

Abstract
Background
The cotton (Gossypium spp.) fiber cell is an important unicellular model for studying cell differentiation. There is evidence suggesting that phosphorylation is a critical post-translational modification involved in regulation of a wide range of cell activities. Nevertheless, the sites of phosphorylation in G. hirsutum and their regulatory roles in fiber cell initiation are largely unknown. In this study, we employed a mass spectrometry-based phosphoproteomics to conduct a global and site-specific phosphoproteome profiling between ovules of a fuzzless-lintless (fl) Upland cotton (G. hirsutum) mutant and its isogenic parental wild type (WT) at -3 and 0 days post-anthesis (DPA).
Results
A total of 830 phosphopeptides and 1,592 phosphorylation sites from 619 phosphoproteins were identified by iTRAQ (isobaric tags for relative and absolute quantitation). Of these, 76 phosphoproteins and 1,100 phosphorylation sites were identified for the first time after searching the P3DB public database using the BLAST program. Among the detected phosphopeptides, 69 were differentially expressed between the fl mutant and its WT in ovules at -3 and 0 DPA. An analysis using the Motif-X program uncovered 19 phosphorylation motifs, 8 of which were unique to cotton. A further metabolic pathway analysis revealed that the differentially phosphorylated proteins were involved in signal transduction, protein modification, carbohydrate metabolic processes, and cell cycle and cell proliferation.
Conclusions
Our phosphoproteomics-based research provides the first global overview of phosphorylation during cotton fiber initiation, and also offers a helpful dataset for elucidation of signaling networks in fiber development of G. hirsutum.
Electronic supplementary material
The online version of this article (doi: 10.1186/1471-2164-15-466) contains supplementary material, which is available to authorized users.
Keywords: Gossypium hirsutum, Fuzzless-lintless mutant, Fiber initiation, Phosphoproteomics
Background
Cotton is an important global economic crop that is widely grown for production of textile fiber materials and cottonseed oil [1]. Cotton fibers are single seed trichomes derived from epidermal cells, and their development occurs in four steps: fiber initiation, elongation, secondary cell-wall biosynthesis, and maturation [2, 3]. About 15–25% of epidermal cells differentiate before or on the day of anthesis, and then develop into lint fibers [4]. In addition to their economic value, cotton fiber cells also serve as a classical biological model system for researching mechanisms of plant cell differentiation and elongation [5]. Great progress has been made in illuminating cotton fiber metabolic pathways and molecular mechanisms [6], but most studies have been focused on gene regulation at the transcriptional and, post-transcriptional levels, and a few at the translational level [7–9].
One well-studied cotton line, the Xuzhou 142 fuzzless-lintless (fl) mutant, serves as a classical genetic material to investigate molecular events specific to fiber differentiation and initiation. Xuzhou 142 fl was found in wild-type Xuzhou 142, a cultivar with lint and fuzz fibers. Although the fiberless phenotype is reportedly controlled by two recessive genes [10], the genetics of cotton fuzzless-lintless fiber production is not well understood. Several studies have been conducted to examine cotton fiber initiation process. Wu et al. [9] used mRNA from 0 day post-anthesis (DPA) ovules of wild-type cotton and six reduced-fiber or fiberless mutants to probe a cotton cDNA microarray covering about 10,410 ovule cDNA clones, and eventually narrowed down the fuzzless-lintless candidate genes to 13. Using Illumina sequencing of transcriptomes of -2 to 1 DPA cotton ovules, Wang et al. [11] compared Xuzhou 142 WT with its fl mutant and identified 130 up-regulated genes and 442 down-regulated genes in the WT. In a proteomics analysis, Liu et al. [12] compared -3 DPA and 0 DPA ovules between Xuzhou 142 and its fl mutant using a two-dimensional electrophoresis and a tandem mass spectrometry (MS/MS) technology. They found 46 differentially expressed proteins between WT and fl ovules. In addition to these studies, as proteomic technology has developed, the number of proteins detectable in a complex protein sample has increased rapidly, and spectral counting in quantitative proteomics has gained recognition [13, 14]. Recent sequencing of the G. raimondii genome has also provided excellent tools and resources to study cotton in a greater depth [15]. The data generated by above studies can assist functional annotation of genes and proteins associated with cotton fiber differentiation and initiation.
Despite the importance of cotton fiber differentiation and initiation, the roles of post-translational modifications, especially the reversible phosphorylation of proteins, remain a mystery. Phosphorylation is one of the most important protein post-translational modifications, and is involved in regulating many biological activities. Phosphorylation of specific intracellular proteins/enzymes by protein kinases and dephosphorylation by phosphatases provide information on both activation and deactivation of critical cellular pathways, including regulatory mechanisms of metabolism, cell division, and cell growth and differentiation [16]. In almost all cases, proteins may be phosphorylated on different residues and their properties altered, leading to activation or down-regulation of their activities, modification of subcellular localization stabilities, and consequently alteration of their functions [17]. Eukaryotic proteins are phosphorylated primarily on serine (Ser), threonine (Thr), and tyrosine (Tyr) residues at a ratio of 1800:200:1 [18]. Numerous research projects on phosphoproteomes have generated a large data collection allowing deeper understanding of phosphorylation events in various species, including yeast [19], mice [20], humans [21], Arabidopsis [22], rice [23], and other organisms [24]. Data resources for plant phosphoproteomics produced by these studies are available from public databases such as PhosPhat for Arabidopsis [25], the Plant Protein Phosphorylation DataBase (P3DB) [26, 27], and the Medicago Phosphoprotein Database [28].
The iTRAQ (isobaric tags for relative and absolute quantitation) approach is a sensitive, accurate technique for both qualitative and quantitative peptide analysis, and has been successfully used in various proteomic studies, including those involving phosphoproteomics [29–33]. To better understand the molecular mechanisms involving phosphorylated proteins (phosphoproteins) and signaling networks during cotton fiber differentiation and initiation, we investigated the cotton ovule phosphoproteome using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) enriched by titanium dioxide (TiO2) affinity chromatography. We used four-plex iTRAQ to compare phosphopeptide levels in ovules at -3 and 0 DPA between the Xuzhou 142 fl mutant and its parental WT. We identified 830 phosphorylated peptides from 619 phosphoproteins, providing both quantitative and qualitative information on cotton phosphorylation between fl and WT. The information obtained from this research provides valuable resources and novel insights into mechanisms of phosphorylation modification during cotton fiber initiation.
