

Abstract
Background
The polyunsaturated, ω-3 fatty acid, docosahexaenoic acid (DHA), claims diverse cytoprotective potentials, although via largely undefined triggers. Thus, we currently first tested the ability of DHA to ameliorate valproate (VPA)-evoked hepatotoxicity, to modulate its anticonvulsant effects, then sought the cellular and molecular basis of such actions. Lastly, we also verified whether DHA may kinetically alter plasma levels/clearance rate of VPA.
Methods and Results
VPA (500 mg/kg orally for 14 days in rats) evoked prominent hepatotoxicity that appeared as a marked rise (2- to 4-fold) in serum hepatic enzymes (γ-glutamyl transferase [γ-GT], alanine aminotransferase [ALT], and alkaline phosphatase [ALP]), increased hepatic lipid peroxide (LPO) and tumor necrosis factor-alpha (TNFα) levels, as well as myeloperoxidase (MPO) activity (3- to 5-fold), lowering of serum albumin (40 %), and depletion of liver reduced-glutathione (GSH, 35 %). Likewise, histopathologic examination revealed hepatocellular degeneration, replacement by inflammatory cells, focal pericentral necrosis, and micro/macrovesicular steatosis. Concurrent treatment with DHA (250 mg/kg) markedly blunted the elevated levels of liver enzymes, lipid peroxides, TNFα, and MPO activity, while raising serum albumin and hepatic GSH levels. DHA also alleviated most of the cytologic insults linked to VPA. Besides, in a pentylenetetrazole (PTZ) mouse convulsion model, DHA (250 mg/kg) markedly increased the latency in convulsion evoked by VPA, beyond their individual responses. Lastly, pharmacokinetic studies revealed that joint DHA administration did not alter serum VPA concentrations.
Conclusions
DHA substantially ameliorated liver injury induced by VPA, while also markedly boosted its pharmacologic effects. DHA manipulated definite cellular machinery to curb liver oxidative stress and inflammation, without affecting VPA plasma levels. Collectively, these protective and synergy profiles for DHA propose a superior VPA-drug combination regimen.
Introduction
An increasing emphasis is being placed on the capacity of dietary supplements to modulate host response to disease, injury, infection, and adverse drug reactions [1–3]. It is estimated that drug-induced adverse reactions account for at least 5–6 % of hospital admissions [4]. Valproate (VPA) is a widely prescribed fatty acid (FA) that has served as a mainstay in the management of epileptic seizures, bipolar and schizoaffective disorders, social phobias, and neuropathic pain [5]. Despite its clinical benefits, VPA has also been a hallmark representative of drug-induced adverse reactions. In particular, patients receiving VPA chronically may well develop hemorrhagic pancreatitis, bone marrow suppression and, more frequently, hepatic injury [6]. Thus, in up to 44 % of patients, chronic dosing with VPA elevates serum liver enzymes and lipid peroxidation during the first months of therapy. Another typical clinical finding of VPA intoxication was the development of fatty liver as microvesicular steatosis in 80 % of patients [7]. Moreover, animal models of VPA toxicity clearly show disruption of FA metabolism, along with accumulation of liver lipid content within 2–4 hours after VPA administration [8]. Histopathologic and biochemical studies also revealed that VPA evokes hepatic necrosis, apoptosis, and oxidative stress [9, 10]. However, VPA toxicity that can lead to death has also been reported. The basis of such paradoxical subacute and idiosyncratic VPA toxicity has remained largely enigmatic [11].
At the molecular level, multiple lines of evidence suggest that hepatic accumulation of 4-en-VPA and its β-oxidation products triggers a cascade of reactions that culminates in hepatic injury. Some such reactions involve lipid peroxidation and glutathione (GSH) depletion [12, 13]. Conceivably, therefore, a big need arises to seek avenues that could either alleviate VPA-induced hepatic injury or reduce its dose down to a safer level, thus possibly improving its overall therapeutic index. Thus far, diverse concepts have been adopted, which focused merely on lessening oxidative stress or disrupted mitochondrial fatty-acyl β-oxidation [14, 15]. Conversely, no attempts have been made to boost the pharmacologic efficacy of VPA so as to reduce its toxicity, while also augmenting its therapeutic efficacy. Docosahexaenoic acid (DHA) is a cold-water-fish-oil-derived omega-3 FA that has demonstrated numerous health benefits against malignant, inflammatory, proliferative, and vascular diseases [16]. Furthermore, we recently demonstrated that DHA can reverse a vicious, fatal, cisplatin-induced nephrotoxicity in rats by ablating oxidative stress and suppressing cytokine-mediated inflammation [17]. As far as central effects are concerned; DHA was effectively used to treat neuronal hyperexcitability models in animals and some neurological disorders in humans [18, 19]. Therefore, we currently envisaged that such responses, along with established hypolipidemic effects elicited mostly at the liver level [20], could make DHA supplementation a superb candidate to blunt toxicity and confer therapeutic synergy with VPA.
Accordingly, this study was marshaled to investigate whether, and how, DHA may abate VPA-induced liver toxicity. To accomplish this, we monitored levels of hepatocellular oxidative stress, inflammatory cytokines, and markers for hepatic integrity/function and for neutrophil infiltration. We further substantiated these results with histopathologic investigation to figure out relevant hepatic subcellular changes. On the other hand, the possibility of pharmacologic synergy with VPA was explored in a pentylenetetrazole (PTZ) mouse convulsion model. Lastly, to verify any role for DHA via kinetic interaction (clearance of VPA), we measured plasma concentrations of VPA in the presence and absence of DHA.
