

Abstract
Unequivocally, genetic variants within the fatty acid desaturase (FADS) cluster are determinants of long chain polyunsaturated fatty acid (LC-PUFA) levels in circulation, cells and tissues. A recent series of papers have addressed these associations in the context of ancestry; evidence clearly supports that the associations are robust to ethnicity. However ∼80% of African Americans carry two copies of the alleles associated with increased levels of arachidonic acid, compared to only ∼45% of European Americans raising important questions of whether gene-PUFA interactions induced by a modern western diet are differentially driving the risk of diseases of inflammation in diverse populations, and are these interactions leading to health disparities. We highlight an important aspect thus far missing in the debate regarding dietary recommendations; we content that current evidence from genetics strongly suggest that an individual's, or at the very least the population from which an individual is sampled, genetic architecture must be factored into dietary recommendations currently in place.
Keywords: polyunsaturated fatty acids, nutrition, genetic variants, fatty acid desaturase (FADS), single nucleotide polymorphisms, arachidonic acid, eicosanoids, inflammation, cardiovascular disease
Introduction
Polyunsaturated fatty acids (PUFAs) and particularly LC-PUFA have a wide range of roles ranging from regulating immunity and inflammation to impacting brain and eye development and function. Key determinants of the efficiency with which long chain polyunsaturated fatty acid (LC-PUFA) are synthesize as well as LC-PUFA levels themselves have been established within the fatty acid desaturase cluster (FADS) on chromosome 11q12.2; these determinants appear to be robust to ethnicity. However, given dramatic differences in the frequencies of these critical determinants of LC-PUFA metabolism, we will highlight an important aspect thus far missing in the debate regarding dietary recommendations: the consideration of these genetic determinants.
Biosynthesis of LC-PUFAs
Figure 1 highlights a set of genes known to play key roles in the biosynthesis and metabolism of LC-PUFAs. In two parallel and competing pathways, enzymatic products from these genes convert 18-carbon (18C) n-6 or n-3 essential PUFAs (abundant in the modern western diet [MWD]) into LC-PUFA. On the n-6 arm of the pathway, arachidonic acid (ARA, 20:4n-6) is synthesized from linoleic acid (LA, 18:2n-6) utilizing three (2 desaturation and 1 elongation) enzymatic steps [1]. The primary n-3 LC-PUFAs, eicosapentaenoic acid (EPA, 20:5n-3), docosapentaenoic acid (DPA, 22:5n-3) and docosahexaenoic acid (DHA, 22:6n-3) can be synthesized from dietary α-linolenic acid (ALA, 18:3n-3). Foe example, it requires seven (3 desaturation, 3 elongation and 1 β-oxidation) enzymatic steps to synthesize DHA. Importantly, both n-6 and n-3 substrates compete for early the enzymatic steps in the pathway (described in detail below). The desaturation steps have long been recognized as the rate-limiting steps in the pathway. The initial desaturation step converts the n-6, 18C PUFA, LA and the n-3, 18C PUFA, ALA to gamma (γ)-linolenic acid (GLA, 18:3, n-6) and stearidonic acid (SDA, 18:4, n-3), respectively, by the enzyme encoded for by the gene fatty acid desaturase 2 (FADS2; chromosome 11q12.2) [2-4]. The second desaturation step is catalyzed by an enzyme encoded by the gene fatty acid desaturase 1 (FADS1; chromosome 11q12.2-q13.1); this step converts DGLA (2-:3, n-6) and ETA (20:4, n-3) to ARA (20:4, n-6) and EPA (20:5, n-3), respectively [4, 5].
Figure 1.
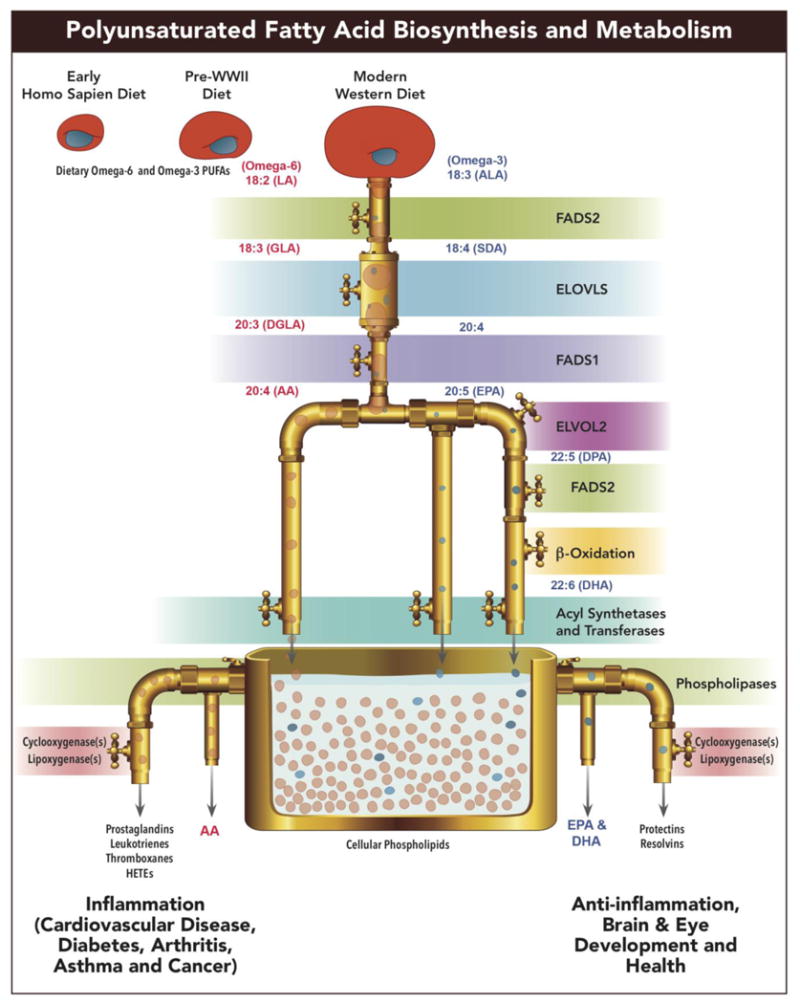
Biosynthetic pathway of n3 and n6 PUFAs highlighting a set of genes known to play key roles in the biosynthesis and metabolism of LC-PUFAs in two parallel and competing pathways. Enzymatic products from these genes convert 18-carbon (18C) n-6 or n-3 essential PUFAs (abundant in the modern western diet [MWD]) into LC-PUFA.