Methods
Plant material and chemicals
Plants of G. hirsutum L. ‘Xuzhou 142’ with normal fuzz and lint fibers and its isogenic fl mutant line were grown in three replications side by side in a field at the Institute of Cotton Research, Chinese Academy of Agricultural Sciences (CAAS), Anyang (E 114°48′, N 36°06′), China. Cotton materials were grown in a normal agronomic field from April to September. Flower buds at -3 DPA and flowers at 0 DPA were collected individually from the 60 plants in morning (9:00–11:00); the ovules were dissected from five bolls cottected in each of 60 plants, frozen in liquid nitrogen, and stored at -80°C until use. For each genotype, we used three biological pools, each with 60 plants grown at similar stages.
An iTRAQ Reagent-4plex Multiplex kit was obtained from Applied Biosystems (Foster City, CA, USA). Ultra-pure HPLC-grade water was produced with a Barnstead Millipore water purification system (Billerica, MA, USA). Titanium dioxide (TiO2) beads were obtained from Shimadzu (Kyoto, Japan). Other chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Scanning electron microscopy (SEM)
SEM was conducted using a modification of a previously reported procedure [34]. In brief, ovules from -3 DPA and 0 DPA were dissected from WT and fl plants, fixed in a solution of 3% formaldehyde and glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4), and rinsed three times in 0.2 M sodium cacodylate buffer (pH 7.4). The ovules were dehydrated in an 30–100% ethanol series for 30 min at each concentration. Ovules were frozen in an Oxford CT 1500 cryotrans system, gold-coated with an ion coater (Eiko IB 3, Tokyo, Japan), and scanned using a Hitachi S-530 scanning electron microscope (Tokyo, Japan) at an accelerating voltage of 15 kV. Ten separate samples were observed and used to produce a representative image.
Protein extraction, digestion, and iTRAQ labeling
Proteins were isolated in accordance with a protocol described by Wiśniewski [35] with modifications. Plant tissues (1 g) were finely ground, and the powders were precipitated in a 10% (w/v) trichloroacetic acid/acetone solution containing 65 mM dithiothreitol (DTT) at -20°C for 1 h. After extraction, the solution was centrifuged at 10,000 × g for 45 min. The supernatant was discarded, and the precipitate was vacuum-dried and solubilized in 1/10 volumes of SDT buffer (4% SDS, 100 mM DTT, and 150 mM Tris–HCl, pH 8.0). The solution was heated in a boiling water bath for 5 min, followed by ultrasonication (10 rounds of 80-W sonication for 10 s with 15-s intervals). Total protein in the supernatant was quantified based on the Bradford method [36].
Protein (300 μg) from three equally pooled biological replicates was diluted with 200 μl UA buffer (8 M Urea and 150 mM Tris–HCl, pH8.0) and subjected to 30-kDa ultrafiltration. Samples were centrifuged at 14,000 × g for 15 min; 200 μl UA buffer was then added, followed by centrifugation for an additional 15 min. After addition of 100 μl iodoacetamide (50 mM in UA) and oscillation for 1 min at 600 rpm, the samples were incubated for 30 min in darkness, and then centrifuged at 14,000 × g for 10 min. The filters were washed twice with 100 μl UA buffer, and 100 μl dissolution buffer (50 mM triethylammonium bicarbonate at pH 8.5) was added to the filters followed by centrifugation for 10 min. This step was repeated twice, and 40 μl trypsin buffer (2 μg trypsin in 40 μl dissolution buffer) was then added to each filter. The samples were incubated at 37°C for 18 h, and the peptides were collected by centrifugation for 10 min at 14,000 × g. This step was repeated twice, and peptide content was determined by spectral density using UV light at 280 nm. About 90 μg of peptides were labeled five times with iTRAQ reagents according to the manufacturer’s protocol (Applied Biosystems). The peptide mixture was vacuum freeze-dried prior to enrichment with TiO2 breads.
An overview of the applied analytical strategy using the iTRAQ-based quantitative phosphoproteomic method is presented in Additional file 1: Figure S1. Protein extracts from -3 and 0 DPA developmental stages of WT were respectively labeled with iTRAQ tags 114 and 115, while those of the fl mutant were labeled with tags 116 and 117.
Phosphopeptide enrichment using TiO2 breads
The four-plex iTRAQ-labeled peptides were subjected to phosphopeptide enrichment using TiO2 beads as described by Larsen et al. [37]. The trypsin-digested peptide mixture was diluted with 1× DHB buffer (0.6% 2, 5-dihydroxybenzoic acid, 16% acetonitrile [ACN], and 0.02% trifluoroacetic acid [TFA]) and added to the TiO2 breads. After 40 min of shaking, the TiO2 beads were packed into a GELoader tip (Eppendorf, Hamburg, Germany). The column was washed three times with 50 μl washing buffer I (30% ACN and 3% TFA) and then three times with 50 μl washing buffer II (80% ACN and 0.3% TFA). The bound peptides were eluted with 50 μl NH4OH, pH 10.5, and then vacuum freeze-dried. The lyophilized phosphopeptides were dissolved in 0.1% formic acid prior to MS analysis.
Mass spectrometry
Phosphopeptides were subjected to capillary LC-MS/MS using an automated Easy-nLC 1000 system coupled to a Q-Exactive mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). A pre-column (20 mm × 100 μm; 5 μm-C18) and an analytical column (250 mm × 75 μm; 3 μm-C18) were used (Thermo Fisher Scientific) with mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in 84% ACN). The phosphopeptides were separated at a flow rate of 250 nl min-1 using the following gradient: 0–55% mobile phase B from 0–220 min, 55–100% mobile phase B from 220–228 min, and 100% mobile phase B from 228–240 min. Data-dependent mass spectra were acquired for 240 min. The full MS surveys were collected over a mass-to-charge ratio (m/z) range of 300–1,800, with the resolution set to 70,000 at m/z 200. For MS/MS, we used a resolution of 17,500 at m/z 200, with an isolation window of 2 m/z.
Database search and quantification
Mascot 2.2 (Matrix Science, Boston, MA, USA) and Proteome Discoverer 1.3 (Thermo Fisher Scientific) software were used to simultaneously identify and quantify phosphoproteins [38–41] based on two combined databases derived from G. raimondii (40,976 entries) [15] and the CGI (cotton gene index) database from DFCI (CGI.release-11.zip; 117,992 entries) [42].