Materials
Drugs and Chemicals
Sodium valproate, a white pure powder, was a gift from Sanofi-synthelabo, Cairo, Egypt, and was dissolved in distilled water. DHA was purchased from Healthspan Co., UK, as capsules; each provides 100 mg of pure DHA. DHA was diluted, as needed, in corn oil, with equivalent amounts of oil given to all animals and the control group. All other chemicals used were of analytical grade, and were obtained from Sigma Aldrich Chemical Co., St. Louis, MO, USA. Kits for reduced GSH, malondialdehyde and γ-glutamyl transferase (γ-GT) were obtained from Bio-Diagnostic, Cairo, Egypt. Kits for alkaline phosphatase (ALP), alanine aminotransferase (ALT) and albumin were obtained from ABC-Diagnostics, Cairo, Egypt. A myeloperoxidase kit was purchased from Northwest Co. (Canada) and a TNFα kit was from DRG Co. (USA). VPA assay ELISA kit was obtained from Dade Behring, Atterbury, Milton Keynes, UK.
Animals Studies
Adult male Sprague–Dawley rats weighing 200–250 g were used in liver toxicity study experiments. Male albino mice weighing 20–25 g were used for PTZ-epilepsy model experiments. All animals were maintained under standard conditions of temperature (30 °C), with a regular 12-hour light/12-hour dark cycle, and allowed free access to standard laboratory food and water. The dose used for DHA, as well as time courses used in this study were in the same range and scope as those of other studies that utilized the same models. This strategy was further confirmed after appropriate preliminary experiments. All animal care and experimental procedures were approved by the Animal Ethics Committee of Mansoura University, Mansoura, Egypt (MUEC-8-91), which is in accordance with the Principles of Laboratory Animals Care (NIH publication No. 85-23, revised 1985).
Rat Liver Toxicity Studies
Experimental Design
Different animal groups, of 6–8 rats each, received the antiepileptic drug (VPA), with and without the DHA, daily for a total period of 2 weeks. Rat groupings and protocols were conducted as detailed:
| Control | Received vehicle for the same period of time |
| VPA | Received VPA alone (500 mg/kg orally [PO], daily) |
| VPA + DHA | VPA (500 mg/kg PO, daily), then after 1 hour received DHA (250 mg/kg PO) |
Animals were anesthetized and blood samples were collected after 1 and 2 weeks of treatment via the orbital sinus. Serum was separated by centrifugation at 2,000 rpm for 10 minutes at 4 °C. All liver markers (in serum) were measured after 1 and 2 weeks of VPA treatment; except for albumin which was monitored only after 2 weeks in virtue of its known long half-life (T½) value that hinders imminent short-term changes in its serum levels. Parameters measured in liver tissue were taken only after the second week of treatment (when animals were killed). Thus, liver was quickly removed and washed in an ice-cold isotonic saline, dissected, weighed, and minced. A 10 % (w/v) homogenate was made in phosphate-buffered saline (PBS) (pH 7.4) for the assay of GSH and liver lipid peroxide (MDA). A consistent piece from each liver was collected in a formalin solution for histopathologic evaluations.
Biochemical Determinations
All enzymes, oxidative stress and hepatic synthesis markers were determined spectrophotometrically using appropriate kits. Protocols used were according to the recommended manufacturer procedures.
Mouse Acute, Pentylenetetrazole (PTZ), Anticonvulsant Studies
Mouse groups, of eight animals each, were randomly constituted. Four such groups received DHA orally, 1 hour before PTZ 85 mg/kg was injected subcutaneously (SC). The positive control group received the ED50 (dose effective in 50 % of tested mice) of VPA (175 mg/kg, PO), as determined by preliminary experiments. PTZ was injected 30 minutes after VPA administration, a time proven to allow peak plasma VPA level to be reached. The combination group received the DHA then VPA doses, respectively, at 30-minute intervals before PTZ was given (see next scheme).
Details for mouse groupings and their drug treatments are tabulated here:
| Negative control | Received equivalent amount of vehicle (corn oil, PO) 1 hour before PTZ (85 mg/kg SC) was injected |
| VPA | Received VPA (175 mg/kg PO) 30 minutes before PTZ (85 mg/kg SC) was injected |
| DHA1 | Received DHA (120 mg/kg PO) 1 hour before PTZ (85 mg/kg SC) was injected |
| DHA2 | Received DHA (200 mg/kg PO) 1 hour before PTZ (85 mg/kg SC) was injected |
| DHA3 | Received DHA (250 mg/kg PO) 1 hour before PTZ (85 mg/kg SC) was injected |
| VPA + DHA | Received DHA (250 mg/kg PO), VPA (175 mg/kg, after 30 minutes), then PTZ was injected after another 30 minutes |
Time Course and Kinetic Parameters for Serum VPA Levels in Rats, in Presence and Absence of DHA
Rats received VPA (200 mg/kg) alone or in combination with DHA (250 mg/kg). DHA was given 1 hour before VPA. Blood samples were collected (from orbital sinus) at 30 minutes, 1 hour, 3 hours, and 6 hours after VPA was given. Samples were centrifuged and the separated serum was used for determination of VPA concentrations by enzyme immunoassay, as detailed next.
Rat Grouping and Treatment Protocols for Pharmacokinetic Studies
| VPA | Received VPA (200 mg/kg PO) |
| VPA + DHA | Received DHA (250 mg/kg PO) and after 1 hour received VPA (200 mg/kg PO) |
Quantitative analysis of VPA was based on a homogeneous enzyme-immunoassay technique that measures both free and protein-bound VPA in serum. The assay is fully automated through a programmed protocol that utilizes a Dad Behring instrument. The results are calculated automatically by the analyzer, based on a standard curve that is constructed concurrently with the assay of samples.
