. The numbers of invasive tumors or metastases were unchanged in the Shb mutant.
It is concluded that the Shb mutant background reduces tumor frequency by chronically suppressing VEGF‐A dependent angiogenesis. However, VEGF‐A independent angiogenesis supports a significant degree of tumor expansion in Shb‐deficient mice, indicating heterogeneity in the mechanisms by which tumor expansion is promoted. Interference with Shb signaling may provide novel means for future cancer therapy.
Keywords: Insulinomas, Shb, Angiogenesis, VEGF
Highlights
Shb is an adapter protein operating downstream of VEGFR‐2.
Shb was presently investigated in the context of RIP‐Tag2 angiogenesis.
Absence of Shb reduces RIP‐Tag2 tumor burden and VEGF‐dependent tumor angiogenesis.
A fraction of tumors escape the angiogenic inhibition imposed by the absence of Shb.
Tumor heterogeneity allows escape from angiogenic restriction by various means.
1. Introduction
The RIP‐Tag2 mouse is an oncogenic mouse strain with rat insulin promoter‐1 driven viral SV40 large T‐antigen (Tag) expression (Hanahan, 1985) that has been extensively used as a model of endogenous tumorigenesis due to beta‐cell specific expression of the Tag oncogene (Atwood and Shah, 2003). This leads to the development of multiple tumors originating from beta‐cells in a time‐predictable manner. The tumors that develop grow as solid adenomas, and may become invasive or seed off metastases. Studies of the RIP‐Tag2 mouse have shown that induction of angiogenesis (“angiogenic switch”) marks the progression from hyperplasia into tumor outgrowth (Folkman et al., 1989), and that pro‐angiogenic factors like VEGFs and FGFs play a prominent role in this process (Compagni et al., 2000; Inoue et al., 2002).
Shb is an adapter protein participating in intracellular signaling pathways leading to diverse cellular responses such as proliferation, differentiation, cell motility or cell death, depending on what context studied (Anneren et al., 2003). Src, Eps8, PLC‐gamma, Grb2, p85 PI3kinase and c‐Abl (Hagerkvist et al., 2007) have all been shown to interact with Shb's proline‐rich motifs, while FAK associates with the phosphotyrosine‐binding domain, and Shb's SH2‐domain binds activated tyrosine kinase receptors such as the PDGFR, FGFR‐1 and VEGFR‐2 (Holmqvist et al., 2004).
In endothelial cells, Shb has been found to associate with and relay signals from activated FGFR‐1 (Cross et al., 2002) and VEGFR‐2 (Holmqvist et al., 2004). Upon FGF‐2‐stimulation of FGFR‐1, Shb mediates proliferative signals through Shp2‐FRS2‐Grb2‐Sos‐Raf‐Ras activation of MAPK, and cytoskeletal rearrangements in concert with Src and FAK (Cross et al., 2002; Holmqvist et al., 2003; Lu et al., 2002). After VEGF‐A mediated VEGFR‐2 activation, Shb stimulates PI3kinase activity, and interacts with Src and FAK, to promote focal adhesion formation and cell migration (Holmqvist et al., 2004).
We have recently generated an Shb knockout mouse, in which viable Shb null mice are born on a mixed genetic background (FVBN/C57Bl6/129SvJ) but not when the mutant allele was bred onto the C57Bl/6 background (Calounova et al., 2010; Kriz et al., 2007). The inheritance of the Shb mutant allele on the mixed genetic background did not follow Mendelian inheritance due to a transmission ratio distortion showing an overrepresentation of +/− and an underrepresentation of −/− and +/+ pups. In addition, an increased frequency of early embryonic malformations was noted as a consequence of Shb deficiency, further reducing the birth frequency of Shb−/− pups. The immune system also exhibits abnormalities in Shb‐deficient mice with reduced numbers of blood leukocytes (lymphocytes and monocytes) and a TH2‐skewing of the response of peripheral CD4+ lymphocytes to T cell receptor stimulation (Gustafsson et al., 2011). Shb +/− or −/− mice show vascular abnormalities that cause impaired Lewis lung carcinoma or T241 tumor cell growth in vivo (Funa et al., 2009). Since Shb has been shown to mediate important cellular processes in endothelial cells we wanted to assess the characteristics of the heritable RIP‐Tag2 insulinoma in Shb mutant mice, with the aim to increase our understanding of the role of Shb in tumor growth, metastasis and angiogenesis.
2. Materials and methods
2.1. Animals
RIP‐Tag2 (Grant et al., 1991; Hanahan, 1985) mice on C57Bl/6 background were mated with Shb allele (Kriz et al., 2007) mice of mixed background (FVB/N, C57Bl/6, and 129/SvJ strains) and Shb heterozygous offspring were bred to obtain Shb+/+ RIP‐Tag2, Shb+/− RIP‐Tag2 and Shb−/− RIP‐Tag2 mice. The mice were earmarked at 3 weeks of age, and that tissue sample was used for genotyping.
RIP‐Tag2 bearing mice received sucrose 5–10% in their drinking water, beginning at 10 weeks of age, to counteract symptoms of hypoglycemia caused by the developing insulinomas. All animal experiments were approved by the animal ethics committee for Uppsala University (permit number C93/9).
2.2. Tumor burden and lesion frequency
Pancreata from 12 weeks mice were fixed in 4% paraformaldehyde, sucrose infiltrated and frozen. Pancreata were then sectioned in their entirety, and five 7 μm sections every 250 μm (level) were taken and mounted on individual microscope slides. To assess total tumor burden and lesion frequency, one slide/level was stained with hematoxylin–eosin and then scanned using a KonicaMinolta Dimage Scan Dual IV photographic scanner. The pictures of the scanned sections were put together into a sequence, and lesions were identified and numbered. Lesions were estimated to have an elliposoidal shape and lesion size was determined with ImageJ v1.38 (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997–2007.).
2.3. Tumor histology, metastasis and transplantation
Frozen sections were stained as described in Funa et al. (2009). The following primary antibodies were used: anti‐mouse CD31 (MEC13.3 cat# 553370, www.bdbiosciences.com) (1:500), mouse monoclonal [DE‐U‐10] to Desmin cat# ab6322 (Abcam, Cambridge, UK) (1:100), goat anti‐mouse VE‐Cadherin cat# AF1002 (www.RnDSystems.com) (1:200), Cleaved caspase‐3 (Asp175) #9661S (Cell Signaling Technologies, www.cellsignal.com) (1:50), rat anti‐mouse CD68 (AbD Serotec #MCA1957, Dusseldorf, Germany) (1:50). All fluorescent secondary antibodies (Alexa Fluor) were from Invitrogen, Molecular Probes, Eugene, Oregon, USA, and used at 1:500 dilution. For staining of Ki67 positive cells the following antibodies were used: Monoclonal Rat anti‐Mouse Ki67 Antigen, clone TEC3, (#M7249 DakoCytomation, Denmark) (1:600), Biotinylated goat anti‐rat, (# BA‐9400 Vector Laboratories Inc., Burlingame CA, USA) (1:300).
Photos were taken with a Nikon Eclipse TE2000‐U fluorescence inverted microscope, with a Nikon D Eclipse C1 camera and Nikon ACT‐1C for DXM1200C v1.02.10 software, or for confocal microscopy, the Nikon microscope with a C1 confocal unit and Nikon EZ‐C1 v3.90 software. Transmission electron microscopy was performed as described in Funa et al. (2009). Invasiveness was scored in a blinded fashion on morphological criteria as indicated in Supporting Figure 8. For liver metastases, livers from 14‐week‐old wild‐type or Shb +/− RT2 mice (five mice of each genotype) were fixated, sectioned in their entirety and stained at three different levels for SV40‐Tag (BD Bioscience, www.bdbiosciences.com). Metastases were scored based on the criterion corresponding to a cluster of at least five cells staining positive for Tag in their nuclei. Average numbers of liver metastases per section were calculated.