The iTRAQ quantification workflow was performed essentially as reported previously [41] (Additional file 1: Figure S1). After estimating the protein concentration of each sample, proteins were digested using a trypsin enzyme to produce proteolytic peptides. Each peptide was labeled with a different iTRAQ reagent and then mixed. The combined peptide mixture was analyzed by LC-MS/MS for both identification and quantification. The sequence of a peptide was determined from the product ions that were generated from cleavage about peptide interresidue bonds using Mascot 2.2. The relative quantity of a peptide among the different samples was determined by comparing the intensities of reporter ion signals also present in the MS/MS scan using the Proteome Discoverer 1.3 software.
The raw files were searched individually with Mascot 2.2 using the following search parameters: selection only of tryptic peptides with two missed cleavages, peptide mass tolerance of ± 20 ppm, and fragment mass tolerance = 0.1 Da. Proteome Discoverer 1.3 software was used to extract the peak intensity of each expected iTRAQ reporter ion from each analyzed fragmentation spectrum. The search parameters were as follows: peptide false discovery rate ≤ 0.01, use only unique peptides, reject all quantification values if not all quantification channels are present, normalize on protein median, normalize all peptide ratios by the median protein ratio, and median protein ratio = 1 after normalization [41]. For each phosphorylation site on the phosphopeptides, Phosphorylation site probabilities were set above 75%, indicating that the site is truly phosphorylated, and Phosphorylation site score was set above 50, indicating a good peptide spectral match [43].
Bioinformatics and motif analyses
To investigate amino acid frequencies around each identified pSer, pThr, and pTyr site, the 12 surrounding amino acids were retrieved to generate a list of “phosphor-13-mers” using a BioPerl script [44]. In the case of C- and N-terminal peptides, the sequences were completed to phosphor-13-mers with the required number of “X”s, where X indicates any amino acid. Phosphor-13-mers amino acid frequencies surrounding the three amino acid phosphorylation sites (phosphosites) were completed using the WebLogo server [45, 46], and their motifs were extracted using the Motif-X algorithm [47]. Phosphopeptide sequences for these phosphosites were pre-aligned using a custom Perl script, and the pre-aligned phosphor-13-mers from cotton (1,592 phosphosites) and nine other species (47,923 phosphosites) were submitted to the Motif-X algorithm as a foreground process. Because of upload restrictions, the database generated by combining 15,070 random protein sequences derived from the nine species (from the P3DB database) and cotton (10 Mb) generated with a custom Perl script was submitted as background. Motif-X default settings of width = 13, occurrence = 20, and significance = 0.000001 were used for pSer and pThr. For pTyr, less stringent settings of width = 13, occurrence = 2, and significance = 0.0005 were used because only 18 peptides contained localized pTyr motifs. Motif identification was carried out using CompariMotif [48] and the PhosphoMotif Finder database [49]. Phosphoproteins and phosphor-13-mer sequences were compared against the P3DB database (9 species, 16,477 phosphoproteins, and 47,923 phosphosites) to assess their novelty.
The Blast2GO suite [50] was applied to annotate identified protein sequences. Phosphoprotein functional classification was conducted using Gene Ontology (GO), GO-Enzyme-Code, and other search tools. The Batch sequence search tool of Pfam 27.0 (14,831 families) was used to obtain phosphoprotein domains [51]. The Plant Transcription Factor database (83 species, 129,288 transcription factors) was downloaded from the Center for Bioinformatics [52] and used to identify transcription factors [53]. To search for homologs, the identified phosphoproteins were analyzed by local blast tool in the software package ncbi-blast-2.2.26 + -win32.exe [54] against a reference database downloaded from P3DB databases. For multiple sequence alignment, ClustalX2 was used with default parameter settings [55]. The aligned sequences were further analyzed using the MEGA5 program [56]. For pathway enrichment analysis, the differentially phosphorylated proteins were mapped to the terms in the KEGG (Kyoto Encyclopedia of Genes and Genomes) database by using the KOBAS 2.0 (KEGG Orthology-Based Annotation System) program [57]. KEGG pathways with corrected p values ≤ 0.05 were considered to be statistically enriched in cotton fiber initiation.
Results
Phosphopeptide identification
When fiber development of the WT and its fl mutant was examined by SEM (Additional file 1: Figure S1), no obvious differences were observed between the WT and fl at -3 DPA. The ovule surfaces were flat, and the epidermal cells were dotted with stomata. On the day of anthesis, numerous cells destined to become fibers had already started to balloon out from the epidermis on the WT; in contrast, the fl mutant had no fiber initiation, as this phenomenon was not observed.
Based on the criteria given in Methods, 830 phosphopeptides and 1,592 individual phosphosites originating from 619 proteins were identified from ovules of fl and its parental WT at -3 and 0 DPA (Table 1). The spectra representing all phosphopeptides and the original data were shown in Additional file 2. Of the 1,592 non-redundant phosphosites, 89.4% were phosphorylated at serine, 9.5% at threonine, and 1.1% at tyrosine residues. This finding is consistent with previous reports in other plants: 88% pSer, 11% pThr, and 1% pTyr in Arabidopsis, and 89.3% pSer, 10.2% pThr, and 0.5% pTyr in soybean [30, 58]. A detailed examination of the phosphoproteomic data revealed that 33.4%, 48.3%, and 18.3% of the 830 unique phosphopeptides were singly, doubly, and multiply phosphorylated, respectively (Additional file 3: Table S1, sheet 1). These values are very different from those reported for Arabidopsis (80.9%, 19.1%, and 0%) and soybean (95%, 5%, and 0%) [22, 30], but are more or less consistent with Saccharomyces cerevisiae (27.8%, 67.1%, and 5.1%) [19]. This difference might be explained by the use of different methodologies or biological systems, where each tissue and organism under a particular environment may have a special phosphoproteome profile.
Table 1.