Statistical Analyses
Distribution of the data was verified to be normal using Tests of Normality (SPSS package). Statistical significance was tested by one-way analysis of variance (ANOVA) followed by Bonferroni post hoc analysis. Statistical significance was predefined at p < 0.05.
Results
Treatment with valproate (500 mg/kg, daily) for 1–2 weeks disrupted liver cell integrity as reflected by marked (2- to 5-fold) rises in serum ALT, γ-GT, and ALP (Fig. 1a–c). Such enzyme levels did not significantly vary when VPA treatment was extended from 1 to 2 weeks. Likewise, as measured by serum albumin level; the hepatocellular synthetic capacity was notably reduced (by 40 %) (Fig. 1d).
Fig. 1.
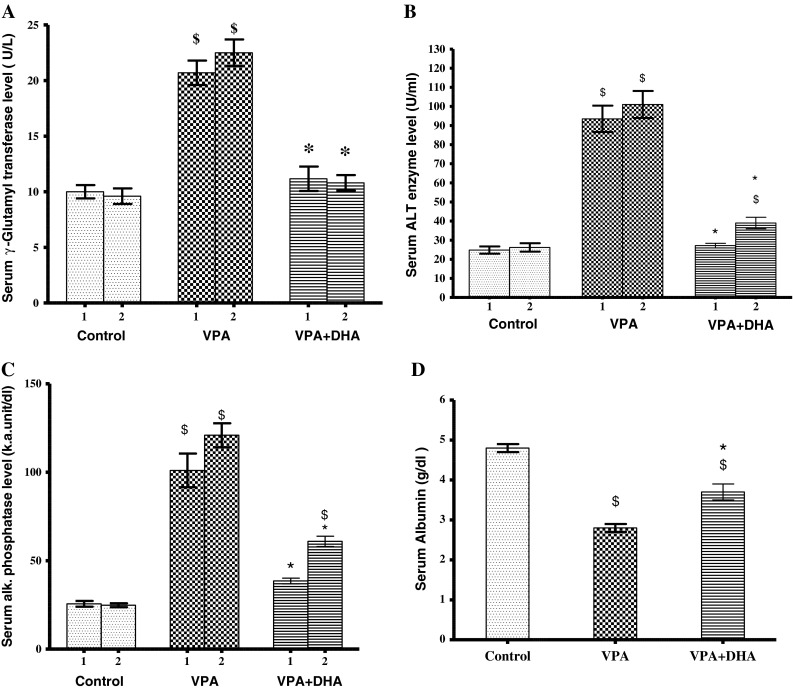
a–d Effect of VPA (500 mg/kg daily/2 weeks) with and without DHA (250 mg/kg/day) on serum hepatic enzyme and albumin levels. DHA was given orally 1 h after VPA, then blood was withdrawn from the orbital sinus for determination of enzymes (a–c; γ-GT, ALT, ALP, respectively) after 1 and 2 weeks, or albumin (d), after 2 weeks. Data represent the mean ± SEM of each group; n = 6–8. Symbols indicate significance against VPA-treated group (asterisks) and normal control group (dollar symbols), γ-GT γ-glutamyl transferase, ALT alanine aminotransferase, ALP alkaline phosphatase, DHA docosahexaenoic acid, VPA valproate
To gain insights into the hepatic molecular and cellular changes occurring following VPA treatment; oxidative stress and endogenous antioxidant levels were monitored, and histopathologic examination of the liver was also conducted. Figure 2a demonstrates that VPA evoked a 3-fold rise in MDA levels. This was also accompanied by 35 % reduction in levels of endogenous cellular protector: reduced GSH, Fig. 2b.
Fig. 2.
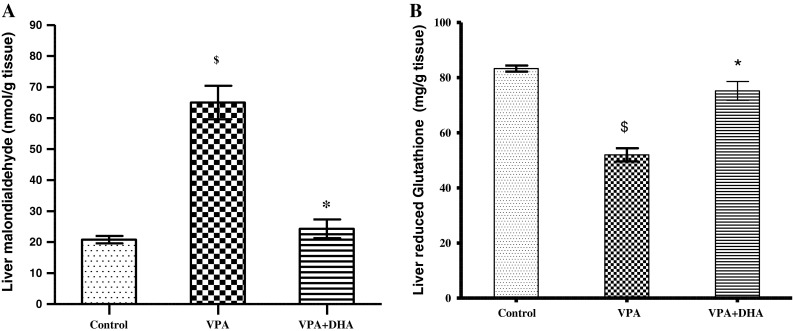
a, b Effect of VPA (500 mg/kg daily/2 weeks) with and without DHA (250 mg/kg/day) on liver lipid peroxide (MDA) (a), and reduced glutathione (GSH) (b) levels. After 2 weeks of treatment, animals were sacrificed and a 10 % W/V liver homogenate was assayed for its content of MDA or GSH. Data represent the mean ± SEM of each group; n = 7. Symbols indicate significance against VPA-treated group (asterisks) and normal control group (dollar symbols), DHA docosahexaenoic acid, VPA valproate
Downstream from hepatocellular disruption and oxidative stress, we also investigated whether VPA liver intoxication had involved inflammatory signals and/or neutrophil infiltration into the liver; and if so, how these signals may be modified by DHA. Accordingly, in liver cell homogenates, VPA upregulated the expression of proinflammatory cytokine TNFα (5-fold, p < 0.05). This was paralleled by a ~ 6.1-fold rise in this cytokine level in the serum (p < 0.05, Fig. 3a, b). Considering time-course dependency, DHA managed to blunt the rise in TNFα effectively, after both 1 and 2 weeks, although effects of DHA were more pronounced after 1 week. Co-treatment with DHA largely suppressed the VPA-induced hepatocytic production of TNFα in both the liver and the serum, implying also that rises in the serum are most likely linked to those in the liver. Moreover, an enzyme marker of neutrophil infiltration with known contributions to both inflammation and oxidative stress, that is myeloperoxidase (MPO), had an appreciably enhanced activity in liver homogenates (4.2-fold; p < 0.05). This response was likewise highly sensitive to co-treatment with DHA (p < 0.05), thus also revealing the versatility whereby DHA protects liver cells against VPA-induced injury.