Pancreata from mice (Shb+/+RT and Shb−/−RT) at 8 weeks of age were collagenase digested and 100 early lesions (hyperplastic islets) were transplanted to the left or right renal capsules, respectively, of a wild‐type littermate lacking the RIP‐Tag oncogene. The mouse was kept for an additional 7 weeks after which it was sacrificed and the kidneys retrieved for immunostaining for CD31.
2.4. Blood vessel density
To assess tumor blood vessel density/endothelial cell proportion, slides were chosen so that the analyzed sections were as close as possible to the center of the tumors of interest, and then stained for CD31. Medium‐sized tumors were chosen and one photo that covered most of the tumor was taken with the 10× objective. Vessel density was assessed blinded on photos using a grid overlay, counting percentage of CD31 positive intersections, on 20 tumors from 5 to 6 mice for each genotype.
2.5. Tumor vascular perfusion and vascular leakage
To measure tumor vascular perfusion, 14‐week‐old Shb+/+RT and Shb+/−RT mice were injected intravenously with 50 μg fluorescein tomato lectin (#FL‐1171, Vector Laboratories Inc., Burlingame, CA, USA), and sacrificed after 30 min. Pancreata were collected, parts with tumors excised and frozen in liquid nitrogen. Frozen sections were stained for CD31 and the fraction FITC‐positive CD31 fluorescence was determined on 4–12 fields/mouse
Tumor vascular leakage assay was performed as in Funa et al. (2009), on 14‐week‐old Shb+/+RT and Shb+/−RT mice. The mice were subjected to intravenous injections with FITC‐conjugated dextran (100 mg/kg body weight, Fluka Analytical, fluorescein isothiocyanate dextran 70,000 conjugate, #46945 Sigma–Aldrich). After 2 h animals were sacrificed, pancreata were collected, parts with tumors rapidly excised and frozen in liquid nitrogen. Photos of frozen sections were taken with a fluorescence microscope, 200× magnification, and the number of vascular leakage/field was counted on 5–10 fields/mouse. The area per field corresponds to approx. 0.14 mm2.
2.6. Micro‐CT imaging
Mice were heparinized with 100 IU undiluted heparin (1000 USP U/ml) after receiving 0.02 ml/g body weight of 2.5% 1 g/ml 2,2,2‐tribromoethanol (Sigma–Aldrich) dissolved in 2‐methyl‐2‐butanol (avertin). Once sacrificed, the descending thoracic aorta was cannulated with a 25‐gauge blunt butterfly needle for aortic retrograde perfusion of phosphate‐buffered saline (PBS) containing 4 mg/l papaverin and 1 g/l adenosine for 3 min, 2% paraformaldehyde for 5 min, and PBS flush for 2 min, followed by 0.2 ml/10 g body weight home‐made 20% Bismuth in 5% gelatin at 0.75 ml/min using an automatic, mechanical injector (Zagorchev et al., 2010). After completion of vascular perfusion, the mouse was covered with wet ice for over 30 min to polymerize the contrast agent. The pancreas was then dissected from the body and stored in 2% paraformaldehyde overnight.
The pancreas vasculature was imaged with a high‐resolution micro‐CT imaging system (GE eXplore Locus SP, GE Healthcare) set to a 0.008‐mm effective detector pixel size (Zagorchev et al., 2010). Micro‐CT was operated at 60‐kVp X‐ray tube voltage, 100‐mA tube current, 2960‐milli‐second per frame, 1 × 1 detector binning model, 720 views, and 0.5° increments per view. This acquisition resulted in a set of contiguous axial VFF‐formatted images through each sample.
2.7. Image reconstruction and quantitative image analysis
With the use of Microview Software (GE Healthcare), the raw data was corrected and reconstructed with voxels of dimensions 16 μm × 16 μm × 16 μm to visualize the whole vascular tree. After the above reconstructed micro‐CT data set was transferred to the Advanced Workstation (version 4.4; GE Healthcare), different post‐processing techniques enabled us to extract the vasculature and re‐batch this reconstructed imaging according to the re‐orientated central line along the long axis of the major vessel using modified method (Zhuang et al., 2006) for quantitation. A volume of interest was reconstructed of the whole tumor (130–350 slices depending on size of the tumor). A semi‐automated algorithm was used to extract detailed morphometric data on the diameter, area, numbers of vessels, and distribution of different sizes of vessels by using modified software (ImageJ; National Institutes of Health, Bethesda, MD). Finally, the total area and volume could be summed up for each tumor. The data was expressed as vascular segment numbers, representing the total numbers of vessels of specified diameters, and counted in all reformatted cross‐sections, or relative numbers (in percent) of vessels observed in the specified size range over the total number of vessels for each tumor.
2.8. In vivo anti‐VEGF‐A treatment
Mice were treated with B20‐4.1.1 anti‐mouse VEGF‐A antibody G6‐31 (kindly provided by Genentech Inc., San Francisco, CA, USA) 5 mg/kg bodyweight in phosphate buffer saline (PBS) (1 mg/ml) or a corresponding volume of PBS. The antibody/PBS were administrated intraperitoneally once a week, with the first dose given at 10 weeks of age. This treatment regimen was that recommended by the manufacturer of the antibody. Mice were killed at 12 weeks of age. The pancreata were collected and tumors were isolated by collagenase digestion (Akerblom et al., 2009). Tumors and smaller lesions were handpicked with forceps under a stereomicroscope and transferred to a Petri dish and photographed. The photos were used to determine tumor volume and to classify tumors as red or white as shown in the figure. “Red” or “white” corresponded to tumors that were more than 50% red or white, respectively. Tumors were subsequently transferred to RNA later™ (Qiagen) for gene expression analysis, or arranged in Tissue‐Tek OCT compound (Sakura Finetek, Zoeterwoude, Netherlands) and frozen on a metal cylinder embedded in dry ice for histological evaluation. Lesion volume was determined with ImageJ. Lesions were estimated to have a spheroid shape.
2.9. RNA isolation and quantitative real‐time RT‐PCR
RNA‐later‐fixed tumors were weighed, then disrupted in RNA‐lysis buffer with a glass tissue grinder pestle, briefly sonicated, and homogenized with syringe and needle. Total RNA was prepared by using the RNeasy Plus mini kit (Qiagen, Hilden, Germany) with on‐column DNase digestion with RNase‐Free DNase set (Qiagen), according to the manufacturer's descriptions.
One‐step quantitative real‐time RT‐PCR was performed with QuantiTect™ SYBR® Green RT‐PCR‐kit (Qiagen) on a LightCycler™ real‐time PCR machine (Roche Diagnostics, Mannheim, Germany). For primer sequences please see Supplemental Table 2. Reverse transcription and cycling conditions were performed following the standard recommendations of the kit. Cycle threshold (C T) values were determined with the LightCycler Software v3.5. Gene expression was normalized by subtracting the corresponding β‐actin C T‐value. Statistical comparisons were made on normalized C T‐values.
2.10. Statistics
Means ± SEM are given for the number of observations. Tests for probabilities of chance differences are given in the legends.
3. Results
3.1. Tumor incidence and burden in Shb +/− and −/− mice at 12 weeks
Shb+/+RIP‐Tag2, Shb+/−RIP‐Tag2 and Shb−/−RIP‐Tag2 mice (designated Shb+/+RT, Shb+/−RT and Shb−/−RT), were used in the experiment. Shb mutant refers to Shb+/−RT and/or Shb−/−RT mice. All RT‐positive mice displayed tumors at 12 weeks of age, whereas Shb+/+, Shb+/− and Shb−/− mice did not show any signs of tumorigenesis (data not shown).