Numbers of phosphoproteins, phosphopeptides, and phosphorylation sites identified in this study
| Listing | Number |
|---|---|
| Phosphoproteins | 619 |
| Phosphopeptides | 830 |
| Phosphopeptides (single:doubleness:mulriple) | 277:401:152 |
| Phosphorylation sites | 1592 |
| Phosphorylation sites (Ser:Thr:Tyr) | 1423:151:18 |
We compared phosphorylation patterns of orthologous phosphosites between cotton and nine other species (from P3DB) to analyze phosphosite conservation. Phosphosites in cotton that were absent from their equivalent phosphoproteins in other plant species were considered to be novel. Among the identified phosphosites, 875 were conserved across the nine species, but only 492 were phosphorylated in their P3DB orthologs (Table 2 and Additional file 4: Table S2). For example, 60s acidic ribosomal protein P0 (RPLP0, D-10009321) considered as an important protein during the fiber initiation was well conserved [12], and its homologs were phosphorylated (Figure 1A). In contrast, another important protein in fiber initiation named phosphoenolpyruvate carboxykinase (PEPCK, TC237491) had two phosphosites [9], each having homologs (Figure 1B). We also identified two previously unknown phosphosites at the Rho GTPase activation protein (RhoGAP, D-10019024) N-terminus: Ser541 and Ser544 [59], which were well conserved across Medicago truncatula but only present in this plant as non-phosphorylated residues. In addition, only cotton phosphopeptide contained all three phosphosites simultaneously (Figure 1C). In total, nearly one-third of orthologs (492; 30.9%) were phosphorylated at equivalent sites that have been previously reported; the majority (1,100; 69.1%) of identified phosphosites, however, were novel. These results also indicate that phosphosites are conserved in a similar manner across different plant species.
Table 2.
Comparison of conserved phosphosites in cotton and species in the P 3 DB database
| Number of phosphorylation sites | |||
|---|---|---|---|
| Conservation of phosphosites (%) | Phosphosites in 9 species counterparts | ||
| Described (%) | Undescribed (%) | ||
| pS | 759 (86.7) | 436 (88.6) | 323 (84.3) |
| pT | 105 (12.0) | 50 (10.2) | 55 (14.4) |
| pY | 11 (1.3) | 6 (1.2) | 5 (1.3) |
| All | 875 (100) | 492 (56.2) | 383 (43.8) |
Figure 1.

Conservation of phosphosites between cotton proteins and homologs in nine other plant species. Phosphosites identified in this study are indicated in red; previously identified phosphosites are represented by lower-case blue letters. A: Phosphosites conserved across the plant 60s acidic ribosomal protein P0 (RPLP0). B: Phosphosites conserved across the plant phosphoenolpyruvate carboxykinase (PEPCK). C: Phosphosites conserved across the plant Rho GTPase activation protein (RhoGAP).
To evaluate whether phosphosites were concentrated in conserved domains, we performed Pfam searches to obtain domain information for 473 of the 619 phosphoproteins (Additional file 5: Table S3, sheet 1). These data suggested that 73.1% of the phosphosites (885 sites) were located outside of conserved domains (Table 3; Additional file 5: Table S3, sheet 2). The finding that most phosphorylation occurs outside of conserved domains is consistent with previous results [60, 61]. Interestingly, nearly one-third (31.9%) of pThrs were found to be located in conserved domains. These data indicate that pThrs may have more impact on domain-associated functions compared with pSers and pTyrs.
Table 3.
Location of phosphorylation sites on characterized phosphoprotein domains
| Number of phosphorylation sites | |||
|---|---|---|---|
| Pfam domain | Total (%) | ||
| ON (%) | OUT (%) | ||
| pS | 284 (26.4) | 792 (73.6) | 1076 (100) |
| pT | 37 (31.9) | 79 (68.1) | 116 (100) |
| pY | 4 (22.2) | 14 (77.8) | 18 (100) |
| All | 325 (26.9) | 885 (73.1) | 1210 (100) |
Phosphoproteome characterization and classification
We used the Blast2GO program to annotate and classify proteins into biological process (BP), cellular component (CC), and molecular function (MF) categories. Sequences were searched against the non-redundant (NR) protein database. Functions and annotations of these predicted phosphoproteins are shown in Additional file 3: Table S1 (sheet 2). The majority of the proteins in our dataset were annotated, with only 29 cases having no annotation information (classified as “unknown”). Among the 619 identified phosphoproteins, information related to BP, CC, and MF was obtained for 351, 370, and 383 phosphoproteins, respectively (Figure 2; Additional file 6: Table S4). In this study, most of the identified phosphoproteins were involved in binding and catalytic activity, consistent with the findings of a previous investigation [58].
Figure 2.
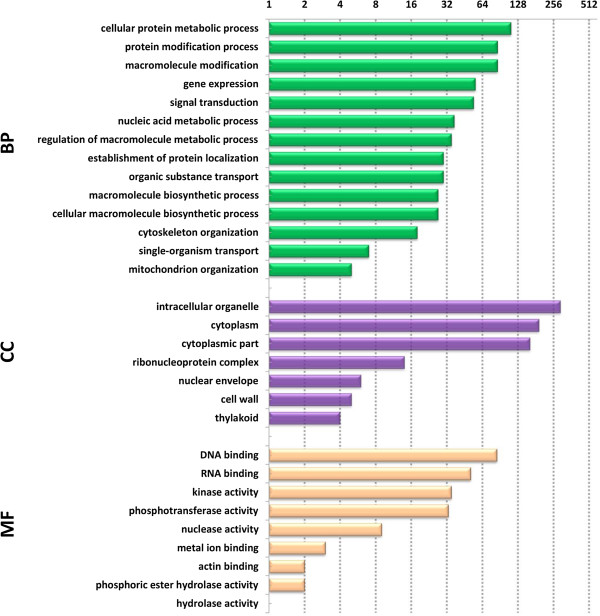
Distribution of phosphoprotein Gene Ontology functional classifications. BP: biological process, CC: Cellular component, MF: molecular function.
To determine if the 619 phosphoproteins identified in our study are commonly found or novel, we searched our dataset against the P3DB database, a repository for plant protein phosphorylation site data. P3DB currently hosts protein phosphorylation data for nine species from 32 experimental studies, comprising 16,477 phosphoproteins and 47,923 phosphosites. Of our 619 identified phosphoproteins, 543 showed homology to phosphoproteins in P3DB, while 76 were novel (Additional file 7: Table S5). These newly identified phosphoproteins may prove useful for identifying components of phosphorylation-dependent signal cascades and for estimating the function of phosphorylation events in response to specific environment signals. The 619 phosphoproteins were also searched against the Plant Transcription Factor Database. As shown in Additional file 8: Table S6, 109 of these phosphoproteins corresponded to transcription factors. This result is consistent with previous studies demonstrating that regulatory proteins, such as transcription factors and kinases, are more often subjected to post-translational regulation via phosphorylation than are metabolic enzymes [60, 62].