Fig. 3.
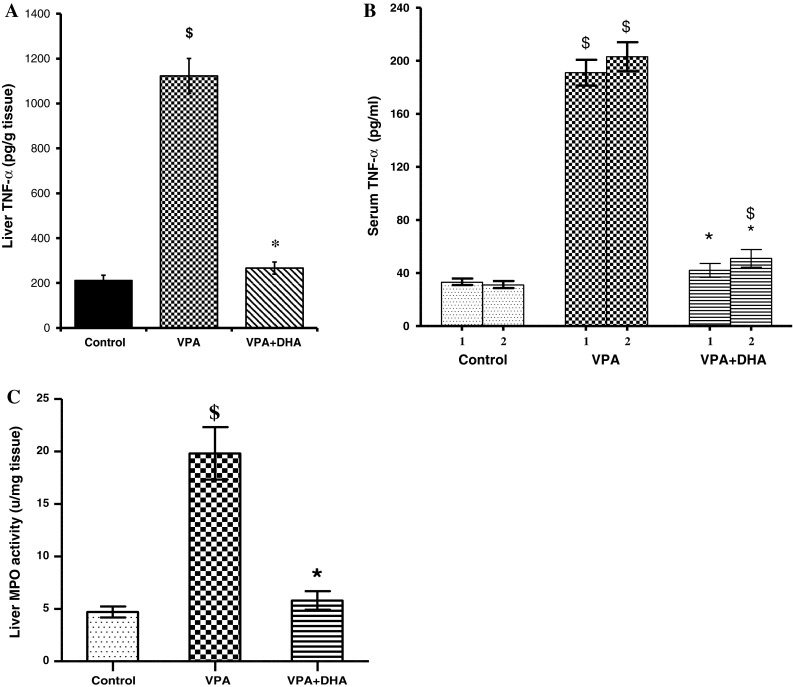
Effect of VPA (500 mg/kg daily/2 weeks) with and without DHA (250 mg/kg/day) on liver and serum levels of TNFα (a, b), and on liver activity of myeloperoxidase (MPO) (c). Blood was withdrawn for determination of TNFα after 1 and 2 weeks of treatments. Animals were sacrificed after 2 weeks and a 10 % W/V liver homogenate was assayed for both parameters. Data represent the mean ± SEM of each group; n = 8. Symbols indicate significance against VPA-treated group (asterisks) and normal control group (dollar symbols), DHA docosahexaenoic acid, TNFα tumor necrosis factor alpha, VPA valproate
Figure 4 represents necropsies of the liver to assess the pathological changes in the studied animals. The negative control group showed average size and color of the liver with no detected histopathologic abnormalities (photos 1, 2). Conversely, the VPA-treated group showed grossly enlarged pale livers with significantly increased weights over control values. Besides, multiple foci of focal lytic necrosis were detected in which replacement by both inflammatory cells and cellular degeneration had occurred (photo 3). Moreover, combined macrovesicular and microvesicular steatosis were evident in the periportal zone of four animals (out of six) of this group (photo 4). Concurrent treatment with DHA significantly alleviated the hepatic cellular and molecular anomalies entailed by VPA treatment. This was manifested as reduced serum liver enzymes (better after 1 than 2 weeks), lipid peroxide generation, and increased levels of hepatic GSH and serum albumin, consonant with promoted liver defensive mechanisms and enhanced protein synthesis. Furthermore, when combined with VPA, DHA showed only minimal small focal necrosis/apoptosis (single cell death) with no evidence of degeneration or steatosis (photo 5); consistent with amelioration of pathologic anomalies by DHA.
Fig. 4.

Necropsies of the liver of studied animals from each group to assess the pathologic changes. Photos 1, 2 are for the negative control group, showing average size/color of the liver with no detected histopathologic abnormalities. Photo 3: VPA control group showing grossly enlarged pale livers with multiple foci of focal lytic necrosis with replacement by inflammatory cells and hepatocyte degeneration. Also, combined macrovesicular and microvesicular steatosis occurring in the periportal zone were evident in four animals in this group (photo 4). DHA when combined with VPA showed only minimal small focal necrosis with no evidence of degeneration (photo 5). DHA docosahexaenoic acid, VPA valproate
Because DHA recently demonstrated some neuroinhibitory effects on its own [18], it was of current interest to also seek possible synergy with anticonvulsant effects of VPA. Figure 5 shows that DHA elicited a dose-responsive increase in latency (onset) of mouse tonic convulsions, with significance from control value elicited at (250 mg/kg, p < 0.05), a response that was also comparable to that evoked by VPA at its ED50 dose (13.8 vs 14.9 min). Combining the two FAs at such lower doses triggered a notable synergy in the latency of convulsion (32.8 min, p < 0.05). Similar effects for DHA were observed for the myoclonic aspect of PTZ convulsion (data not shown).
Fig. 5.