Tumor lesion incidence and total burden was studied in 12‐week‐old RT‐mice, a time point at which multiple tumors have arisen. Assessment of the size distribution of the tumor lesions in the +/+, +/− and −/− genotypes (Figure 1A) revealed a reduction in the number of lesions primarily with a size corresponding to early angiogenic tumors, Ø 1.0–1.5 mm (Lopez and Hanahan, 2002), in Shb mutant mice (Shb+/−RT and Shb−/−RT). Shb+/−RT and Shb−/−RT mice both demonstrated a reduced total number of lesions with a diameter >0.5 mm (Figure 1B) and tumor burden (Figure 1C) compared with the +/+ control. The findings suggest that Shb deficiency impairs tumor formation and expansion primarily at a stage corresponding to early angiogenic tumors.
Figure 1.
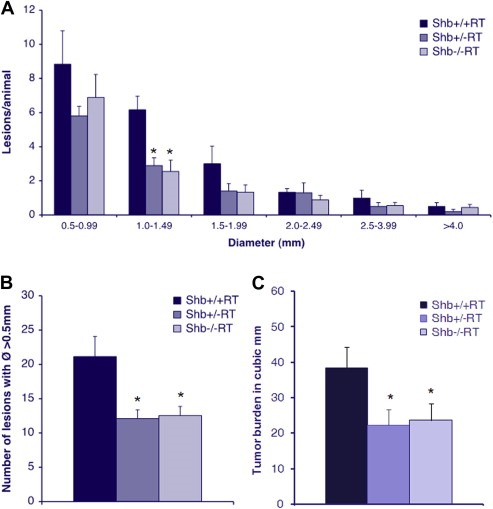
(A) Lesion size distribution in 12‐week Shb+/+RT, Shb+/−RT and Shb−/−RT mice. Number of animals Shb+/+RT n = 6, Shb+/−RT n = 10, Shb−/−RT n = 9, * denotes p‐value < 0.05 Student's t‐test when compared wild‐type control. (B) Number of lesions with a diameter >0.5 mm in 12‐week Shb+/+RT, Shb+/−RT and Shb−/−RT mice. (C) Tumor burden in 12‐week Shb+/+RT, Shb+/−RT and Shb−/−RT mice, measured as total volume of lesions with a diameter >0.5 mm. n = 16–24.
3.2. Microvasculature of Shb +/− and −/− tumors
The reduction in the number of early angiogenic tumors prompted an investigation of the vasculature in Shb+/−RT or Shb−/−RT tumors. Tumors were stained with CD31 to visualize the microvasculature. The proportion endothelial cell area in Shb+/−RT and Shb−/−RT tumors was reduced when compared with the corresponding Shb+/+RT tumors (Figure 2A, B and E), indicating a lower tumor vessel density upon reduced expression of Shb. The difference in endothelial area between Shb−/−RT and Shb+/+RT became less apparent when large tumors were inspected (Figure 2C and D). It is thus clear that the Shb+/−RT and Shb−/−RT phenotypes are indistinguishable with respect to tumor incidence, burden and vascular density. The microvasculature was further analyzed by staining for VE‐cadherin/desmin (Figure 3a–f) and CD31/desmin (Figure 3g–l). In addition to the reduced vascular density in the Shb+/−RT or Shb−/−RT tumors (apparent in the VE‐cadherin panels, Figure 3a vs Figure 3d), the Shb+/−RT VE‐cadherin and CD31 staining patterns were less distinct with a punctate and diffuse appearance (See Supplemental Figure 1 for higher magnification). The wild‐type tumors exhibited staining for the pericyte marker desmin that in part was immediately adjacent to the VE‐cadherin‐ or CD31‐positive (endothelial) cells but not directly overlapping (Figure 3c and i). Consequently, the desmin‐positive cells appeared to form a cover or sheath around the endothelial cells that partly cased these. Co‐localization (yellow) of VE‐cadherin or CD31 and desmin was only detected at the boundaries between these two compartments (Figure 3c and i). In addition, desmin‐positive cells not immediately adjacent to endothelial cells could be detected (Figure 3b and h). The most apparent characteristic of the Shb mutant insulinoma staining pattern besides the indistinct staining of the endothelial markers VE‐cadherin and CD31 was the presence of cellular conglomerates with largely overlapping staining (yellow) for the endothelial markers (VE‐cadherin or CD31) and the pericyte marker (desmin) (Figure 3f and l). Contrary to the wild‐type situation, this staining was not primarily localized to the narrow interface between the endothelial and pericyte compartments. Instead it was detected in larger areas appearing to comprise most of the corresponding cell bodies (Figure 3f and l, exemplified by arrows). The currently observed endothelial‐staining pattern strongly resembles that observed in subcutaneous tumors or normal heart tissue in Shb mutant mice (Funa et al., 2009). Scanning electron microscopy (SEM) of tumor venules revealed in the Shb mutant an irregular endothelium with partly overlapping cells that were organized in a shingle‐like manner (Supplemental Figure 2). The ultrastructure as visualized by transmission electron microscopy (TEM) of the endothelial cells in the Shb mutant insulinomas was also different from that of wild‐type control tumors, with an expanded endothelial cytoplasm (Supplemental Figure 2) and fewer capillaries that exhibited fenestrae. Fenestrations were observed among 16 of 20 wild‐type tumor endothelial cells and 8 of 22 Shb mutant tumor endothelial cells analyzed (p < 0.05 by Chi‐square test). The basal membrane appeared to form invaginations. The tumor vasculature was also analyzed by micro‐CT to visualize arteries/arterioles larger than 16 μm in diameter (Figure 4). Both large (Figure 4A, b) and small (Figure 4A, d) Shb+/−RT insulinomas appeared to possess a less evenly distributed vascular network. Their vascular density tended to be diminished in each of the size intervals (Figure 4B), and when combining the data over all size ranges, the difference became significant (p < 0.001 paired Student's t‐test). However, the relative frequency of tumor blood vessels in the 48–96 μm size interval (Figure 4C) was slightly, but significantly increased, and this effect could have hemodynamic consequences for blood supply to Shb mutant tumors. The function of the Shb+/−RT insulinoma vasculature with respect to perfusion and leakage was also evaluated. Shb+/− tumors showed similar levels of vascular perfusion and leakage as wild‐type tumors (Supplemental Table 1). The observed aberrations in the Shb mutant vascular system as visualized by micro‐CT could at least partly influence vascular perfusion and leakage.
Figure 2.
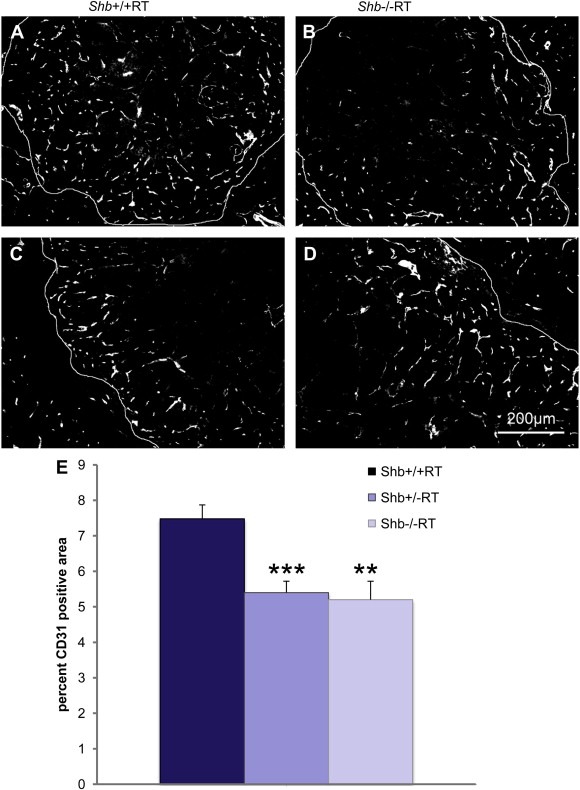
(A–D) CD31 staining of Shb+/+ (A, C) or Shb−/− (B, D) RIP‐Tag2 tumors and tumor vascular density (E). Small (A, B) and large (C, D) tumors are shown at 12 weeks of age. Large tumors, were defined as having a Feret's diameter >2.0 mm or a V > 2.0mm3, and small tumors, were defined as having a Feret's diameter approx. 1–1.5 mm. Original magnification 100×. * indicates p < 0.05 with a Student's t‐test.