Phosphopeptide motif discovery
To evaluate sequence conservation at phosphosites, we used WebLogo to generate sequence logos that were graphical representations of patterns within multiple sequence alignments (Figure 3A; Additional file 9: Table S7). The frequency of amino acid residue occurrence at three key positions — n + 1 (proline: P and aspartic: D), n + 2 (aspartic: D, serine: S and Glutamic acid: E), and n + 3 (Glutamic acid: E and aspartic: D) — reached 51%, 50%, and 33%, respectively. We therefore deduced that the sequence pSer/pThr-P/D-D/S/E-E/D is the conserved phosphosite motif. This result may serve as a clue for identifying targets of protein kinases in large-scale phosphopeptide analyses.
Figure 3.
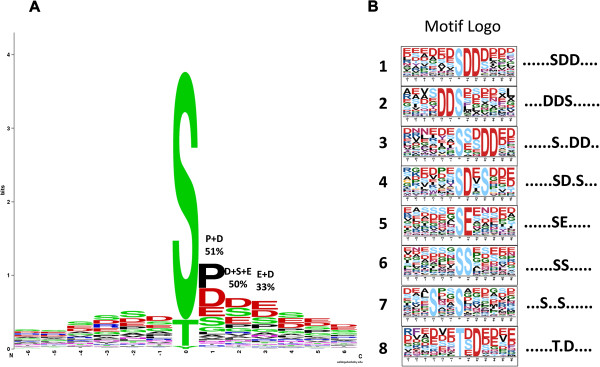
WebLogo analysis of identified phosphorylation sites and extraction-enriched phosphorylation motifs. A: Frequency distribution of amino acid residues surrounding phosphorylation sites at positions -6 to +6. B: Motifs extracted from the phosphopeptide dataset.
Given that protein phosphorylation seems to be the most important proteotype feature, motifs associated with localized phosphosites were identified using Motif-X. To compare potential consensus sequences among different plant species, all phosphopeptides from our study, as well as phosphopeptides from nine other species in P3DB, were used for motif extraction against a background database generated by combining 15,070 random protein sequences derived from the genomes of the 10 species (Additional file 10: Table S8, sheet 6). Compared with the 189 phosphorylation motifs detected in the other nine species, 17 pSer motifs and 2 pThr motifs were identified in our cotton dataset. All identified phosphorylation motifs are listed in Additional file 10: Table S8. The 17 pSer motifs could be divided into three major categories: pro-directed, basic, and acidic. Acidic motifs accounted for 11 of the 17 identified pSer motifs, and nearly 65% of Ser phosphopeptides possessed this motif (Table 4). This result was confirmed by a WebLogo alignment of all identified phosphor-13-mer phosphosites (Figure 3A). These results suggest that acidic kinases may be the major kinase group involved in phosphorylation of the identified phosphoproteins during cotton fiber initiation. Among 19 phosphorylation motifs identified in our data, we found 7 distinct motifs in the PhosphoMotif Finder database and 8 motifs unique to cotton (Figure 3B). Four motifs, ......SDD...., ....DDS......, ......S..DD.., and ......SE....., belonged to casein kinase 2. Casein kinase 2 is involved in cell cycle control, DNA repair, circadian rhythm regulation, and other metabolic pathways [63]. The ......SD.S… phosphosite motif resembled a known motif in transforming growth factor-β (TGFB) receptor kinase, which is involved in cell growth, cell differentiation, apoptosis, cellular homeostasis, and other cellular functions [64]. The ......SS..... motif showed high similarity to a motif found in AKT kinase (also known as protein kinase B) family members, which are serine/threonine-specific protein kinases that play key roles in multiple cellular processes such as glucose metabolism, apoptosis, cell proliferation, transcription, and cell migration [65]. The …S..S...... motif was similar to that of extracellular-signal-regulated kinases (ERKs), which are classical mitogen-activated protein kinases (MAPs) involved in various functions, including regulation of meiosis, mitosis, and post-mitotic processes in differentiated cells [66].
Table 4.
The description of the identified phosphorylation motifs
| NO | Cotton motif | Novel or not | Motif pattern | Motif class |
|---|---|---|---|---|
| 1 | ......SP..... | No | sP | Pro-directed |
| 2 | ......SD.E… | No | sX[D/E] | Acidic |
| 3 | ......SDD.... | Novel | sX[D/E] | Acidic |
| 4 | ......SD.D… | No | sX[D/E] | Acidic |
| 5 | ......SE.E… | No | sX[D/E] | Acidic |
| 6 | ......S.D.E.. | No | sXX[D/E] | Acidic |
| 7 | ....DDS...... | Novel | [D/E]s | Acidic |
| 8 | ......S..DD.. | Novel | sXX[D/E] | Acidic |
| 9 | ......S.....X | No | unknown | unknown |
| 10 | .....DS...... | No | [D/E]s | Acidic |
| 11 | …R..S...... | No | RXXs | Basic |
| 12 | ......SD.S… | Novel | s[D/E] | Acidic |
| 13 | ......S.D.... | No | sX[D/E] | Acidic |
| 14 | ......SE..... | Novel | s[D/E] | Acidic |
| 15 | ......S.SP… | No | unknown | unknown |
| 16 | ......SS..... | Novel | unknown | unknown |
| 17 | …S..S...... | Novel | unknown | unknown |
| 18 | ......TP..... | No | unknown | unknown |
| 19 | ......T.D.... | Novel | unknown | unknown |
Analysis of differentially phosphorylated proteins
Phosphopeptides were considered to be significantly differentially regulated when their levels differed based on a cutoff value of p < 0.05 with Student t-test and ANOVA analysis. The fold-change cutoff value for up-regulated or down-regulated phosphorylation activity was 1.5-fold. In the four datasets, we found that quantities of phosphoproteins were differentially expressed between -3 DPA WT and -3 DPA fl, and between 0 DPA WT and 0 DPA fl. Between -3 DPA WT and 0 DPA WT, 66 phosphoproteins were differently expressed. Of these, 44 were up-regulated and 22 were down-regulated in 0 DPA WT compared with -3 DPA WT, suggesting that many phosphoproteins were enriched during the transition from fiber differentiation to initiation. In contrast, 31 were up-regulated and 34 were down-regulated in 0 DPA fl compared with -3 DPA fl. Similarly, between -3 DPA and 0 DPA in WT and fl, 4 and 50 phosphoproteins, respectively, were up-regulated in the WT (Figure 4A; Additional file 11: Table S9). We further classified differential patterns of phosphoprotein expression using the following diagrams (Figure 4B; Figure 4C). For example, the abundances of 69 phosphoproteins were found to change significantly at one or two time points between WT and fl (i.e., -3 DPA WT vs. -3 DPA fl and 0 DPA WT vs. 0 DPA fl). Among these 69 phosphoproteins, the abundances of 3 were significantly changed at both -3 and 0 DPA in both genotypes, whereas the abundance of 9 and 57, respectively, varied significantly only at either -3 DPA or 0 DPA (Figure 4C; Additional file 11: Table S9). Of the 3 phosphoproteins differentially expressed at both time points, 2 were up-regulated at 0 DPA but down-regulated at -3 DPA in the WT; the rest phosphoprotein exhibited the same trend at both time points. The 69 differentially phosphorylated proteins were classified into 6 functional categories based on their predicted molecular functions (Additional file 12: Figure S2 and Additional file 13: Table S10). The largest functional categories were DNA binding, Nucleotide binding, RNA binding, Kinase activity, Phosphotransferase activity and Metal ion binding. The significantly enriched pathways were also analyzed by KOBAS software suite. Four biochemical pathways, such as RNA transport, Herpes simplex infection, Spliceosome and Gap junction, were enriched (Corrected P-Value < 0.05) during the fiber initiation period (Additional file 14: Table S11).