Individual and combined effects of VPA (175 mg/kg) and DHA (100–250 mg/kg) on onset of tonic convulsion (min) evoked by PTZ (85 mg/kg). PTZ was injected 30 min after VPA administration. The combination groups received DHA then VPA, respectively; at 30 min intervals, before PTZ was given. Data represent mean ± SEM of times recorded for each group (8 animals). Symbols indicate significance against VPA-treated group (asterisks) and normal control group (dollar symbols), DHA docosahexaenoic acid, PTZ pentylenetetrazole, VPA valproate
It was next worthwhile investigating whether the protective and synergistic effects of DHA involve pharmacokinetic interaction with VPA, that is, alteration of VPA clearance rate. To this end, plasma VPA levels were determined over a time frame of 6 hours in both the presence and absence of DHA (250 mg/kg), a dose that was proven protective in earlier toxicological studies. Various kinetic parameters such as area under the curve (AUC) and volume of distribution (V d) are displayed in Table 1. As judged by statistical analyses, neither the peak/trough values nor the magnitude of other measured points was altered in animals given a combination of DHA and VPA, as compared with those given VPA alone. These findings unequivocally exclude the possibility of pharmacokinetic interaction and, instead, indicate peculiar dynamic effects for DHA.
Table 1.
Computed pharmacokinetic parameters following administration of VPA (200 mg/kg, PO) alone or in combination with DHA (250 mg/kg PO) in rats
| Group | AUC (mg.h/L) | C max (mg/L) | T max (h) | T ½ (h) | V d/F (L/kg) | Cl/F (L/h/kg) |
|---|---|---|---|---|---|---|
| VPA | 404.3 ± 22.1 | 107.6 ± 6.6 | 0.5 | 2.11 ± 0.1 | 1.518 ± 0.11 | 0.505 ± 0.03 |
| VPA + DHA | 409.6 ± 12.8 | 110.1 ± 3.2 | 0.5 | 2.04 ± 0.12 | 1.436 ± 0.07 | 0.491 ± 0.02 |
AUC area under serum concentration–time curve, C max maximum plasma concentration, Cl clearance, DHA docosahexaenoic acid, F oral availability, PO orally, T ½ elimination half-life, T max time needed to attain C max, V d apparent volume of distribution, VPA valproate
Discussion
This study reports a prominent protection by DHA against VPA-induced hepatic dysfunction, cellular anomalies, necrosis and steatosis. Likewise, it reveals that DHA enhances the anticonvulsant effects of VPA in a PTZ animal-convulsion model. These favorable effects for DHA do not target the kinetic profiles or distribution pattern of VPA, but rather trigger specific dynamic mechanisms.
Because the liver is the main drug/xenobiotic metabolic engine of the body, it is very much vulnerable to drug toxicity [21, 22]. In particular, antiepileptic drugs (AED) have many such serious untoward reactions, as seen with VPA, phenytoin, and carbamazepine. Though relatively rare, when compared with other consistently known hepatotoxic drugs, the consequences encountered with AED can cause death or an acute liver failure that would require liver transplantation. The molecular underpinnings of these anomalies to AED pertain either to production of reactive toxic metabolite/s or to induction of paradoxical immunoallergic reactions [23].
Aspects of hepatotoxicity associated with VPA have been fully unfolded [10]. Type I VPA-mediated hepatic injury is associated with a dose-dependent rise in serum liver enzymes and decline in plasma albumin. Type II VPA-mediated hepatotoxicity is a fatal, irreversible idiosyncratic reaction that is characterized by microvesicular steatosis and necrosis [11]. Although the mechanisms involved are not fully characterized, a large body of evidence suggests that reactive VPA metabolites (i.e., 4-ene-VPA and its subsequent metabolite, 2,4-diene-VPA) may mediate the hepatotoxicity by inhibiting mitochondrial β-oxidation of FAs. Further, excessive generation of reactive oxygen species (ROS) (such as peroxides and hydroxyl radical) may follow the toxicity of VPA as a consequence of disrupting the liver antioxidant machinery [10, 24, 25].
Although DHA has demonstrated protection against some drug-induced systemic toxicity [17], its impact on VPA-induced liver injury has never been sought. These views prompted us to evaluate whether, and how, DHA may obliterate VPA hepatotoxicity. Accordingly, when DHA was jointly given with VPA, serum liver marker enzyme levels (ALP, ALT and γ-GT) significantly declined, thereby suggesting the utility of DHA in protecting liver cell integrity and maintaining healthy biliary outflow. Further, DHA raised serum albumin levels, consonant with restoration of liver protein synthetic capacity. More such clues were provided from the present histopathologic studies, which depicted the capacity of DHA to ameliorate VPA-evoked hepatocellular degeneration, infiltration of inflammatory cells, induction of focal pericentral necrosis, and micro/macrovesicular steatosis.
Next, it was both worthy and intriguing to unravel the cellular and molecular means whereby DHA abates VPA-evoked liver injury. Thus, DHA markedly replenished hepatic GSH levels to near baseline and blunted lipid peroxide (MDA) levels, thereby alleviating VPA-induced oxidative stress. In support, in animal models of alcohol fatty liver, DHA terminated oxidative stress and mitochondrial dysfunction [25]. Besides, human nutritional studies in prevention of heart diseases revealed that supplementation with a daily 200–800 mg DHA enhanced its incorporation into LDL, thereby reducing its susceptibility to oxidation and accumulation of lipid peroxides [26, 27].