Figure 3.
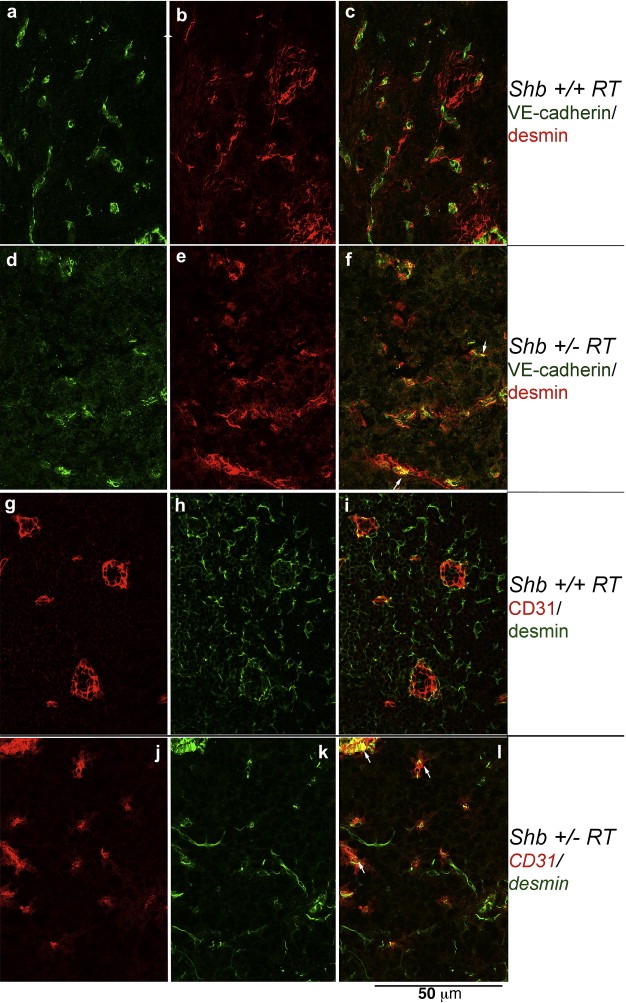
Staining of Shb+/+RT (a–c, g–i) and Shb+/−RT (d–f, j–l) tumors for pericyte (desmin, b‐c, e‐f, h‐i, k‐l) and vascular (CD31 in g, i, j, l and VE‐cadherin in a, c, d, f) markers. Confocal images (0.1 μm slice thickness) are shown. Arrows indicate structures with co‐localization of the vascular and pericyte markers. Original magnification 600×.
Figure 4.
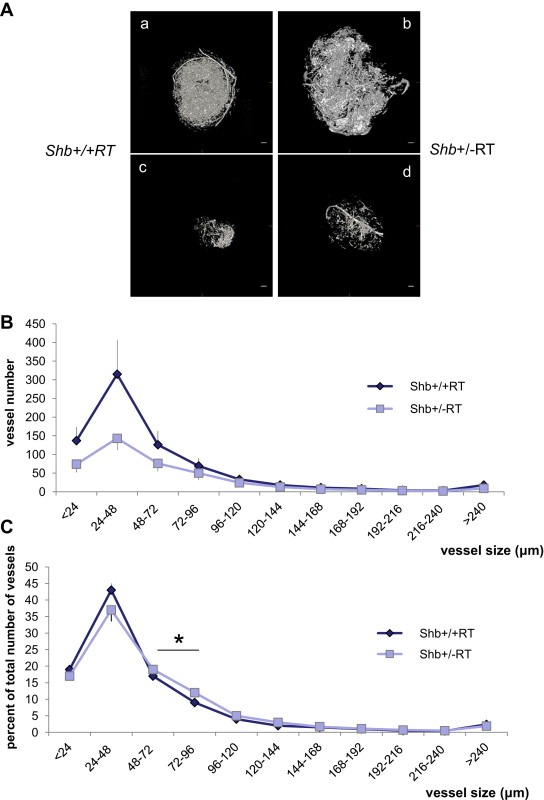
Micro‐CT of RT2 tumors in Shb+/+RT and Shb+/−RT mice. (A) Examples of afferent vascular networks of large (a, b) and small (c, d) tumors of each genotype (a, c for +/+ and b, d for +/−). Scale bar indicates 400 μm. (B) Vascular density in each size range for Shb+/+ (n = 3) and Shb+/− (n = 5) tumors. Means ± SEM are given. And values are vessel diameter in μm. p < 0.001 when comparing the vascular density over all size ranges. (C) Relative size distribution. For each tumor, the percentage vascular density in each size interval was determined in percent of total vascular density for that tumor. Means ± SEM are given. * indicates p < 0.05 when compared with wild‐type control by a paired Student's t‐test.
3.3. Shb−/− tumor cell proliferation and apoptosis
It has been shown that cell proliferation in the RIP‐Tag2 model increases when hyperplasia progresses to early angiogenic tumors, but cell proliferation is not further increased in large tumors. The rate of apoptosis also increases from hyperplasia to early angiogenic tumor, however apoptosis then becomes less frequent in large tumors, making down regulation of apoptosis likely to be the deciding factor necessary for tumor mass expansion at this stage (Naik et al., 1996). To elucidate whether this pattern was altered in Shb−/−RT mice, tumors were stained for Ki67 to assess cell proliferation (Gerdes et al., 1984) and for cleaved caspase‐3 to determine apoptotic rates (Nicholson et al., 1995). Neither cell proliferation nor cell apoptosis differed significantly between Shb+/+RT and Shb−/−RT tumors (Supplemental Figure 3) suggesting that these were not primarily responsible for the decreased tumor incidence in the Shb−/−RT tumors. Another finding suggesting that the angiogenic response was indeed responsible for the differences in tumor vascularization and expansion was that after transplantation of Shb+/+RT and Shb−/−RT insulinomas to wild‐type recipients, the tumor vasculature was comparable in the two groups (Supplemental Figure 4).
3.4. Tumor burden after treatment with a VEGF‐A blocking antibody
To assess the dependence of tumor angiogenesis on VEGF‐A, mice were treated for 2 weeks (week 10–12) with a VEGF‐A blocking antibody after which tumor volume was determined. The protocol was modified in two respects from that in Figure 1. Firstly, the pancreata were collagenase digested by a standard procedure for islet isolation. Retrieved tumors were photographed and their volumes were determined. By implementing this protocol, it became apparent that the tumors could be classified as “red” or “white” (Figure 5). That white tumors indeed were insulinomas and not lymphatic tissue was confirmed by staining for T‐antigen and low expression of lymphoid markers (Supplemental Figure 5). Secondly, the data were collected from Shb+/−RT mice since all the analyses (tumor lesions and burden and vascular density) showed identical effects in the Shb+/− and Shb−/− situations. In addition, it became very difficult to obtain matched Shb−/−RT mice for the antibody treatment regimen due to a low frequency of born Shb−/− mice, an observation in‐line with that previously reported (Kriz et al., 2007). Indeed, the validity of this experimental strategy was further supported by the fact that when the Shb+/−RT and Shb−/−RT data in control experiments were compared, identical results for the two groups were obtained (Supplemental Figure 6).