Considerations for Making Dietary PUFA Recommendations in Diverse Human Populations
There are several important considerations when determining dietary recommendations for PUFAs in diverse human populations. First it is important to understand how and what quantities of PUFAs enter the biosynthetic pathways from the background human diet. There has been a dramatic (∼3-fold) increase in the dietary access to the 18C n-6 PUFA, LA (currently 6-8% of calories) in the MWD in the past 50 years with the increase in LA-containing vegetable oil products (soybean, corn, palm, and canola oils as well as margarine and shortenings [6]). In contrast, ALA levels (typically 0.5-1.5% of calories) found in green plants, nuts and botanical oils, such as flax seed oil have remained relatively constant over that same period of time (Figure 1). Given that humans ingest much lower quantities of LC-PUFAs (typically less than 200mg/day) than 18C PUFAs, LA and ALA, the capacity to synthesize LC-PUFAs from these 18C PUFAs is critical in determining circulating and cellular levels of LC-PUFAs. Additionally, the dramatic increase of dietary LA has not only increased total PUFAs in the diet, but has dramatically shifted the ratio of LA to ALA that enters the pathway [6, 7]. Because LA and ALA compete in early steps of the pathway (Figure 1) and there is a limited synthetic capacity of the pathway, both human and animal models indicate that this shift has markedly reduced the synthesis and bioavailablity of n-3 LC-PUFAs [6, 8, 9]. It is important to note that the current levels and ratios of 18C PUFAs in the MWD are in line in in many ways resulted from with recommendations by the American Heart Association to consume 5-10% of daily calories as PUFAs [6]. For the MWD, that means these calories from PUFAs will be primarily LA and have a typical LA to ALA ratio of > 6 and more typically 10.
The second consideration for recommendations is the biological functions of n-6 and n-3 LC-PUFAs and subsequent molecular product that can result from these products. For example while a few oxidation products of ARA have been shown to be anti-inflammatory, hundreds of laboratories around the world have been able to find eicosanoids (prostaglandins and leukotrienes) at concentrations that are pharmacologically active as pro-inflammatory, and this has resulted in thousands of publications establishing the role of these ARA metabolites in inflammation [10]. Importantly there is the emerging story of n-3 LC-PUFAs and their products as anti-inflammatory, “pro-resolution” metabolites termed resolvins, protectins, and maresins [11-13]. Consequently, there is a consistent scientific literature that supports the concept that n-6 and n-3 LC-PUFAs and their products have not only different, but often opposing effects, with regard to inflammation. Many in the field of eicosanoid biology believe that this component has been underappreciated in the current recommendations by groups such as the AHA. Additionally, LC-PUFAs serve as important molecular components for disease risk factors such as circulating phospholipids, triglycerides and cholesterol esters found in lipoproteins, and thus their levels can play an important role in determining amounts and impact of these lipoproteins.
The third consideration is the type of data used to make the recommendations. For example, the current AHA dietary recommendations have been made largely based on several randomized controlled trials and population cohort studies that measured cardiovascular disease biomarkers such as serum lipids and lipoproteins [14-16]. These data show that PUFAs can have cardiovascular benefits when viewed from the perspective of measuring cardiovascular disease biomarkers such as serum lipids and lipoproteins. However, recent sets of data have raised important questions about this approach. Ramsden and colleagues recently re-examined studies utilized to support this recommendation and found that many of the oils used in the aforementioned clinical trials were mixtures of n-6 and n-3 PUFAs. Their data suggests that only substituting n-6 PUFAs for saturated and trans-fatty acids actually trended toward increased risk of death from all causes [17, 18]. This same group also recently reexamined the Sydney Diet Heart Study (458 men 30-59 with a recent coronary event), which replaced dietary saturated fatty acids with a high LA-containing life the aforementioned studies found that the LA intervention group had lower levels of total cholesterol; however, the n-6 PUFA group had unexpectedly higher rates of death than controls [19]. These data raise the important consideration of which disease biomarker should be used to reliably predict disease and mortality.
Finally, the critical fourth consideration and the topic of this review that has not been appreciated is the potential for dietary 18C PUFAs to impact LC-PUFA biosynthesis, levels of intermediate risk factors and the incidence of human disease in dramatically different ways in diverse human populations. Most dietary PUFA recommendations made to date have been based on a simple assumption that there is a limited capacity (somewhere between 2-3% of calories) of humans to synthesize LC-PUFAs from dietary 18C PUFAs such as LA and ALA. This line of reasoning assumes that higher (than 3% calories) dietary quantities of 18C PUFAs are irrelevant since it is beyond the capacity of the PUFA biosynthetic pathway to utilize them. The second critical assumption in this line of reasoning is that the biosynthetic capacity is equivalent for all human populations. This review examines the veracity of these important assumptions by examining rapidly emerging genetic studies that point to population-associated genetic variation within or near FADS genes in the FADS cluster on chromosome 11q12-13.1 as being critical to an individual's levels of LC-PUFAs, disease biomarkers and the risk of the diseases themself.
The role of FADS genetic variants in the Biosynthesis of LC-PUFAs in the context of population diversity
As mentioned above, the desaturase enzymes encoded by genes in the FADS cluster are consistently recognized as a bottleneck that determines the levels of LC-PUFA through the biosynthetic pathway. There have been several excellent reviews on the role of FADS genetic variants as genetic determinants of PUFA levels [20, 21] and we present a birds-eye-view of the consistency of this data in Figure 2. The FADS cluster comprising three genes (FADS1, FADS2 and FADS3) is essentially a single large expanse of high linkage disequilibrium (LD) in populations of European ancestry which explains the numerous single nucleotide polymorphisms (SNPs) in the literature that have been shown to have exceedingly strong effects on PUFA metabolism (Figure 2) [22]. Subsequent to the first candidate gene study by Shaeffer et al. [23] there have been close to 20 candidate gene studies [22-43] lending support to the role these variants play, with a particular note that the dramatic effects are not only for the inter-individual variation in a single PUFA level (e.g., ARA or DGLA) but rather in those product-precursor ratios such as ARA/DGLA that specifically serve a surrogate markers for the efficiency by which PUFAs are moving through a given step (FADS1 in this case) within the biosynthetic pathway. In fact, these effects described by candidate genes studies have found strong support in the genome-wide association (GWA) approach as well; the peak associated SNP, rs174537, yielded a p-value of p = 5.95 × 10-46 and accounted for 18.6% of the additive variance in ARA, perhaps one the strongest GWAS-identified allelic effects to date [44]. Figure 2 documents these effects these SNPs have on both arms of the LC-PUFA metabolism pathway across numerous SNPs in the region.