Figure 4.
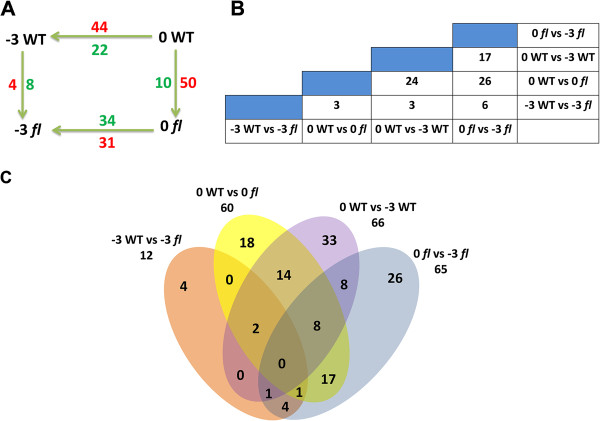
Summary of differentially phosphorylated proteins identified in this study. A: Number of phosphoproteins differentially expressed during fiber development within and between WT and fl ovules. Numbers above and below arrows denote numbers of phosphoproteins differentially expressed for the specified comparison. For example, between stages -3 and 0 DPA in the WT, 44 phosphoproteins were up-regulated (red) and 22 were down-regulated (blue) at 0 DPA. Similarly, between WT and fl at -3 DPA, 4 phosphoproteins were up-regulated and 8 phosphoproteins were down-regulated in the WT. B: Numbers at column-row intersections are the number of differentially phosphorylated proteins common to the four tissues. C: Venn diagram showing the number of differentially phosphorylated proteins shared between phosphoproteomes of the different four tissues.
Discussion
Differentially phosphorylated proteins involved in signal transduction
We found four differentially phosphorylated proteins (CK640461, TC229918, D-10009824, and D-10019024) related to signal transduction, of which three were up-regulated and one was down-regulated in the WT. CK640461 belongs to the small-GTPase superfamily, comprising Rab, Ran, Arf, and Rho GTPases in Arabidopsis. The different Rab GTPases are localized to the cytosolic face of specific intracellular membranes, where they function as regulators of distinct steps in membrane traffic pathways. The run and tbc1 domain-containing protein (CK640461) has Rab GTPase activity and may regulate vesicle formation, actin- and tubulin-dependent vesicle movement, and membrane fusion [67, 68]. Genetic evidence suggests that GTPases of the Rho class (also called ROPs) are involved in spatial regulation of reactive oxygen species (ROS) production and growth [69]. We also found one Rho GTPase activation protein (D-10019024); the activities of Rho-GTPases are negatively controlled by a group of proteins called Rho-GDP dissociation inhibitors (RhoGDIs) [70]. Loss of function of one member of this family results in both spatially deregulated ROS accumulation and hair outgrowth. However, a constitutively active form of a cotton small GTPase highly expressed during cotton fiber development has been shown to induce ROS production in cultured Arabidopsis and soybean cells [59]. This result also suggests that Rho GTPase can perform its role during the rapid elongation of cotton fibers. IQ-domain 32-like protein (TC229918) may be involved with calmodulin specificity. The IQ-domain is approximately 25 amino acids in length and forms an amphiphilic seven-turn α-helix capable of binding calmodulin in a Ca2+-independent manner. Calcium-mediated singnal transduction plays crucial roles in plant growth, especially in tip growth [5]. A highly Ca2+ concentration can be observed in the tips of root hairs [71]. Obvious inhibition of fiber growth also occurs when cotton ovules are cultured in the absence of exogenous Ca2+ ions [72].Transcription of protein-coding genes by RNA polymerase II (RNAP II) is facilitated by a multitude of transcription factors [73]. Elongation is highlighted as a central process that coordinates multiple stages during mRNA biogenesis and maturation [74]. Transcription elongation factors (D-10009824) are thought to increase RNAP II productivity by facilitating chromatin passage and mRNA processing.
Differentially phosphorylated proteins associated with protein modification
A protein is made up of amino acid chains, i.e., one or more polypeptides. Post-translational modification, including polypeptide folding, cleavage, and other processes, is one of the steps of protein biosynthesis. A novel phosphoprotein, compared with those of other examined species, was identified in this study: E3 ubiquitin-protein ligase HOS1 phosphoprotein (CO096048). This protein negatively regulates cold signal transduction, and its mutant flowers early [75]. Cyclophilin-like peptidyl-prolyl cis-trans isomerase family protein (D-10005130, PPIase) can accelerate protein folding, and also has protein chaperone-like functions. This protein contains a common cyclophilin-like domain (CLD) and other domains, which are important for selection of protein substrates and subcellular compartmentalization [76]. The formation of a decapping complex comprising Decapping 5 (TC240176, DCP5), which occurs inside processing P-bodies, represents an irreversible step in mRNA degradation. Knockdown mutants of DCP5 share abnormalities in postembryonic development [77]. DCP5 may therefore play an important role in mRNA decapping during postembryonic development. We also identified two protein kinases and two histone deacetylase, all of which were up-regulated in the WT: serine threonine-protein kinase prp4 (D-10033485), g-type lectin s-receptor-like serine threonine-protein kinase sd2-5-like (D-10037115) and histone deacetylase (TC230548, TC232936). A serine/threonine protein kinase is a kinase enzyme that phosphorylates the OH group of serine or threonine. Serine threonine-protein kinase prp4 plays a role in the regulation of pre-mRNA splicing codes and also is essential for yeast growth [78]. Histone deacetylases are a class of enzymes that allow the histones to wrap the DNA more tightly and then regulate DNA expression by acetylation and de-acetylation. In Arabidopsis, histone deacetylase 18 can exhibit altered hair and non-hair epidermal cell patterning [79].