Figure 5.
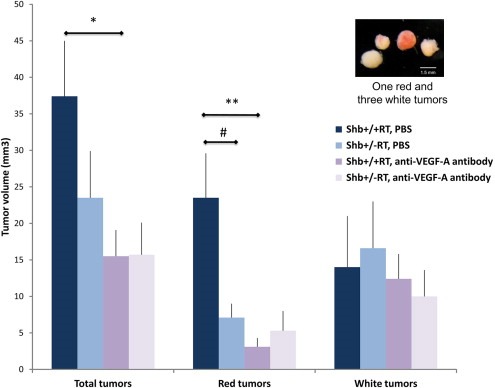
Effect of VEGF‐A blocking treatment on tumor volume. Shb+/+RT or Shb+/−RT mice were treated with PBS (PBS) or VEGF‐A blocking antibody (ab) during weeks 10–12. Tumors were collagenase isolated and classified as “red” or “white” according to the insert. Tumor volumes were estimated and given as means ± SEM. n = 13 for +/+ PBS; n = 14 for +/− PBS; n = 10 for +/+ ab; n = 11 for +/− ab. * p < 0.05 and ** p < 0.01 with Student's t‐test, # p < 0.05 with Mann–Whitney U‐test due to failure of normality test.
Shb+/−RT mice had a total tumor burden that was slightly reduced (Figure 5). This is a finding in‐line with that in Figure 1C. In Shb+/+RT, most of the tumors were red (highly vascularized and/or tumors with extravasated erythrocytes) whereas in the Shb+/−RT group, the “red” fraction of tumors was significantly lower than that of the corresponding wild‐type control. Treatment with VEGF‐A blocking antibody strongly reduced Shb+/+RT tumor burden and this effect was primarily confined to the “red” category of tumors, whereas the Shb mutant mice displayed an identical tumor load as the wild‐type group after subject to this regimen. The “red” tumor volume was barely (less than 5 mm3) detectable after anti‐VEGF‐A treatment in both wild‐type and Shb +/− mice. This corresponded to a major reduction (87%) in “red” tumor burden for the wild‐type mice whereas the effect (25%) in the Shb+/− situation was insignificant. Genotype or anti‐VEGF‐A treatment had no significant influence on the “white” tumor volumes. The tumor vascular morphology (Supplemental Figure 7, A–H) after VEGF‐A blocking treatment showed no obvious differences from that of the non‐treated tumors (Figure 3). The vascular density (CD31‐positive area) showed a trend (p = 0.19) towards a decrease after VEGF‐blocking treatment (Supplemental Figure 7I) in the wild‐type tumors whereas it was comparable to that of the untreated tumors in Shb+/−RT, providing support for the notion that the vascular density data agree roughly with the tumor burden data under these conditions. The data suggest that Shb deficiency impairs VEGF‐A dependent tumor angiogenesis and that a certain fraction of the wild‐type and Shb mutant tumors possess VEGF‐A independent strategies to promote vascularization and expansion, a process that is relatively increased in Shb mutant RIP‐Tag2 mice.
3.5. Tumor invasiveness, liver metastasis and intestinal lesions
Tumor invasiveness was assessed by morphological criteria on HE‐stained pancreatic sections in which the numbers of tumors infiltrating the surrounding exocrine tissue was scored (Supplemental Figure 8). By such analysis, 34% of Shb+/−RT showed invasive features whereas the corresponding figure for the wild‐type was 16%. However, since tumor incidence was reduced in the Shb+/− situation, the actual numbers of infiltrating tumors were very similar in both groups (Figure 6A). It is thus concluded that tumor invasiveness is regulated by an Shb‐independent process.
Figure 6.
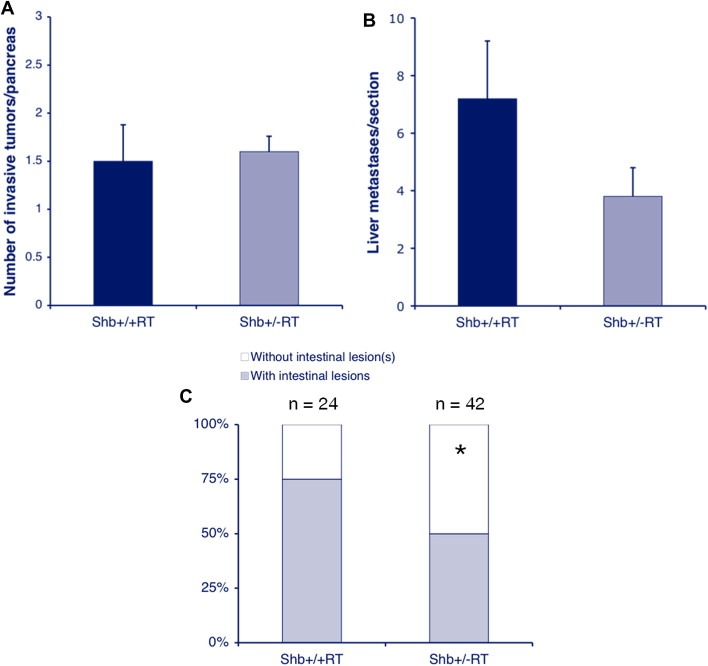
Tumor invasiveness (A), liver metastasis (B) and intestinal lesions (C) of wild‐type and Shb+/−RT2 tumors. Means ± SEM for invasiveness and liver metastasis and number of mice with intestinal lesions are given. For invasiveness, values are from 6 to 10 mice and for liver metastasis 15 sections from 5 mice of each genotype were analyzed. * indicates p < 0.05 with chi‐square test.
In addition, the degree of liver metastasis was evaluated (Figure 6B). Shb+/−RT mice showed a number of liver metastases that tended to be lower than that of the wild‐type controls (p = 0.15). Consequently, the data argue against the notion that the vascular abnormalities in the Shb mutant situation would promote tumor metastasis.
The occurrence of intestinal lesions has previously been reported as a characteristic of the RIP‐Tag2 mouse strain (Grant et al., 1991) and consequently we decided to determine the frequency of intestinal lesions as a consequence of Shb deficiency. These intestinal lesions (Supplemental Figure 8) are likely to be primary tumors arising at sites outside the pancreas (Grant et al., 1991). The relative frequency of Shb+/−RT mice with intestinal lesions was decreased compared with that in Shb+/+RT mice (Figure 6C) suggesting a similar degree of Shb‐dependency for intestinal lesions as for insulinoma incidence.
3.6. Tumor gene expression of microvascular markers
Tumors were analyzed by real‐time RT‐PCR for the expression of genes of relevance to microvascular function (Table 1). Comparing Shb mutant tumors with wild‐type revealed reduced expression of the angiogenic factor VEGF‐A. All other genes were found to be unchanged in the Shb mutant situation. The reduction of VEGF‐A gene expression is in further agreement with VEGF‐A independent angiogenesis predominating in the Shb mutant situation. We also examined E‐cadherin expression but were unable to detect any difference in its expression between wild‐type and Shb‐deficient tumors (results not shown). One observation was that with the exception of VEGF‐A, individual tumors showed a considerable variation in their expression of the microvascular markers. Such a variation in gene expression was investigated further by assessing the signaling profiles for different tumors (Supplemental Figure 9). Each individual tumor showed a unique signal‐transduction signature with respect to Akt and ERK phosphorylation/activation that did not correlate with the genotype (Supplemental Figure 9), further reinforcing the view of tumor heterogeneity with respect to signaling characteristics and gene expression profiles. HIF‐1A protein levels (results not shown) and gene expression levels of the ischemic sensor PGC‐1 alpha (Table 1) (Shoag and Arany, 2010) were unchanged, making differences in tumor oxygenation unlikely causes for the effects on VEGF‐A gene expression or the differences in signaling characteristics. Taken together, the data support a loss in Shb mutant tumors of the default VEGF‐A dependent angiogenic pathway that then becomes compensated for by diverse and tumor‐unique means.