Figure 2.
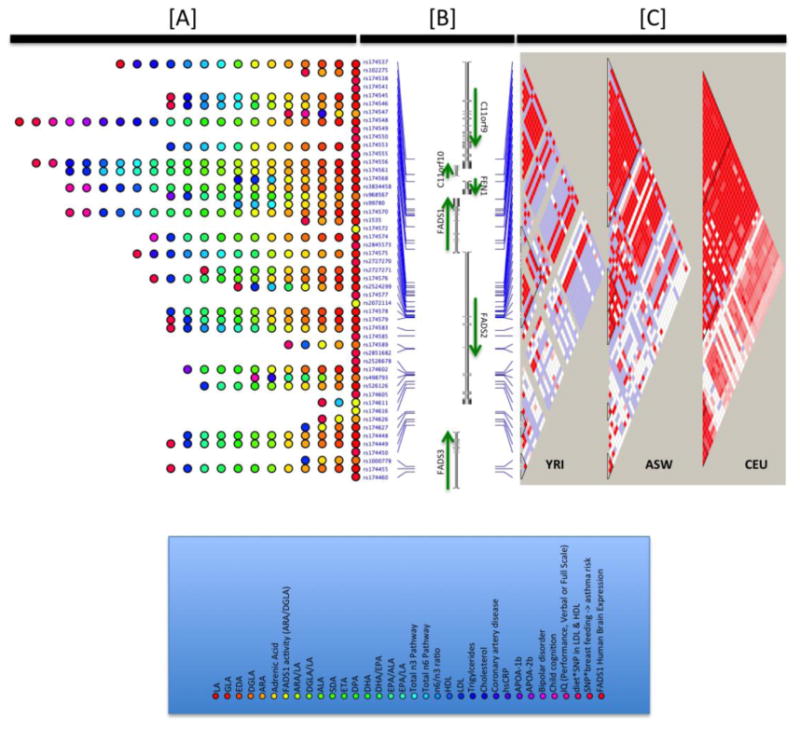
Illustration of the associations observed between genetic variants within and near the FADS locus and phenotypes/traits of relevance. Panel A shows the orientation of genes in this regions and the SNPs implicated across the range of studies ([20-43, 56-60, 86-93]), phenotypes presented are those with p<0.05 in at least one reviewed study. Panel B illustrates the strength of linkage disequilibrium in three populations from the Thousand Genomes Project [94], including the European ancestry CEU samples, African YRI samples and the African American ASW samples. The strength of LD between variants in this region reveals an extensive block of high LD (red regions) in the European ancestry samples and smaller blocks in the African ancestry samples. Panel C indicates the range of phenotypes with p<0.05 at each SNP under consideration.
In this review, these well-established effects are not the focus. Rather, we turn to the differences noted across populations. Tragically, as is typical for most diseases and traits that have been the focus of our attention in the past decade [45, 46], PUFA metabolism has had a paucity of literature and attention in populations of African and Hispanic ancestry. For example, a recent review by Johnson and Fritsche concluded that there was little evidence that addition of LA to the diet increases the concentration of inflammatory markers in healthy humans [47]. However, this review had several critical limitations, but most importantly, only one of the fifteen studies may have utilized subjects other than European or European ancestry populations; even this one South African study did not reveal the racial composition of subjects. There have been only two recent studies in African Americans, both of which document a dramatic difference in LC-PUFA levels and product-precursor ratios (pathway efficiency) in total plasma lipids between African Americans and European Americans in the United States [22, 24], with the evidence pointing to enhanced FADS1 efficiency (p=9.80×10-11 for DGLA, p=1.35×10-48 for ARA, and p=2.06×10-38 for ARA/DGLA) [22] as being a pivotal component to these differences. DGLA is lower in African Americans in contrast to ARA, which is higher in African Americans compared to European Americans [22, 24]. Similar trends were recently reported within the Multi-Ethnic Study of Atherosclerosis Risk (MESA) cohort when examining plasma phospholipids [48].
These studies confirm that variants within the region of expansive LD encompassing the FADS cluster are also key determinants of LC-PUFA metabolism in African Americans, but also highlight two critical points: (1) the studies indicate remarkably similar allelic effects in both racial group; and (2) the studies were the first insight into dramatic differences in allele frequencies between the two groups. We first showed that 79-82% of African Americans carry two copies of the G allele compared to only 42-45% of European Americans, where the G allele is the allele that has increased levels of ARA, decreased levels of DGLA and increased enzymatic efficiency (ARA/DGLA ratio) at rs174537.
There appears to be compelling evidence that the G allele at rs174537 is the derived allele and swept to fixation within African, is maintained at intermediate levels in Europe and European ancestry populations and at very low frequencies in Native American in the US [49]. Ameur et al. discuss that a common haplotype associated with the enhanced enzymatic efficiency of FADS1 is specific to humans appearing after the split of the common ancestor of humans and Neanderthals. Similar to our work [49], they also demonstrate that this haplotype shows evidence of positive selection in African populations [50]. Nonetheless, no matter the processes that led to these dramatic evolutionary pressures, speculated by both groups to be related to the proportionally large human brain relative to body size that is unique among primates, the present geographic distribution of the LC-PUFA enhancing alleles/haplotypes is unequivocally higher in populations of African ancestry. Coincidentally (or not), African Americans represent a fraction of the population in the United States that bears a disproportional burden of chronic diseases of inflammation [51-53]. We Believe these observations warrant a redirection of the ongoing debate surrounding the AHA's current dietary recommendations on PUFA intake away from a one-toned discussion on its appropriateness within a ‘one size fits all approach’, to a more personalized argument given an individual's innate ability to synthesize LC-PUFA (with resulting molecular and clinical phenotypes at a more rapid rate given their specific genetic architecture.
The wide impact of FADS genetic variants
While it is clear that FADS variants have an important impact on LC-PUFA levels and ratios of PUFA products to precursors, it is likely that resulting additional molecular and clinical phenotypes associated with FADS variation place individuals at varying risk. For example, there are studies that document the role FADS variants in complex lipid and inflammatory phenotypes and coronary artery disease (CAD) [20, 21]. Over the past decade, genome-wide association studies (GWAS) have identified a number of genetic polymorphisms that convey increased risk for coronary artery disease, diabetes, cancer, and other common diseases, including some that implicate FADS variants [54, 55]. Associations have also been documented between FADS variants and traditional markers of cardiovascular disease, including LDL-cholesterol, triglycerides, HDL-cholesterol, and total cholesterol levels [56-59]. A recent study including ∼8,000 African Americans [60] from five cohorts leveraged the LD patterns in African Americans to identify SNPs more strongly associated with these phenotypes than previously reported GWAS SNPs in the Caucasian studies above, including for FADS variants, confirming FADS importance in CAD and its associated phenotypes across populations.