Differentially phosphorylated proteins involved in carbohydrate metabolism
Two phosphoproteins in this study, both down-regulated in the WT, were identified as especially enriched in the sucrose metabolism pathway. Generally, sucrose synthase (SUS) is a key player in plant sucrose catabolism, and, like invertases (D-10036040), catalyzes the reversible conversion of sucrose and UDP into fructose and UDP-glucose [80]. Sucrose has recently been recognized to have important hormone-like functions as a primary messenger in signal transduction. Additionally, sucrose molecules regulate gene expression at transcriptional and post-transcriptional levels [81, 82]. Increasing evidence indicates that SUS is essential for cell wall thickening and cotton fiber cell development [83–85]. Sus is preferentially expressed in elongating fiber cells, and antisense suppression of Sus expression reduces hexose levels, leading to a fiberless phenotype [86, 87]. UDP-glucose 6-dehydrogenase (TC263488) is an enzyme encoded by the UGDH gene. This enzyme converts UDP-glucose to UDP-glucuronate, which is readily incorporated into the pectin fraction of cell wall preparations and can significantly improve fiber growth during ovule culture [88].
Differentially phosphorylated proteins related to the cell cycle and cell proliferation
Seven up-regulated phosphoproteins identified in the WT—serrate RNA effector molecule-like (D-10014027), vacuolar import/degradation protein (D-10025850), lysine-specific histone demethylase 1 isoform 3 (D-10036681), uncharacterized protein (D-10037099), ROP interactive partner 5 isoform 4 (DW225237), WD repeat-containing protein 70-like (ES794679) and pre-rRNA processing protein esf1-like (TC233572)—are involved in the regulation of cell cycle patterns and cell proliferation development.
Serrate RNA effector molecule protein belongs to the ARS2 family and contains one C2H2-type zinc finger domain. This protein expresses in shoot meristems and in emerging organ primordia throughout development. Its mutant displays defects in shoot and leaf development or death during embryogenesis in Arabidopsis [89]. Vacuolar import/degradation protein is involved in the negative regulation of gluconeogenesis. This protein is required for both proteosome-dependent and vacuolar catabolite degradation of fructose-1,6-bisphosphatase (FBPase), where it probably regulate FBPase targeting from the FBPase-containing vesicles to the vacuole [90]. Lysine-specific demethylase 1 protein is a flavin-dependent monoamine oxidase, which can demethylate mono- and di-methylated lysines. This protein is a component of several histone deacetylase complexes, though it silences genes by functioning as a histone demethylase. In a certain yeast strain, Saccharomyces cerevisiae, to become methylated causes a delay in the mitotic cell cycle [91]. ROP interactive partner 5 isoform 4 is a putative Rho protein effector, interacting specifically with the active form of ROPs (Rho proteins of plants), expresses in the root and shoot apex in Arabidopsis [92]. WD repeat-containing protein 70-like protein can express with the pollen germination and tube growth in Arabidopsis [93]. Pre-rRNA processing protein esf1-like protein is involved in embryo sac egg cell differentiation and karyogamy [94].
Conclusions
Our study, which has demonstrated that iTRAQ is a powerful technique for performing quantitative phosphoproteomic analyses, represents the first comprehensive phosphoproteomic analysis of cotton fiber differentiation and initiation using a WT and its fl mutant. A total of 619 phosphoproteins, including 76 new phosphoproteins, were identified. The 69 differentially phosphorylated proteins were found to be involved in signal transduction, protein modification, carbohydrate metabolic processes, and cell cycle and cell proliferation. Our analysis of WT and fl cotton ovule phosphoproteomes sheds light on the post-translational modification role of protein phosphorylation, a major regulator of various biological processes during fiber differentiation and initiation.
Availability of supporting data
All the raw mass spectra files in LC-MS/MS have been deposited into the publicly accessible database PeptideAtlas and now are available with dataset Identifier PASS00508 (http://www.peptideatlas.org/PASS/PASS00508).
Electronic supplementary material
Additional file 1: Figure S1: Cotton quantitative phosphoproteomic analysis workflow. WT -3 DPA and WT 0 DPA: Ovules from -3 and 0 DPA developmental stages of Xuzhou 142 WT. fl -3 DPA and fl 0 DPA: Ovules from -3 and 0 DPA developmental stages of Xuzhou 142 fl mutant. A: Peptide sequence identification from peptide backbone fargment ions. B: Quantification from iTRAQ reporter ions. Scale bars: WT -3 DPA and fl -3 DPA, 200 μm; WT 0 DPA and fl 0 DPA, 20 μm. (TIFF 757 KB)
Additional file 2: The file contains all original MS/MS spectra of the 830 phosphopeptides identified in this research. (ZIP 15 MB)
Additional file 3: Table S1: Two sheets were included. Sheet 1: Detected phosphorylated sites. Sheet 2: Phosphoprotein annotations. (XLSX 367 KB)
Additional file 4: Table S2: Comparison of phosphosites conserved between cotton and species in the P3DB database. (XLSX 233 KB)
Additional file 5: Table S3: Phosphoprotein Pfam domain information (sheet 1) and phosphosites location in characterized protein domains (sheet 2). (XLSX 232 KB)
Additional file 6: Table S4: Three sheets were included. Distribution of identified phosphoproteins in cellular component (sheet 1), biological process (sheet 2), and molecular function (sheet 3) categories. (XLSX 46 KB)
Additional file 7: Table S5: Novel phosphoproteins (sheet 1) and phosphoproteins with homologs in other species in P3DB (sheet 2). (XLSX 9 MB)
Additional file 8: Table S6: Number of transcription factors among the identified phosphoproteins. (XLSX 17 KB)
Additional file 9: Table S7: Sequence alignment of phosphorylation sites and extraction of significantly enriched phosphorylation motifs. (XLSX 61 KB)
Additional file 10: Table S8: Six sheets were included. Motifs identified via Motif-X surrounding localized pSer in nine plant species (sheet 1); pThr residues in nine species (sheet 2); pSer in cotton (sheet 3); pThr residues in cotton (sheet 4); prealigned “phosphor-13-mers” in cotton and nine other species (sheet 5); random protein sequences derived from the genomes of cotton and the nine other species (sheet 6). (XLSX 12 MB)
Additional file 11: Table S9: Five sheets were included. Sheet 1: quantitative analysis of 830 phosphopeptides. Sheet 2: Differentially phosphorylated proteins between WT and fl -3 DPA ovules. Sheet 3: Differentially phosphorylated proteins between WT and fl 0 DPA ovules. Sheet 4: Differentially phosphorylated proteins between WT 0 DPA and fl -3 DPA ovules. Sheet 5: Differentially phosphorylated proteins between fl 0 DPA and fl -3 DPA ovules. (XLSX 257 KB)
Additional file 12: Figure S2: Molecular functional classification of identified differentially phosphorylated proteins (-3 DPA WT vs. -3 DPA fl, and 0 DPA WT vs. 0 DPA fl). (TIFF 190 KB)
Additional file 13: Table S10: Molecular functional classification of the 69 differentially phosphorylated proteins identified in this study. (XLSX 13 KB)
Additional file 14: Table S11: The significantly enriched pathways were identified by KOBAS. (XLSX 14 KB)
Acknowledgements
The authers wish to thank the 973 Program of China (grant no. 2010CB126006) and the 863 Project of China (grant no. 2012AA101108 and grant no. 2013AA102601) for the financial support provided to this project. We thank Edanz group for manuscript editing and Shanghai Applied Protein Technology Co. Ltd. for providing technical support.