Table 1.
Gene expression of vascular markers in wild‐type and Shb mutant tumors
| Shb+/+RT (Ct gene–Ct actin) | Shb+/−RT (Ct gene–Ct actin) | Shb+/− expression relative Shb+/+ (%) | |
|---|---|---|---|
| CD31 | 7.10 ± 0.25 | 7.03 ± 0.23 | 105 |
| VEGFR‐2 | 6.88 ± 0.33 | 6.73 ± 0.25 | 111 |
| VEGFR‐3 | 9.45 ± 0.15 | 9.94 ± 0.50 | 71 |
| VE‐cadherin | 6.70 ± 0.28 | 6.89 ± 0.25 | 88 |
| NG‐2 | 7.25 ± 0.59 | 6.76 ± 0.16 | 140 |
| α‐Smooth muscle actin | 6.50 ± 0.27 | 6.75 ± 0.33 | 119 |
| Desmin | 10.24 ± 0.40 | 10.73 ± 0.37 | 140 |
| VEGF‐A | 4.40 ± 0.17 | 6.27 ± 0.56* | 27 |
| VEGF‐B | 7.25 ± 0.09 | 7.15 ± 0.24 | 107 |
| VEGF‐C | 9.02 ± 0.24 | 8.42 ± 0.55 | 152 |
| VEGF‐D | 9.78 ± 0.45 | 9.88 ± 0.35 | 93 |
| PlGF | 10.68 ± 0.41 | 11.13 ± 0.41 | 73 |
| FGF‐2 | 11.22 ± 0.31 | 11.84 ± 0.69 | 66 |
| Angiopoietin‐1 | 11.52 ± 0.23 | 11.17 ± 0.33 | 127 |
| Angiopoietin‐2 | 8.80 ± 0.29 | 9.29 ± 0.47 | 71 |
| Tie‐1 | 8.92 ± 0.10 | 9.35 ± 0.36 | 74 |
| Tie‐2 | 8.88 ± 0.25 | 9.01±0.35 | 91 |
| PDGF‐AA | 8.07 ± 0.13 | 8.34 ± 0.23 | 84 |
| PDGF‐BB | 11.65 ± 0.30 | 12.27 ± 0.35 | 65 |
| PDGF‐CC | 8.93 ± 0.21 | 9.41 ± 0.20 | 72 |
| TGF‐β1 | 6.20 ± 0.27 | 5.73 ± 0.21 | 139 |
| TGF‐β2 | 11.48 ± 0.32 | 11.52 ± 0.36 | 97 |
| TGF‐β3 | 8.60 ± ±0.33 | 8.35 ± 0.23 | 119 |
| BMP‐2 | 10.08 ± 0.73 | 10.96 ± 0.51 | 54 |
| BMP‐4 | 11.52 ± 0.41 | 11.78 ± 0.16 | 84 |
| Activin‐A | 9.08 ± 0.42 | 9.79 ± 0.21 | 61 |
| PGC‐1‐α | 13.5 ± 0.71 | 12.95 ± 1.07 | 146 |
Values are mean Ct after normalization for beta‐actin ±SEM for 4–7 observations. Relative changes (given in percent) in expression were calculated by the formula 2 exp (ΔCt+/+ − Ct+/−) (alternatively expressed as 2(ΔCt+/+ − Ct+/−).
Significant differences have been highlighted with bold numbers.
indicates p < 0.05 with a Student's t‐test.
4. Discussion
The present study demonstrates the partial dependence of RT2 tumor incidence and expansion on the Shb adapter protein and that the predominant mechanism for this effect appears to be via angiogenic restriction. RT2 tumors are highly vascularized, and consequently inhibition of angiogenesis and/or VEGF signaling will reduce tumor incidence and/or expansion (Bergers et al., 1999, 2000; Casanovas et al., 2005). The relevance of angiogenic restriction in the case of Shb deficiency is inferred from the finding that Shb deficiency does not affect tumor cell proliferation or apoptosis, but that it causes microvascular abnormalities and that these were normalized when tumors from mutant mice were transplantated to wild‐type recipient hosts. Shb deficiency appears to primarily reduce VEGF‐A dependent tumor angiogenesis since treatment with a VEGF‐A blocking antibody had no effect on insulinoma expansion in Shb mutant mice. The current use of an inheritable tumor model provides increased support for the notion that Shb is important for tumor angiogenesis. Another observation is that loss of one Shb allele is sufficient to impose the full restrictive effect of Shb deficiency on tumor angiogenesis. We previously noted reduced tumor cell (T241 and Lewis lung carcinoma) growth in Shb+/− mice (Funa et al., 2009).
The findings also suggest tumor heterogeneity with respect to their dependence on VEGF‐A. This can be demonstrated by the “white” versus “red” tumor phenotype in which blockage of VEGF‐A signaling reduced expansion of the “red” tumor phenotype whereas it had no effect on the growth of the “white” tumors. The data suggest that the most common tumor phenotype is to grow as a “red” tumor in the presence of abundant VEGF‐A supply but in case VEGF‐A angiogenesis is impaired (VEGF‐A blockage or Shb deficiency) the “white” tumor phenotype can still expand, thus explaining resistance to VEGF‐A blocking treatment. In a recent study in which a VEGFR‐2 inhibitory antibody was administrated, it was noted that the suppressive effects were transient and that resistance eventually developed in the VEGFR‐2 blocked tumors by a hypoxia‐induced expression of other angiogenic factors (Casanovas et al., 2005). Our data confirm those observations and add the notion that the “white” tumor phenotype is always present but under normal conditions only constitutes a minority of the tumors. Another sign of heterogeneity among RT2 tumors can be noted in their ability to become invasive and seed off metastases. In these instances Shb deficiency was without an effect. Invasiveness has been previously noted in subsets of tumors and has been linked to low expression to E‐cadherin and other cell adhesion markers (Chun and Hanahan, 2010; Hakansson et al., 2005; Herzig et al., 2007). We thus examined E‐cadherin expression but were unable to detect any difference in its expression between wild‐type and Shb‐deficient tumors. This suggests that invasiveness is a trait independent of Shb expression. However, since invasiveness was not reduced as a consequence of Shb deficiency despite a reduction in total tumor incidence, this indicates that the tumors that grow in the absence of Shb contain those that show invasive characteristics. The number of intestinal tumor lesions was reduced in Shb‐deficient mice to a similar degree as those in the pancreas and it seems plausible that this reflects the same process of angiogenic restriction as that in the pancreas. Intestinal lesions are likely to be primary tumors occurring due to expression of the oncogene in intestinal endocrine cells and not metastases from the pancreas (Grant et al., 1991).