The possible second molecular trigger for hepatic protection by DHA is an anti-inflammatory and lipotropic effect. Inflammation and hepatic accumulation of triglycerides can foster/exacerbate oxidative stress and liver cell damage. DHA reportedly gets incorporated into liver cells, and can evidently suppress hepatic gene expression of proinflammatory cytokines [16, 20, 28]. Because hepatic necrosis, apoptosis, and steatosis were recently linked with TNFα and its TNFR1 receptor in response to B-virus hepatitis [29, 30], it was of present interest to assess levels of this cytokine in VPA-intoxicated animals. Additionally, ω-3 FAs can specifically activate the peroxisome proliferator-activated receptor-α (PPARα), a transcriptional activator of FA oxidation in peroxisomes and mitochondria [31]. Thus, current evaluations of TNFα were further substantiated by the reported interaction between TNFα and PPARα [32]. In this vein, TNFα was implicated in downregulating PPARα, thereby inducing hepatic steatosis [33]. We detected several-fold rises in hepatic TNFα levels following VPA treatment, a response that was appreciably blocked with DHA, implying that this ω-3 FA also protects the liver via a specific anti-inflammatory mechanism. Because we also showed here the capacity of DHA (a PPARα agonist) to suppress expression of TNFα and reduce hepatic inflammation/steatosis, these findings further establish a concept of ‘cross-talk’ between the TNFα and PPARα systems in VPA-intoxicated liver cells. Further, DHA blunted the activity of a neutrophil-specific pro-inflammatory/pro-oxidant enzyme (MPO). Together, these findings demonstrate new effector players that are recruited by VPA to induce hepatic injury, while also attest to the diversity of the molecular basis whereby DHA can reverse these insults to ultimately elicit liver protection.
An additional objective in this study was to evaluate the possibilities of DHA synergy with anticonvulsant effects of VPA, so as to infer whether lower doses of VPA (certainly less toxic) can be therapeutically applied. Thus far, clinically, DHA is recognized to be essential for normal growth and development, and has demonstrated therapeutic benefits against some central disease states/models [16]. More recently, in a rat model, DHA was shown to raise the threshold of convulsion, suggesting its utility in the management of epilepsy. Likewise, supplementation with ω-3 FAs was efficacious in the amelioration of depressive symptoms in elderly patients [18, 19].
Therefore, we first demonstrated that DHA evoked dose-responsive anticonvulsant effects against PTZ-induced seizures when given alone at 250 mg/kg. Furthermore, when co-administered with VPA, the latency in onset of convulsion was greater than their individual responses, thereby revealing a superb synergic response. Thus, these current findings suggest the use of less hepatotoxic concentrations of VPA, while preserving its pharmacologic efficacy. At the molecular level, though neuroinhibitory targets for DHA are still incompletely defined, evidence suggests that ω-3 FAs can cause inhibition of sodium and calcium voltage-gated ion channels. Additionally, the production of anti-inflammatory metabolites, like neuroprotectin-D1, has also been suggested to reduce neuroinflammation, thereby raising the seizure threshold and abating convulsions in response to ω-3 FAs [34, 35].
Collectively, the above-mentioned effects for DHA clearly depict the involvement of cellular ‘dynamic’ effectors in protection against VPA-induced liver injury. However, currently these findings cannot exclude the involvement of metabolic/kinetic means whereby DHA may modulate plasma levels/clearance of VPA. This view is also supported by earlier findings that both DHA and VPA can individually evoke kinetic interactions with many other drugs, thereby altering their efficacies [35–38]. Hence, it was indeed both challenging and intriguing to probe these possibilities for the present combination regimen (DHA/VPA). We found that co-treatment with DHA had no effect on serum VPA concentration at different time intervals, as compared with animals that had received VPA only. Likewise, no significant statistical difference was observed in the VPA pharmacokinetic parameters generated in the presence and absence of DHA, thus unequivocally indicating that DHA had no effect on clearance rate of VPA. Although the hepatoprotective effects of DHA were observed with another drug, paracetamol [39], this study not only revealed some molecular underpinnings and synergy effects for DHA actions, but also ruled out any sort of kinetic interactions with VPA, an important drug efficacy aspect.
Conclusively, DHA is an ideal aide in synergy with VPA that acts via dynamic mechanisms to abate VPA-induced hepatic injury, while also largely enhancing its anticonvulsant effects, thus potentially allowing lower doses of VPA to be applied. Notably also, the known kinetic profiles and safety reports on DHA largely support these findings.
Accordingly, it becomes evident that a rational design/exploitation of synergy via the use of phytomedicals should enrich modern pharmacotherapy enough to revolutionize the management of vicious adverse drug reactions, as typically exemplified here by VPA-evoked hepatic injury [40]. Clinically, data from this study suggest a fruitful drug regimen to reduce hepatic injury. This is governed by the capacity of DHA to restore normal liver function and integrity, and to synergize with neuroinhibitory (antiepileptic) effects to enable lower doses of VPA. Together, this combined drug regimen should augment the overall therapeutic index of VPA.
Acknowledgments
This study was supported in part by a postgraduate fellowship award to (M.A.E1) from Mansoura University, Egypt; and by an American Heart Association SDG grant to (A.A.E-M2).
Footnotes
S. A. Said: Deceased.
References
- 1.El-Mowafy AM, Al-Gayyar MM, El-Mesery ME, Salem HA, Darweish MM. Novel chemotherapeutic and renal protective effects for the green-tea (EGCG): role of oxidative stress and inflammatory-cytokine signaling. Phytomedicine. 2010;17:1067–1075. doi: 10.1016/j.phymed.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Calder PC. n-3 Fatty acids, inflammation, and immunity relevance to postsurgical and critically ill patients. Lipids. 2004;39(12):1147–1161. doi: 10.1007/s11745-004-1342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Mowafy AM, Alkhalaf M. Resveratrol activates adenylyl-cyclase in human breast-cancer cells: a novel, estrogen receptor-independent cytostatic mechanism. Carcinogenesis. 2003;24(5):869–873. doi: 10.1093/carcin/bgg015. [DOI] [PubMed] [Google Scholar]
- 4.Einarson TR. Drug-related hospital admissions. Ann Pharmacother. 1993;27(7–8):832–840. doi: 10.1177/106002809302700702. [DOI] [PubMed] [Google Scholar]
- 5.Johannessen CU, Johannessen SI. Valproate: past, present, and future. CNS Drug Rev. 2003;9(2):199–216. doi: 10.1111/j.1527-3458.2003.tb00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bedry R, Parrot F. Severe valproate poisoning. Réanimation. 2004;13:324–333.