Associations have also been documented for phospholipid metabolites with four double bonds (i.e. ARA) for all major phospholipid species (98). Perhaps it is not surprising that strong associations are noted with FADS SNPs and LC-PUFA-containing glycerolipids as well as total cholesterol, LDL-cholesterol, HDL-cholesterol and triglycerides given that PUFA-containing gylcerolipids are key molecular components (intermediate phenotypes) of lipoprotein particles. However, there are also now several studies that indicate specific FADS haplotypes favoring high desaturase activity lead to enhanced levels of inflammatory and CAD biomarkers including oxidative products of ARA (104; 116; 117). In one of the earliest studies in this area, Martinelli and colleagues demonstrated that a higher ARA/LA ratio (presumably representing more conversion of LA to ARA via the FADS cluster) is an independent risk factor for CAD. Additionally, they linked ARA/LA ratios to systemic inflammation by showing that concentrations of high-sensitivity C-reactive protein (CRP) increased progressively across tertiles of ARA/LA. Importantly, increases in CRP concentrations and CAD risk were related to the FADS haplotypes associated with higher levels ARA and ARA/LA ratios. (119) The ARA/LA ratio was also found to be higher in CAD patients and the low efficiency/capacity T allele at rs174537 was associated with a lower risk of CAD in a Chinese population. Contradictory evidence however does exist (120) which may result from the interaction between the balance of the n-6 to n-3 PUFA dietary environment and specific genotypes in the FADS cluster of the cohort being examined.
And then there is the important issue of gene-gene interactions between variants in the FADS cluster and variants in other genes that control LC-PUFA metabolism to eicosanoids (Figure 1). There are now numerous studies that support the concept that specific variants in genes whose activity are responsible for releasing ARA from membrane phospholipids (such as cytosolic PLA2 alpha; PLA2G4) or metabolizing ARA to eicosanoids (such as 5-lipoxygenase; ALOX5) may further be a source of gene-diet interactions and predispose certain individuals and populations exposed to high amounts of n-6 PUFAsto enhanced inflammation and cardiovascular disease [61-63]. For example, once ARA is released from glycerolipids via a PLA2 cleavage, 5-lipoxygenase (ALOX5) becomes the rate-limiting step in leukotriene generation from ARA (Figure 2). While this group of eicosanoids were originally been identified with inflammation and bronchoconstriction associated with asthma and allergies, a large body of evidence has emerged over the past decade that demonstrates that leukotrienes play a key pro-athrogenic role particularly in CVD [64-66].
The importance of FADS variation extends well beyond the central arguments around cardiovascular disease and recommendations by the AHA, to include cancer, mental health and brain development. For example, there has been an intense focus on its role in levels of n-3 and n-6 LC-PUFAs in human milk and cognitive development in children.[67-69] [70]. Lattka et al provide an excellent review of the contradictory nature of the literature on prenatal and neonatal maternal diet and/or supplementation regarding LC-PUFA [21] and its consequent effects on measures of intelligence in the child. Many of the contradictions are speculated to be a result of the lack of consideration in maternal and child genotype at key determinants of LC-PUFA synthesis and metabolism, including FADS, an argument supported by gene*environment interaction studies in the field [71]. Associations of FADS variants have also been reported with attention deficit/hyperactivity disorder [72] mono- and bi-polar depression disorder [73], all of which have had previous linkage scans [74-76] that highlight this chromosomal region in families with the disorder. Pre-clinical animal and some clinical studies have provide compelling evidence that LC-PUFA levels and ARA metabolism to eicosanoids are important factors several cancers including colon, prostate and breast cancers. Interestingly, these are many of the same cancers where major disparities between African and European Americans are observed with incidence and severity being higher in African Americans. However, overall examination of epidemiologic studies provides an inconsistent picture of the associations between dietary PUFAs and cancer. Genetic variation in LC-PUFA biosynthesis and particularly within the FADS cluster have the potential to help clarify when and how LC-PUFAs play an important role in cancer.
The interplay between genes and diet
So what are we to make of all of these data and what are the major missing pieces of information necessary to make meaningful recommendations? As mentioned above, the single biggest problem in this regard is there have been few studies utilizing populations other than European and more recently Asian ancestry. For example, the question of capacity through the PUFA biosynthetic pathway needs to be addressed in African ancestry and other populations. There are at least 5 very excellent studies that show that increasing dietary LA intake above 3% of energy has no impact on circulating and tissue ARA levels [77-81]. However, these too were carried out in small numbers of volunteers from Europe (Netherlands, Spain, Germany), Canada (British Columbia) and Australia and in at least one of the studies, there were major outliers (68). We hypothesize that significantly higher concentrations of LA will be converted to ARA in African Ancestry populations where genetic variants are much more favorable (>80% carry GG at rs174537) for enhanced ARA levels. What if 6-8% of energy from LA were converted to ARA in African Americans? Would this change the dietary recommendations for PUFAs? We certainly believe it should given the role ARA and its eicosanoid product plays in human diseases and the link between FADS variants to a variety of human disease and particularly CAD. Given the amount of data implicating variants in the rate limiting steps (enzymes encoded for in the FADS cluster) in LC-PUFA biosynthesis to human disease, the interplay between dietary PUFAs exposure and variants in genes that synthesize and metabolize LC-PUFAs must be better understood. Until we better understand this interplay, the association studies outlined in this review, the recent meta analyses showing substituting n-6 PUFAs (not mixed with n-3 PUFAs) for saturated and trans fatty acids actually trends toward an increase risk of death, the reexamined the Sydney Diet Heart which showed that although the LA intervention group had lower levels of total cholesterol, it had unexpectedly higher rates of death than controls, jointly indicate that we should observe great caution in making n-6 PUFA reccommendations. Understanding the relationship between PUFA exposure and FADS gene variants will likely provide clarity for not only cardiovascular disease, but also a range of human diseases (cognitive, mental, inflammatory and proliferative [cancer]) diseases.