We currently observe a vascular phenotype in Shb‐deficient tumors that consists of diminished and irregular vascular supply, lower microvascular density, diffuse and punctate endothelial staining for CD31 and VE‐cadherin combined with ultrastructural alterations. The irregularities clearly complicate the vascular phenotype in that it will have effects on vascular perifusion and leakage. This can be inferred from the relative increase in the fraction of tumor vessels with a diameter of 48–96 μm, since hemodynamic laws predict that an increased vessel diameter allows augmented blood flow. Thus, the absence of effects on perifusion and leakage in the Shb mutant situation is non‐informative since there might be microvascular normalization that cannot be detected functionally due to aberrations in the arteriolar supply. The punctate and diffuse staining of endothelial cells in RT2 insulinomas resembles the Shb knockout mouse microvascular phenotype showing endothelial cells with an abnormal morphology, reduced VEGF‐A stimulated vascular permeability and impaired angiogenesis (Funa et al., 2009). An abnormal cytoskeleton with increased stress fibers was noted that could explain the cytoplasmic extensions observed, and these changes could reflect aberrant VEGFR‐2 signaling (Funa et al., 2009). In addition, we currently observe cells that co‐stain for endothelial and pericyte markers. The most likely explanation for this is the structural alterations of the endothelial cells that will allow cytoplasmic interdigitation with adjacent pericytes forming structures that appear to co‐express endothelial and pericyte markers. Endothelial–mesenchymal transition has been described (Medici et al., 2010) and relates to the TGF‐β system but our gene expression analysis revealed no differences in the expression of relevant factors or receptors. In addition, the Shb mutation reduces the number of tumor endothelial fenestrations and causes the endothelial cells in venules to grow in an overlapping manner. Taken together it is suggested that the endothelial abnormalities in the Shb knockout mice will render a dysfunctional vasculature that reduces tumor growth and is not particularly permissive for tumor metastasis. The latter finding is of relevance since treatment with VEGFR‐2 blocking antibody increased RIP‐Tag2 liver metastasis (Paez‐Ribes et al., 2009), suggesting that Shb dependent angiogenic restriction, albeit VEGF‐A dependent, confers endothelial aberrations that do not promote metastasis.
Shb has also been shown to affect cell proliferation (Anneren, 2002; Cross et al., 2002) and apoptosis (Davoodpour et al., 2007; Funa et al., 2008; Karlsson and Welsh, 1996) and modulation of these processes due to the absence of Shb could potentially participate in achieving the reduced tumor frequency presently observed. In addition, islets isolated from Shb knockout mice display reduced glucose‐stimulated insulin secretion (Akerblom et al., 2009) and beta‐cell death in response to cytokines (Mokhtari et al., 2009). Determination of DNA synthesis and rates of apoptosis revealed no significant differences between wild‐type and knockout, arguing against the possibility that these processes are major primary determinants in the Shb tumor response.
Gene expression analysis reveals a reduction in VEGF‐A mRNA levels in Shb mutant tumors. Since the gene expression levels of the sensor of ischemia PGC‐1 alpha (Shoag and Arany, 2010) and HIF‐1A protein levels were unchanged, differences in tumor oxygenation seem not to be a likely cause for the effects on the angiogenic growth factor. Islets of Langerhans from Shb‐deficient mice also displayed reduced VEGF‐A gene expression (Akerblom et al., 2009), suggesting that this primarily is a consequence of the altered gene expression profile that absence of Shb causes. VEGF‐A expression is dependent on the local presence of numerous growth factors (Ferrara, 2004), making such alterations more likely explanations to the reduction in VEGF‐A. The reduced VEGF‐A gene expression further supports the notion that in Shb‐deficient RIP‐Tag2 mice VEGF‐A independent mechanisms for angiogenesis predominate.
In summary, we have observed that Shb deficiency restricts tumor expansion in an inheritable tumor model through reduced angiogenesis. RT2 insulinomas are heterogeneous and a few tumors escape this angiogenic restriction. Elucidation of the mechanisms responsible for this effect is of future interest for understanding tumor resistance to anti‐angiogenic therapy. Since Shb deficiency reduced tumor incidence and burden without causing a concomitant increase in tumor metastasis, it is conceivable that interference with Shb signaling may provide novels means for future treatment of cancer.
Author contribution
BÅ, GZ, ZZ, MS and MW conceived and designed the experiments. BÅ, GZ, ZZ and GC performed the experiments. BÅ, GZ, ZZ, MS and MW analyzed the data. BÅ and MW wrote the paper. All authors reviewed the paper.
Competing interests
The authors declare that no competing interests exist.
Supporting information
The following are the Supplementary material related to this article
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Acknowledgements
We are grateful to Dr. Kristian Pietras, LICR, Karolinska Institutet, Stockholm, Sweden, for providing the RIP‐Tag2 breeding mice, to Anders Ahlander and Marianne Ljungkvist for expert technical assistance with electron microscopy, and we thank Dr. Olof Åkerblom for the usage of the KonicaMinolta photographic scanner. Discussions with Drs. J. Arbiser, L. Claesson‐Welsh and K. Pietras are also greatly appreciated. The VEGF‐A blocking antibody (B20‐4.1.1 anti‐VEGF mAB) was kindly provided by Genentech. The work was supported by the Swedish Cancer Fund (100494), Swedish Research Council (68X‐10822), Swedish Diabetes Association (2009‐006), Wallenberg Foundation and Family Ernfors Fund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary material 1.
1.1.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molonc.2012.01.006
Åkerblom Björn, Zang Guangxiang, Zhuang Zhen W., Calounova Gabriela, Simons Michael, Welsh Michael, (2012), Heterogeneity among RIP‐Tag2 insulinomas allows vascular endothelial growth factor‐A independent tumor expansion as revealed by studies in Shb mutant mice: Implications for tumor angiogenesis, Molecular Oncology, 6, doi: 10.1016/j.molonc.2012.01.006.
References
- Akerblom, B. , Barg, S. , Calounova, G. , Mokhtari, D. , Jansson, L. , Welsh, M. , 2009. Impaired glucose homeostasis in Shb−/− mice. J. Endocrinol.. [DOI] [PubMed] [Google Scholar]
- Anneren, C. , 2002. Dual role of the tyrosine kinase GTK and the adaptor protein SHB in beta-cell growth: enhanced beta-cell replication after 60% pancreatectomy and increased sensitivity to streptozotocin. J. Endocrinol.. 172, 145–153. [DOI] [PubMed] [Google Scholar]
- Anneren, C. , Lindholm, C.K. , Kriz, V. , Welsh, M. , 2003. The FRK/RAK-SHB signaling cascade: a versatile signal-transduction pathway that regulates cell survival, differentiation and proliferation. Curr. Mol. Med.. 3, 313–324. [DOI] [PubMed] [Google Scholar]
- Atwood, W. , Shah, K. , 2003. Polyomaviruses ENCYCLOPEDIA Of LIFE SCIENCES Copyright © 2003 John Wiley & Sons, Ltd; 10.1038/npg.els.0001082 http://www.els.net [Google Scholar]
- Bergers, G. , Brekken, R. , McMahon, G. , Vu, T.H. , Itoh, T. , Tamaki, K. , Tanzawa, K. , Thorpe, P. , Itohara, S. , Werb, Z. , Hanahan, D. , 2000. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol.. 2, 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers, G. , Javaherian, K. , Lo, K.M. , Folkman, J. , Hanahan, D. , 1999. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science. 284, 808–812. [DOI] [PubMed] [Google Scholar]