- 7.Zimmerman RI, Ishak KG. Valproate-induced hepatic injury. Analyses of 23 fatal cases. Hepatology. 1982;2:591–597. doi: 10.1002/hep.1840020513. [DOI] [PubMed] [Google Scholar]
- 8.Cotarlu D, Zaldman JL. Valproic acid and the liver. Clin Chem. 1988;34(5):890–897. [PubMed] [Google Scholar]
- 9.Graf WD, Oleinik OE, Glauser T. Altered antioxidant enzyme activities in children with a serious adverse experience related to valproic acid therapy. Neuropediatrics. 1998;29:195–201. doi: 10.1055/s-2007-973560. [DOI] [PubMed] [Google Scholar]
- 10.Tong V, Thomas KH, Frank S. Valproic acid. Time course of lipid peroxidation biomarkers, liver toxicity, and valproic acid metabolite levels in rats. Toxicol Sci. 2005;86(2):427–435. doi: 10.1093/toxsci/kfi184. [DOI] [PubMed] [Google Scholar]
- 11.Spiller HA, Krenzelok EP, Klein-Schwartz W, Winter ML, Webe JA, Sollee DR, et al. Multicenter case series of valproic acid ingestion: serum concentrations and toxicity. J Toxicol Clin Toxicol. 2000;38(7):755–760. doi: 10.1081/CLT-100102388. [DOI] [PubMed] [Google Scholar]
- 12.Tang W, Borel AG, Fujimiya T, Abbott FS. Fluorinated analogues as mechanistic probes in valproic acid hepatotoxicity: Hepatic microvesicular steatosis and glutathione status. Chem Res Toxicol. 1995;8(5):671–682. doi: 10.1021/tx00047a006. [DOI] [PubMed] [Google Scholar]
- 13.Raza M, Al-Bekairi AM, Ageel AM, Qureshi S. Biochemical basis of sodium valproate hepatotoxicity and renal tubular disorder. Pharmacol Res. 1997;35(2):153–157. doi: 10.1006/phrs.1997.0134. [DOI] [PubMed] [Google Scholar]
- 14.Buchi KN, Gray PD, Rollins DE, Tolman KG. Protection against sodium valproate injury in isolated hepatocytes by alpha-tocopherol and N,N’-diphenyl-p-phenylenediamine. J Clin Pharmacol. 1984;24(4):148–154. doi: 10.1002/j.1552-4604.1984.tb01823.x. [DOI] [PubMed] [Google Scholar]
- 15.Lheureux PE, Hantson P. Carnitine in the treatment of valproic acid-induced toxicity. Clin Toxicol (Phila) 2009;47(2):101–111. doi: 10.1080/15563650902752376. [DOI] [PubMed] [Google Scholar]
- 16.Simopoulos AP. Essential fatty acids in health and chronic diseases. Forum Nutr. 2003;56:67–70. [PubMed] [Google Scholar]
- 17.El-Mesery ME, Al-Gayyar MM, Salem HA, Darweish MM, El-Mowafy AM. Chemopreventive and renal protective effects for docosahexaenoic acid (DHA): implications of CRP and lipid peroxides. Cell Div. 2009;4(1):6. doi: 10.1186/1747-1028-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taha AY, Jeffrey MA, Taha NMY, Bala S, Burnham WM. Acute administration of docosahexaenoic acid increases resistance to pentylenetetrazole-induced seizures in rats. Epilepsy Behav. 2010;17:336–343. doi: 10.1016/j.yebeh.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Rondanelli M, Giacosa A, Opizzi A, Pelucchi C, La Vecchia C, Montorfano G, Negroni M, Berra B, Politi P, Rizzo AM. Effect of omega-3 fatty acids supplementation on depressive symptoms and on health-related quality of life in the treatment of elderly women with depression: a double-blind, placebo-controlled, randomized clinical trial. J Am Coll Nutr. 2010;29(1):55–64. doi: 10.1080/07315724.2010.10719817. [DOI] [PubMed] [Google Scholar]
- 20.Ishii H, Horie Y, Ohshima S, Anezaki Y, Kinoshita N, Dohmen T, et al. Eicosapentaenoic acid ameliorates steatohepatitis and hepatocellular carcinoma in hepatocyte-specific Pten-deficient mice. J Hepatol. 2009;50(3):562–571. doi: 10.1016/j.jhep.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 21.Bissell DM, Gores GJ, Laskin DL, Hoofnagle JH. Drug-induced liver injury: mechanisms and test systems. Hepatology. 2001;33:1009–1013. doi: 10.1053/jhep.2001.23505. [DOI] [PubMed] [Google Scholar]
- 22.Pessayre D, Berson A, Fromenty B. Mitochondria in steatohepatitis. Semin Liver Dis. 2001;21:57–69. doi: 10.1055/s-2001-12929. [DOI] [PubMed] [Google Scholar]
- 23.Björnsson E. Hepatotoxicity associated with antiepileptic drugs. Acta Neurol Scand. 2008;118(5):281–290. doi: 10.1111/j.1600-0404.2008.01009.x. [DOI] [PubMed] [Google Scholar]
- 24.Szalowska E, van der Burg B, Man HY, Hendriksen PJ, Peijnenburg AA. Model steatogenic compounds (amiodarone, valproic acid, and tetracycline) alter lipid metabolism by different mechanisms in mouse liver slices. PLoS One. 2014;9(1):e86795. doi:10.1371/journal.pone.0086795. [DOI] [PMC free article] [PubMed]