Conclusions
Much of the discussion above has focused on the impact of genetic variation on LC-PUFA biosynthesis. A major limitation of the field as it currently exists is that little is known about how these SNPs work to regulate PUFA levels, shunting down and between the two arms of the pathway illustrated in Figure 1. While numerous studies use terms such as desaturase activities or efficiencies, these are not true activities (as would be measured with an isolated enzyme or subcellular fraction), but are typically ascertained by measuring PUFA precursor/product ratios within circulating or cellular lipids. There are postulated mechanisms that connect LC-PUFA exposure to the transcriptional machinery of the PUFA biosynthetic pathway [82], variants in this region have been identified as eQTLS [49], and the idea that gene expression is epigenetically modulated cannot ignored [83, 84]. For example, it is interesting to note that two recent studies show that methylation of in a promoter region of ELOVL2 to be among top 3 or 4 hits in the entire human genome with regard to association with age [85]. ELOVL2 is the first step that shows selectivity for n-3 substrates in the PUFA biosynthetic pathway. These data suggest as have other human and animal studies that age may play a critical role in LC-PUFA biosynthesis. We also know that other factors (such as sex and pregnancy status) impacts LC-PUFA biosynthesis. These raise important questions as to how these factors can be incorporated into PUFA recommendations.
The key point to highlight with this review is that the impact of genetic variants are likely to be population-specific given that frequency of the variants in PUFA biosynthetic genes and the dietary precursor PUFA environment maybe different between populations. For example, higher dietary exposure to ALA relative to LA together with high efficiency/capacity variants may markedly shift the balance from ARA towardn-3 LC-PUFAs (DHA, DPA, and EPA) and a wide array of protective n-3 containing eicosanoids and endocannabinoids (intermediate phenotypes). Alternatively, higher dietary exposure to LA relative to ALA together with a high frequency of high efficiency/capacity variants likely shifts the balance back to ARA, proinflammatory eicosanoids and endocannabinoids, inflammation and associated human disease. And if these high efficiency/capacity variants give rise to higher levels of FADS gene expression (as preliminary studies suggest) and protein levels, then the capacity to make ARA may increase well past 2-3% of energy because the enzymatic capacity within cells and tissues have risen with more protein. This is the most likely explanation for what is being observed with the very strong associations between these variant and markedly higher ARA levels (particularly in populations of African ancestry). The major point of this review is that it is highly unlikely that simple dietary recommendations can be applied to diverse populations given the immense complexity and differences in genetic variation of the PUFA biosynthetic pathway in different populations. Even more important, it would be tragic if the need for simplicity drove dietary recommendations that actually harmed certain populations.
Acknowledgments
This work was supported by a grant from the National Institutes of Health, P50 AT002782.
Abbreviations
- ALA
α-linolenic acid
- ARA
arachidonic acid
- COX
cyclooxygenase
- DGLA
dihomo γ-linolenic acid
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- ETA
eicosatetraenoic acid
- CAD
coronary artery disease
- CVD
cardiovascular disease
- FADS
fatty acid desaturase
- GLA
γ-linolenic acid
- GWAS
genome wide association study
- IMT
intima-media thickness
- LA
linoleic acid
- Lc
long chain
- LD
linkage disequilibrium
- LTB4
leukotriene B4
- C18
18 carbon
- MWD
modern western diet
- NSAID
non-steroidal anti-inflammatory drug
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PI
phosphatidylinositol
- PS
phosphatidylserine
- PLA2
phospholipase A2
- PUFA
polyunsaturated fatty acid
- SDA
stearidonic acid
Footnotes
Compliance with Ethics Guidelines: Conflict of Interest: Rasika A. Mathias declares that she has no conflict of interest.
Vrindarani Pani declares that she has no conflict of interest.
Floyd H. Chilton is an unpaid consultant for Gene Smart Health and receives no compensation or equity in this role. This information has been disclosed to Wake Forest University Health Sciences (WFUHS) and outside sponsors, as appropriate, and is institutionally managed.
Human and Animal Rights and Informed Consent: This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Park WJ, et al. An alternate pathway to long-chain polyunsaturates: the FADS2 gene product Delta8-desaturates 20:2n-6 and 20:3n-3. J Lipid Res. 2009;50(6):1195–202. doi: 10.1194/jlr.M800630-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sprecher H. Biochemistry of essential fatty acids. Prog Lipid Res. 1981;20:13–22. doi: 10.1016/0163-7827(81)90009-6. [DOI] [PubMed] [Google Scholar]
- 3.Poisson JP, et al. Evidence that liver microsomes of human neonates desaturate essential fatty acids. Biochim Biophys Acta. 1993;1167(2):109–13. doi: 10.1016/0005-2760(93)90149-4. [DOI] [PubMed] [Google Scholar]
- 4.Horrobin DF. Fatty acid metabolism in health and disease: the role of delta-6-desaturase. Am J Clin Nutr. 1993;57(5 Suppl):732S–736S. doi: 10.1093/ajcn/57.5.732S. discussion 736S-737S. [DOI] [PubMed] [Google Scholar]
- 5.el Boustani S, et al. Direct in vivo characterization of delta 5 desaturase activity in humans by deuterium labeling: effect of insulin. Metabolism. 1989;38(4):315–21. doi: 10.1016/0026-0495(89)90117-0. [DOI] [PubMed] [Google Scholar]
- 6.Blasbalg TL, et al. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr. 2011;93(5):950–62. doi: 10.3945/ajcn.110.006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simopoulos AP. Importance of the omega-6/omega-3 balance in health and disease: evolutionary aspects of diet. World Rev Nutr Diet. 2011;102:10–21. doi: 10.1159/000327785. [DOI] [PubMed] [Google Scholar]