- Calounova, G. , Livera, G. , Zhang, X.Q. , Liu, K. , Gosden, R.G. , Welsh, M. , 2010. The Src homology 2 domain-containing adapter protein B (SHB) regulates mouse oocyte maturation. PLoS One. 5, e11155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanovas, O. , Hicklin, D.J. , Bergers, G. , Hanahan, D. , 2005. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 8, 299–309. [DOI] [PubMed] [Google Scholar]
- Chun, M.G. , Hanahan, D. , 2010. Genetic deletion of the desmosomal component desmoplakin promotes tumor microinvasion in a mouse model of pancreatic neuroendocrine carcinogenesis. PLoS Genet.. 6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagni, A. , Wilgenbus, P. , Impagnatiello, M.A. , Cotten, M. , Christofori, G. , 2000. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res.. 60, 7163–7169. [PubMed] [Google Scholar]
- Cross, M.J. , Lu, L. , Magnusson, P. , Nyqvist, D. , Holmqvist, K. , Welsh, M. , Claesson-Welsh, L. , 2002. The Shb adaptor protein binds to tyrosine 766 in the FGFR-1 and regulates the Ras/MEK/MAPK pathway via FRS2 phosphorylation in endothelial cells. Mol. Biol. Cell. 13, 2881–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoodpour, P. , Landstrom, M. , Welsh, M. , 2007. Reduced tumor growth in vivo and increased c-Abl activity in PC3 prostate cancer cells overexpressing the Shb adapter protein. BMC Cancer. 7, 161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara, N. , 2004. Vascular endothelial growth factor: basic science and clinical progress. Endocr. Rev.. 25, 581–611. [DOI] [PubMed] [Google Scholar]
- Folkman, J. , Watson, K. , Ingber, D. , Hanahan, D. , 1989. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 339, 58–61. [DOI] [PubMed] [Google Scholar]
- Funa, N.S. , Kriz, V. , Zang, G. , Calounova, G. , Akerblom, B. , Mares, J. , Larsson, E. , Sun, Y. , Betsholtz, C. , Welsh, M. , 2009. Dysfunctional microvasculature as a consequence of Shb gene inactivation causes impaired tumor growth. Cancer Res.. 69, 2141–2148. [DOI] [PubMed] [Google Scholar]
- Funa, N.S. , Reddy, K. , Bhandarkar, S. , Kurenova, E.V. , Yang, L. , Cance, W.G. , Welsh, M. , Arbiser, J.L. , 2008. Shb gene knockdown increases the susceptibility of SVR endothelial tumor cells to apoptotic stimuli in vitro and in vivo. J. Invest. Dermatol.. 128, 710–716. [DOI] [PubMed] [Google Scholar]
- Gerdes, J. , Lemke, H. , Baisch, H. , Wacker, H.H. , Schwab, U. , Stein, H. , 1984. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol.. 133, 1710–1715. [PubMed] [Google Scholar]
- Grant, S.G. , Seidman, I. , Hanahan, D. , Bautch, V.L. , 1991. Early invasiveness characterizes metastatic carcinoid tumors in transgenic mice. Cancer Res.. 51, 4917–4923. [PubMed] [Google Scholar]
- Gustafsson, K. , Calounova, G. , Hjelm, F. , Kriz, V. , Heyman, B. , Gronvik, K.O. , Mostoslavsky, G. , Welsh, M. , 2011. Shb deficient mice display an augmented TH2 response in peripheral CD4+ T cells. BMC Immunol.. 12, 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerkvist, R. , Mokhtari, D. , Lindholm, C. , Farnebo, F. , Mostoslavsky, G. , Mulligan, R.C. , Welsh, N. , Welsh, M. , 2007. Consequences of Shb and c-Abl interactions for cell death in response to various stress stimuli. Exp. Cell Res.. 313, 284–291. [DOI] [PubMed] [Google Scholar]
- Hakansson, J. , Xian, X. , He, L. , Stahlberg, A. , Nelander, S. , Samuelsson, T. , Kubista, M. , Semb, H. , 2005. Neural cell adhesion molecule-deficient beta-cell tumorigenesis results in diminished extracellular matrix molecule expression and tumour cell-matrix adhesion. Tumour Biol.. 26, 103–112. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , 1985. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 315, 115–122. [DOI] [PubMed] [Google Scholar]
- Herzig, M. , Savarese, F. , Novatchkova, M. , Semb, H. , Christofori, G. , 2007. Tumor progression induced by the loss of E-cadherin independent of beta-catenin/Tcf-mediated Wnt signaling. Oncogene. 26, 2290–2298. [DOI] [PubMed] [Google Scholar]
- Holmqvist, K. , Cross, M. , Riley, D. , Welsh, M. , 2003. The Shb adaptor protein causes Src-dependent cell spreading and activation of focal adhesion kinase in murine brain endothelial cells. Cell Signal.. 15, 171–179. [DOI] [PubMed] [Google Scholar]
- Holmqvist, K. , Cross, M.J. , Rolny, C. , Hagerkvist, R. , Rahimi, N. , Matsumoto, T. , Claesson-Welsh, L. , Welsh, M. , 2004. The adaptor protein Shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J. Biol. Chem.. 279, 22267–22275. [DOI] [PubMed] [Google Scholar]
- Inoue, M. , Hager, J.H. , Ferrara, N. , Gerber, H.P. , Hanahan, D. , 2002. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell. 1, 193–202. [DOI] [PubMed] [Google Scholar]
- Karlsson, T. , Welsh, M. , 1996. Apoptosis of NIH3T3 cells overexpressing the Src homology 2 domain protein Shb. Oncogene. 13, 955–961. [PubMed] [Google Scholar]
- Kriz, V. , Mares, J. , Wentzel, P. , Funa, N.S. , Calounova, G. , Zhang, X.Q. , Forsberg-Nilsson, K. , Forsberg, M. , Welsh, M. , 2007. Shb null allele is inherited with a transmission ratio distortion and causes reduced viability in utero. Dev. Dyn.. 236, 2485–2492. [DOI] [PubMed] [Google Scholar]
- Lopez, T. , Hanahan, D. , 2002. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 1, 339–353. [DOI] [PubMed] [Google Scholar]
- Lu, L. , Holmqvist, K. , Cross, M. , Welsh, M. , 2002. Role of the Src homology 2 domain-containing protein Shb in murine brain endothelial cell proliferation and differentiation. Cell Growth Differ.. 13, 141–148. [PubMed] [Google Scholar]
- Medici, D. , Shore, E.M. , Lounev, V.Y. , Kaplan, F.S. , Kalluri, R. , Olsen, B.R. , 2010. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med.. 16, 1400–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokhtari, D. , Kerblom, B. , Mehmeti, I. , Wang, X. , Funa, N.S. , Olerud, J. , Lenzen, S. , Welsh, N. , Welsh, M. , 2009. Increased Hsp70 expression attenuates cytokine-induced cell death in islets of Langerhans from Shb knockout mice. Biochem. Biophys. Res. Commun.. [DOI] [PubMed] [Google Scholar]
- Naik, P. , Karrim, J. , Hanahan, D. , 1996. The rise and fall of apoptosis during multistage tumorigenesis: down-modulation contributes to tumor progression from angiogenic progenitors. Genes Dev.. 10, 2105–2116. [DOI] [PubMed] [Google Scholar]
- Nicholson, D.W. , Ali, A. , Thornberry, N.A. , Vaillancourt, J.P. , Ding, C.K. , Gallant, M. , Gareau, Y. , Griffin, P.R. , Labelle, M. , Lazebnik, Y.A. , 1995. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 376, 37–43. [DOI] [PubMed] [Google Scholar]
- Paez-Ribes, M. , Allen, E. , Hudock, J. , Takeda, T. , Okuyama, H. , Vinals, F. , Inoue, M. , Bergers, G. , Hanahan, D. , Casanovas, O. , 2009. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 15, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoag, J. , Arany, Z. , 2010. Regulation of hypoxia-inducible genes by PGC-1 alpha. Arterioscler. Thromb. Vasc. Biol.. 30, 662–666. [DOI] [PubMed] [Google Scholar]
- Zagorchev, L. , Oses, P. , Zhuang, Z.W. , Moodie, K. , Mulligan-Kehoe, M.J. , Simons, M. , Couffinhal, T. , 2010. Micro computed tomography for vascular exploration. J. Angiogenes Res.. 2, 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang, Z.W. , Gao, L. , Murakami, M. , Pearlman, J.D. , Sackett, T.J. , Simons, M. , de Muinck, E.D. , 2006. Arteriogenesis: noninvasive quantification with multi-detector row CT angiography and three-dimensional volume rendering in rodents. Radiology. 240, 698–707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the Supplementary material related to this article
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data