- 25.Higgins S, Carroll YL, McCarthy SN. Susceptibility of LDL to oxidative modification in healthy volunteers supplemented with low doses of n-3 polyunsaturated fatty acids. Br J Nutr. 2001;85:23–31. doi: 10.1079/BJN2000220. [DOI] [PubMed] [Google Scholar]
- 26.Song BJ, Moon KH, Olsson NU, Salem N., Jr Prevention of alcoholic fatty liver and mitochondrial dysfunction in the rat by long-chain polyunsaturated fatty acids. J Hepatol. 2008;49(2):262–273. doi: 10.1016/j.jhep.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calzada C, Colas R, Guillot N, Guichardant M, Laville M, Véricel E, et al. Subgram daily supplementation with docosahexaenoic acid protects low-density lipoproteins from oxidation in healthy men. Atherosclerosis. 2009;208(2):467–472. doi: 10.1016/j.atherosclerosis.2009.07.049. [DOI] [PubMed] [Google Scholar]
- 28.Schmocker C, Weylandt KH, Kahlke L, Wang J, Lobeck H, Tiegs G. Omega-3 fatty acids alleviate chemically induced acute hepatitis by suppression of cytokines. Hepatology. 2007;45:864–869. doi: 10.1002/hep.21626. [DOI] [PubMed] [Google Scholar]
- 29.Kim JY, Song EH, Lee HJ, Oh YK, Choi KH, Yu DY, Park SI, Seong JK, Kim WH. HBx-induced hepatic steatosis and apoptosis are regulated by TNFR1- and NF-kappaB-dependent pathways. J Mol Biol. 2010;397(4):917–931. doi: 10.1016/j.jmb.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 30.Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. Cell Mol Med. 2004;8(4):445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cullingford TE, Dolphin CT, Sato H. The peroxisome proliferator-activated receptor alpha-selective activator ciprofibrate upregulates expression of genes encoding fatty acid oxidation and ketogenesis enzymes in rat brain. Neuropharmacology. 2002;42(5):724–730. doi: 10.1016/S0028-3908(02)00014-X. [DOI] [PubMed] [Google Scholar]
- 32.Beier K, Völkl A, Fahimi HD. TNF-alpha downregulates the peroxisome proliferator activated receptor-alpha and the mRNAs encoding peroxisomal proteins in rat liver. FEBS Lett. 1997;412(2):385–387. doi: 10.1016/S0014-5793(97)00805-3. [DOI] [PubMed] [Google Scholar]
- 33.Vreugdenhil M, Bruehl C, Voskuyl RA, Kang JX, Leaf A, Wadman WJ. Polyunsaturated fatty acids modulate sodium and calcium currents in CA1 neurons. Proc Natl Acad Sci USA. 1996;93(22):12559–12563. doi: 10.1073/pnas.93.22.12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bazan NG. Omega-3 fatty acids, pro-inflammatory signaling and neuroprotection. Curr Opin Clin Nutr Metab Care. 2007;10(2):136–141. doi: 10.1097/MCO.0b013e32802b7030. [DOI] [PubMed] [Google Scholar]
- 35.Hirunpanich V, Sato H. Docosahexaenoic acid (DHA) inhibits saquinavir metabolism in-vitro and enhances its bioavailability in rats. J Pharm Pharmacol. 2006;58(5):651–658. doi: 10.1211/jpp.58.5.0010. [DOI] [PubMed] [Google Scholar]
- 36.Hirunpanich V, Katagi J, Sethabouppha B, Sato H. Demonstration of docosahexaenoic acid as a bioavailability enhancer for CYP3A substrates: in vitro and in vivo evidence using cyclosporin in rats. Drug Metab Dispos. 2006;34(2):305–310. doi: 10.1124/dmd.105.007088. [DOI] [PubMed] [Google Scholar]
- 37.Phua LC, New LS, Goh CW, Neo AH, Browne ER, Chan EC. Investigation of the drug–drug interaction between alpha-lipoic acid and valproate via mitochondrial beta-oxidation. Pharm Res. 2008;25(11):2639–2649. doi: 10.1007/s11095-008-9681-5. [DOI] [PubMed] [Google Scholar]
- 38.Chung JY, Cho JY, Yu KS, Kim JR, Lim KS, Sohn DR, et al. Pharmacokinetic and pharmacodynamic interaction of lorazepam and valproic acid in relation to UGT2B7 genetic polymorphism in healthy subjects. Clin Pharmacol Ther. 2008;83(4):595–600. doi: 10.1038/sj.clpt.6100324. [DOI] [PubMed] [Google Scholar]
- 39.Meganathan M, Madhana MG, Sasikala P, Mohan J, Gowdhaman N, Balamurugan K, et al. Evaluation of antioxidant effect of Omega 3-fatty acid against paracetamol-induced liver injury in albino rats. Global J Pharmacol. 2011;5(1):50–53. [Google Scholar]
- 40.Wagner H, Ulrich-Merzenich G. Synergy research: approaching a new generation of phytopharmaceuticals. Phytomedicine. 2009;16(2–3):97–110. doi: 10.1016/j.phymed.2008.12.018. [DOI] [PubMed] [Google Scholar]