- 8.Gibson RA, et al. Docosahexaenoic acid synthesis from alpha-linolenic acid is inhibited by diets high in polyunsaturated fatty acids. Prostaglandins Leukot Essent Fatty Acids. 2013;88(1):139–46. doi: 10.1016/j.plefa.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Hibbeln JR, et al. Healthy intakes of n-3 and n-6 fatty acids: estimations considering worldwide diversity. Am J Clin Nutr. 2006;83(6 Suppl):1483S–1493S. doi: 10.1093/ajcn/83.6.1483S. [DOI] [PubMed] [Google Scholar]
- 10.Smith WL. The eicosanoids and their biochemical mechanisms of action. Biochem J. 1989;259(2):315–24. doi: 10.1042/bj2590315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serhan CN, et al. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their endogenous aspirin-triggered epimers. Lipids. 2004;39(11):1125–32. doi: 10.1007/s11745-004-1339-7. [DOI] [PubMed] [Google Scholar]
- 12.Serhan CN, et al. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: an overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat. 2004;73(3-4):155–72. doi: 10.1016/j.prostaglandins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Serhan CN. Novel eicosanoid and docosanoid mediators: resolvins, docosatrienes, and neuroprotectins. Curr Opin Clin Nutr Metab Care. 2005;8(2):115–21. doi: 10.1097/00075197-200503000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb. 1992;12(8):911–9. doi: 10.1161/01.atv.12.8.911. [DOI] [PubMed] [Google Scholar]
- 15.Harris WS, et al. Omega-6 fatty acids and risk for cardiovascular disease: a science advisory from the American Heart Association Nutrition Subcommittee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Cardiovascular Nursing; and Council on Epidemiology and Prevention. Circulation. 2009;119(6):902–7. doi: 10.1161/CIRCULATIONAHA.108.191627. [DOI] [PubMed] [Google Scholar]
- 16.Siguel E. A new relationship between total/high density lipoprotein cholesterol and polyunsaturated fatty acids. Lipids. 1996;31(Suppl):S51–6. doi: 10.1007/BF02637051. [DOI] [PubMed] [Google Scholar]
- 17.Ramsden CE, Hibbeln JR, Majchrzak-Hong SF. All PUFAs are not created equal: absence of CHD benefit specific to linoleic acid in randomized controlled trials and prospective observational cohorts. World Rev Nutr Diet. 2011;102:30–43. doi: 10.1159/000327789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramsden CE, et al. n-6 fatty acid-specific and mixed polyunsaturate dietary interventions have different effects on CHD risk: a meta-analysis of randomised controlled trials. Br J Nutr. 2010;104(11):1586–600. doi: 10.1017/S0007114510004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramsden CE, et al. Use of dietary linoleic acid for secondary prevention of coronary heart disease and death: evaluation of recovered data from the Sydney Diet Heart Study and updated meta-analysis. BMJ. 2013;346:e8707. doi: 10.1136/bmj.e8707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glaser C, Heinrich J, Koletzko B. Role of FADS1 and FADS2 polymorphisms in polyunsaturated fatty acid metabolism. Metabolism. 2010;59(7):993–9. doi: 10.1016/j.metabol.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 21.Lattka E, et al. Do FADS genotypes enhance our knowledge about fatty acid related phenotypes? Clin Nutr. 2010;29(3):277–87. doi: 10.1016/j.clnu.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 22••.Mathias RA, et al. The impact of FADS genetic variants on omega6 polyunsaturated fatty acid metabolism in African Americans. BMC Genet. 2011;12:50. doi: 10.1186/1471-2156-12-50. This article provides the first insight into profound differences in LA-PUFA metabolism and levels between African Americans and European Americans with accompanying differences in genetic determinants of the same. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schaeffer L, et al. Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum Mol Genet. 2006;15(11):1745–56. doi: 10.1093/hmg/ddl117. [DOI] [PubMed] [Google Scholar]
- 24.Sergeant S, et al. Differences in arachidonic acid levels and fatty acid desaturase (FADS) gene variants in African Americans and European Americans with diabetes or the metabolic syndrome. Br J Nutr. 2012;107(4):547–55. doi: 10.1017/S0007114511003230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathias RA, et al. FADS genetic variants and omega-6 polyunsaturated fatty acid metabolism in a homogeneous island population. J Lipid Res. 2010;51(9):2766–74. doi: 10.1194/jlr.M008359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malerba G, et al. SNPs of the FADS gene cluster are associated with polyunsaturated fatty acids in a cohort of patients with cardiovascular disease. Lipids. 2008;43(4):289–99. doi: 10.1007/s11745-008-3158-5. [DOI] [PubMed] [Google Scholar]
- 27.Rzehak P, et al. Evidence for an association between genetic variants of the fatty acid desaturase 1 fatty acid desaturase 2 (FADS1 FADS2) gene cluster and the fatty acid composition of erythrocyte membranes. Br J Nutr. 2009;101(1):20–6. doi: 10.1017/S0007114508992564. [DOI] [PubMed] [Google Scholar]
- 28.Xie L, Innis SM. Association of fatty acid desaturase gene polymorphisms with blood lipid essential fatty acids and perinatal depression among Canadian women: a pilot study. J Nutrigenet Nutrigenomics. 2009;2(4-5):243–50. doi: 10.1159/000255636. [DOI] [PubMed] [Google Scholar]
- 29.Xie L, Innis SM. Genetic variants of the FADS1 FADS2 gene cluster are associated with altered (n-6) and (n-3) essential fatty acids in plasma and erythrocyte phospholipids in women during pregnancy and in breast milk during lactation. J Nutr. 2008;138(11):2222–8. doi: 10.3945/jn.108.096156. [DOI] [PubMed] [Google Scholar]
- 30.Porenta SR, et al. Interaction of fatty acid genotype and diet on changes in colonic fatty acids in a Mediterranean diet intervention study. Cancer Prev Res (Phila) 2013;6(11):1212–21. doi: 10.1158/1940-6207.CAPR-13-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong SH, et al. Association of polymorphisms in FADS gene with age-related changes in serum phospholipid polyunsaturated fatty acids and oxidative stress markers in middle-aged nonobese men. Clin Interv Aging. 2013;8:585–96. doi: 10.2147/CIA.S42096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harslof LB, et al. FADS genotype and diet are important determinants of DHA status: a cross-sectional study in Danish infants. Am J Clin Nutr. 2013;97(6):1403–10. doi: 10.3945/ajcn.113.058685. [DOI] [PubMed] [Google Scholar]
- 33.Li SW, et al. FADS gene polymorphisms confer the risk of coronary artery disease in a Chinese Han population through the altered desaturase activities: based on high-resolution melting analysis. PLoS One. 2013;8(1):e55869. doi: 10.1371/journal.pone.0055869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillingham LG, et al. Dietary oils and FADS1-FADS2 genetic variants modulate [13C]alpha-linolenic acid metabolism and plasma fatty acid composition. Am J Clin Nutr. 2013;97(1):195–207. doi: 10.3945/ajcn.112.043117. [DOI] [PubMed] [Google Scholar]
- 35.Freemantle E, et al. Age and haplotype variations within FADS1 interact and associate with alterations in fatty acid composition in human male cortical brain tissue. PLoS One. 2012;7(8):e42696. doi: 10.1371/journal.pone.0042696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lattka E, et al. Umbilical cord PUFA are determined by maternal and child fatty acid desaturase (FADS) genetic variants in the Avon Longitudinal Study of Parents and Children (ALSPAC) Br J Nutr. 2013;109(7):1196–210. doi: 10.1017/S0007114512003108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steer CD, et al. Polyunsaturated fatty acid levels in blood during pregnancy, at birth and at 7 years: their associations with two common FADS2 polymorphisms. Hum Mol Genet. 2012;21(7):1504–12. doi: 10.1093/hmg/ddr588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morales E, et al. Genetic variants of the FADS gene cluster and ELOVL gene family, colostrums LC-PUFA levels, breastfeeding, and child cognition. PLoS One. 2011;6(2):e17181. doi: 10.1371/journal.pone.0017181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lattka E, et al. Genetic variants in the FADS gene cluster are associated with arachidonic acid concentrations of human breast milk at 1.5 and 6 mo postpartum and influence the course of milk dodecanoic, tetracosenoic, and trans-9-octadecenoic acid concentrations over the duration of lactation. Am J Clin Nutr. 2011;93(2):382–91. doi: 10.3945/ajcn.110.004515. [DOI] [PubMed] [Google Scholar]
- 40.Koletzko B, et al. Genetic variants of the fatty acid desaturase gene cluster predict amounts of red blood cell docosahexaenoic and other polyunsaturated fatty acids in pregnant women: findings from the Avon Longitudinal Study of Parents and Children. Am J Clin Nutr. 2011;93(1):211–9. doi: 10.3945/ajcn.110.006189. [DOI] [PubMed] [Google Scholar]
- 41.Kwak JH, et al. FADS gene polymorphisms in Koreans: association with omega6 polyunsaturated fatty acids in serum phospholipids, lipid peroxides, and coronary artery disease. Atherosclerosis. 2011;214(1):94–100. doi: 10.1016/j.atherosclerosis.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 42.Rzehak P, et al. Variants of the FADS1 FADS2 gene cluster, blood levels of polyunsaturated fatty acids and eczema in children within the first 2 years of life. PLoS One. 2010;5(10):e13261. doi: 10.1371/journal.pone.0013261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bokor S, et al. Single nucleotide polymorphisms in the FADS gene cluster are associated with delta-5 and delta-6 desaturase activities estimated by serum fatty acid ratios. J Lipid Res. 2010;51(8):2325–33. doi: 10.1194/jlr.M006205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka T, et al. Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI Study. PLoS Genet. 2009;5(1):e1000338. doi: 10.1371/journal.pgen.1000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenberg NA, et al. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010;11(5):356–66. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bustamante CD, Burchard EG, De la Vega FM. Genomics for the world. Nature. 2011;475(7355):163–5. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson GH, Fritsche K. Effect of dietary linoleic acid on markers of inflammation in healthy persons: a systematic review of randomized controlled trials. J Acad Nutr Diet. 2012;112(7):1029–41. 1041 e1–15. doi: 10.1016/j.jand.2012.03.029. [DOI] [PubMed] [Google Scholar]
- 48.Steffen BT, et al. Ethnicity, plasma phospholipid fatty acid composition and inflammatory/endothelial activation biomarkers in the Multi-Ethnic Study of Atherosclerosis (MESA) Eur J Clin Nutr. 2012;66(5):600–5. doi: 10.1038/ejcn.2011.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49•.Mathias RA, et al. Adaptive evolution of the FADS gene cluster within Africa. PLoS One. 2012;7(9):e44926. doi: 10.1371/journal.pone.0044926. This article provides compelling evidence that genetic determinants of LS-PUFA metabolism differ across populations, is particularly important in populations of African ancestry, and have undergone strong effects of selective pressure within Africa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.Ameur A, et al. Genetic adaptation of fatty-acid metabolism: a human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am J Hum Genet. 2012;90(5):809–20. doi: 10.1016/j.ajhg.2012.03.014. This article provides compelling evidence that genetic determinants of LS-PUFA metabolism differ across populations, is particularly important in populations of African ancestry, and have undergone strong effects of selective pressure within Africa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Braveman P. Health disparities and health equity: concepts and measurement. Annu Rev Public Health. 2006;27:167–94. doi: 10.1146/annurev.publhealth.27.021405.102103. [DOI] [PubMed] [Google Scholar]
- 52.Sankar P, Cho MK, Mountain J. Race and ethnicity in genetic research. Am J Med Genet A. 2007;143A(9):961–70. doi: 10.1002/ajmg.a.31575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuzawa CW, Sweet E. Epigenetics and the embodiment of race: developmental origins of US racial disparities in cardiovascular health. Am J Hum Biol. 2009;21(1):2–15. doi: 10.1002/ajhb.20822. [DOI] [PubMed] [Google Scholar]
- 54.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106(23):9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hindorff LA, et al. A Catalog of Published Genome-Wide Association Studies. cited 2014 Available from: http://www.genome.gov/gwastudies.
- 56.Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40(2):189–97. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Willer CJ, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40(2):161–9. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aulchenko YS, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009;41(1):47–55. doi: 10.1038/ng.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kathiresan S, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41(1):56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lettre G, et al. Genome-wide association study of coronary heart disease and its risk factors in 8,090 African Americans: the NHLBI CARe Project. PLoS Genet. 2011;7(2):e1001300. doi: 10.1371/journal.pgen.1001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Helgadottir A, et al. Association between the gene encoding 5-lipoxygenase-activating protein and stroke replicated in a Scottish population. Am J Hum Genet. 2005;76(3):505–9. doi: 10.1086/428066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Helgadottir A, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet. 2004;36(3):233–9. doi: 10.1038/ng1311. [DOI] [PubMed] [Google Scholar]
- 63.Lohmussaar E, et al. ALOX5AP gene and the PDE4D gene in a central European population of stroke patients. Stroke. 2005;36(4):731–6. doi: 10.1161/01.STR.0000157587.59821.87. [DOI] [PubMed] [Google Scholar]
- 64.Funk CD. Leukotriene modifiers as potential therapeutics for cardiovascular disease. Nat Rev Drug Discov. 2005;4(8):664–72. doi: 10.1038/nrd1796. [DOI] [PubMed] [Google Scholar]
- 65.Riccioni G, Back M, Capra V. Leukotrienes and atherosclerosis. Curr Drug Targets. 2010;11(7):882–7. doi: 10.2174/138945010791320881. [DOI] [PubMed] [Google Scholar]
- 66.Mehrabian M, et al. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ Res. 2002;91(2):120–6. doi: 10.1161/01.res.0000028008.99774.7f. [DOI] [PubMed] [Google Scholar]
- 67.Marangoni F, et al. Polyunsaturated fatty acids in maternal plasma and in breast milk. Prostaglandins Leukot Essent Fatty Acids. 2002;66(5-6):535–40. doi: 10.1054/plef.2002.0396. [DOI] [PubMed] [Google Scholar]
- 68.Marangoni F, et al. Cigarette smoke negatively and dose-dependently affects the biosynthetic pathway of the n-3 polyunsaturated fatty acid series in human mammary epithelial cells. Lipids. 2004;39(7):633–7. doi: 10.1007/s11745-004-1276-5. [DOI] [PubMed] [Google Scholar]
- 69.Smit EN, et al. Estimated biological variation of the mature human milk fatty acid composition. Prostaglandins Leukot Essent Fatty Acids. 2002;66(5-6):549–55. doi: 10.1054/plef.2002.0398. [DOI] [PubMed] [Google Scholar]
- 70.Carnielli VP, et al. The very low birth weight premature infant is capable of synthesizing arachidonic and docosahexaenoic acids from linoleic and linolenic acids. Pediatr Res. 1996;40(1):169–74. doi: 10.1203/00006450-199607000-00029. [DOI] [PubMed] [Google Scholar]
- 71.Caspi A, et al. Moderation of breastfeeding effects on the IQ by genetic variation in fatty acid metabolism. Proc Natl Acad Sci U S A. 2007;104(47):18860–5. doi: 10.1073/pnas.0704292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brookes KJ, et al. Association of fatty acid desaturase genes with attention-deficit/hyperactivity disorder. Biol Psychiatry. 2006;60(10):1053–61. doi: 10.1016/j.biopsych.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 73.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arcos-Burgos M, et al. Attention-deficit/hyperactivity disorder in a population isolate: linkage to loci at 4q13.2, 5q33.3, 11q22, and 17p11. Am J Hum Genet. 2004;75(6):998–1014. doi: 10.1086/426154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ogdie MN, et al. Attention deficit hyperactivity disorder: fine mapping supports linkage to 5p13, 6q12, 16p13, and 17p11. Am J Hum Genet. 2004;75(4):661–8. doi: 10.1086/424387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fallin MD, et al. Genomewide linkage scan for bipolar-disorder susceptibility loci among Ashkenazi Jewish families. Am J Hum Genet. 2004;75(2):204–19. doi: 10.1086/422474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ferrucci L, et al. Relationship of plasma polyunsaturated fatty acids to circulating inflammatory markers. J Clin Endocrinol Metab. 2006;91(2):439–46. doi: 10.1210/jc.2005-1303. [DOI] [PubMed] [Google Scholar]
- 78.Montoya MT, et al. Fatty acid saturation of the diet and plasma lipid concentrations, lipoprotein particle concentrations, and cholesterol efflux capacity. Am J Clin Nutr. 2002;75(3):484–91. doi: 10.1093/ajcn/75.3.484. [DOI] [PubMed] [Google Scholar]
- 79.Chan JK, et al. Effect of dietary alpha-linolenic acid and its ratio to linoleic acid on platelet and plasma fatty acids and thrombogenesis. Lipids. 1993;28(9):811–7. doi: 10.1007/BF02536235. [DOI] [PubMed] [Google Scholar]
- 80.Thijssen MA, Mensink RP. Small differences in the effects of stearic acid, oleic acid, and linoleic acid on the serum lipoprotein profile of humans. Am J Clin Nutr. 2005;82(3):510–6. doi: 10.1093/ajcn.82.3.510. [DOI] [PubMed] [Google Scholar]
- 81.Liou YA, et al. Decreasing linoleic acid with constant alpha-linolenic acid in dietary fats increases (n-3) eicosapentaenoic acid in plasma phospholipids in healthy men. J Nutr. 2007;137(4):945–52. doi: 10.1093/jn/137.4.945. [DOI] [PubMed] [Google Scholar]
- 82.Wijendran V, et al. Dietary arachidonic acid and docosahexaenoic acid regulate liver fatty acid desaturase (FADS) alternative transcript expression in suckling piglets. Prostaglandins Leukot Essent Fatty Acids. 2013;89(5):345–50. doi: 10.1016/j.plefa.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoile SP, et al. Maternal fat intake in rats alters 20:4n-6 and 22:6n-3 status and the epigenetic regulation of Fads2 in offspring liver. J Nutr Biochem. 2013;24(7):1213–20. doi: 10.1016/j.jnutbio.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Burdge GC, Lillycrop KA. Fatty acids and epigenetics. Curr Opin Clin Nutr Metab Care. 2013 doi: 10.1097/MCO.0000000000000023. [DOI] [PubMed] [Google Scholar]
- 85.Garagnani P, et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell. 2012;11(6):1132–4. doi: 10.1111/acel.12005. [DOI] [PubMed] [Google Scholar]
- 86.Lu Y, et al. Dietary n-3 and n-6 polyunsaturated fatty acid intake interacts with FADS1 genetic variation to affect total and HDL-cholesterol concentrations in the Doetinchem Cohort Study. Am J Clin Nutr. 2010;92(1):258–65. doi: 10.3945/ajcn.2009.29130. [DOI] [PubMed] [Google Scholar]
- 87.Standl M, et al. FADS gene cluster modulates the effect of breastfeeding on asthma. Results from the GINIplus and LISAplus studies. Allergy. 2012;67(1):83–90. doi: 10.1111/j.1398-9995.2011.02708.x. [DOI] [PubMed] [Google Scholar]
- 88.Rizzi TS, et al. Genetic Variance in Combination with Fatty Acid Intake Might Alter Composition of the Fatty Acids in Brain. PLoS One. 2013;8(6):e68000. doi: 10.1371/journal.pone.0068000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aslibekyan S, et al. Fatty Acid desaturase gene variants, cardiovascular risk factors, and myocardial infarction in the costa rica study. Front Genet. 2012;3:72. doi: 10.3389/fgene.2012.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Molto-Puigmarti C, et al. Genetic variation in FADS genes and plasma cholesterol levels in 2-year-old infants: KOALA Birth Cohort Study. PLoS One. 2013;8(5):e61671. doi: 10.1371/journal.pone.0061671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gieger C, et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008;4(11):e1000282. doi: 10.1371/journal.pgen.1000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hellstrand S, et al. Intake levels of dietary long-chain PUFAs modify the association between genetic variation in FADS and LDL-C. J Lipid Res. 2012;53(6):1183–9. doi: 10.1194/jlr.P023721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Steer CD, et al. Maternal fatty acids in pregnancy, FADS polymorphisms, and child intelligence quotient at 8 y of age. Am J Clin Nutr. 2013;98(6):1575–82. doi: 10.3945/ajcn.112.051524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]