Abbreviations
- fl
Fuzzless-lintless
- WT
Wild type
- DPA
Days post-anthesis
- iTRAQ
Isobaric tags for relative and absolute quantitation
- MS/MS
Tandem mass spectrometry
- P3DB
The plant protein phosphorylation dataBase
- LC-MS/MS
Liquid chromatography-tandem mass spectrometry
- TiO2
Titanium dioxide
- SEM
Scanning electron microscopy
- DTT
Dithiothreitol
- ACN
Acetonitrile
- TFA
Trifluoroacetic acid
- m/z
Mass-to-charge ratio
- CGI
Cotton gene index
- GO
Gene ontology
- KEGG
Kyoto encyclopedia of genes and genomes
- KOBAS
KEGG orthology-based annotation system
- RPLP0
60s acidic ribosomal protein P0
- PEPCK
Phosphoenolpyruvate carboxykinase
- RhoGAP
Rho GTPase activation protein
- BP
Biological process
- CC
Cellular component
- MF
Molecular function
- NR
Non-redundant
- TGFB
Transforming growth factor-β receptor kinase
- ERKs
Extracellular-signal-regulated kinases
- MAPs
Mitogen-activated protein kinases
- ROS
Reactive oxygen species
- RhoGDIs
Rho-GDP dissociation inhibitors
- RNAP II
RNA polymerase II
- PPIase
Peptidyl-prolyl cis-trans isomerase
- CLD
Cyclophilin-like domain
- DCP5
Decapping 5
- SUS
Sucrose synthase
- FBPase
Fructose-1,6-bisphosphatase
- ROPs
Rho proteins of plants.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SXY and JWY designed the experiments. WFP, HJL and XLL performed the field cultivation of cotton plants and ovules collection. JFZ and MW conceived the study, participated in its design, and drafted and amended the manuscript. QFM performed the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Qifeng Ma, Email: maqifeng1985@163.com.
Man Wu, Email: wuman2004@163.com.
Wenfeng Pei, Email: peiwenfeng1988@163.com.
Haijing Li, Email: leo812002@163.com.
Xingli Li, Email: liyd@cricaas.com.cn.
Jinfa Zhang, Email: jinzhang@nmsu.edu.
Jiwen Yu, Email: yujw666@hotmail.com.
Shuxun Yu, Email: yu@cricaas.com.cn.
References
- 1.Lee JJ, Woodward AW, Chen ZJ. Gene expression changes and early events in cotton fibre development. Ann Bot-London. 2007;100(7):1391–1401. doi: 10.1093/aob/mcm232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.BAsRA AS, Malik C. Development of the cotton fiber. Int Rev Cytol. 1984;89(1):65–113. doi: 10.1016/S0074-7696(08)61300-5. [DOI] [Google Scholar]
- 3.Kim HJ, Triplett BA. Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant Physiol. 2001;127(4):1361–1366. doi: 10.1104/pp.010724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tiwari SC, Wilkins TA. Cotton (Gossypium hirsutum) seed trichomes expand via diffuse growing mechanism. Can J Bot. 1995;73(5):746–757. doi: 10.1139/b95-081. [DOI] [Google Scholar]
- 5.Qin YM, Zhu YX. How cotton fibers elongate: a tale of linear cell-growth mode. Curr Opin Plant Biol. 2011;14(1):106–111. doi: 10.1016/j.pbi.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Samuel Yang S, Cheung F, Lee JJ, Ha M, Wei NE, Sze SH, Stelly DM, Thaxton P, Triplett B, Town CD. Accumulation of genome‒specific transcripts, transcription factors and phytohormonal regulators during early stages of fiber cell development in allotetraploid cotton. Plant J. 2006;47(5):761–775. doi: 10.1111/j.1365-313X.2006.02829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang ZM, Xue W, Dong CJ, Jin LG, Bian SM, Wang C, Wu XY, Liu JY. A comparative miRNAome analysis reveals seven fiber initiation-related and 36 novel miRNAs in developing cotton ovules. Mol Plant. 2012;5(4):889–900. doi: 10.1093/mp/ssr094. [DOI] [PubMed] [Google Scholar]
- 8.Li YJ, Zhang XY, Wang FX, Yang CL, Liu F, Xia GX, Sun J. A comparative proteomic analysis provides insights into pigment biosynthesis in brown colored fiber. J Proteomics. 2012;78(1):374–388. doi: 10.1016/j.jprot.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y, Machado AC, White RG, Llewellyn DJ, Dennis ES. Expression profiling identifies genes expressed early during lint fibre initiation in cotton. Plant Cell Physiol. 2006;47(1):107–127. doi: 10.1093/pcp/pci228. [DOI] [PubMed] [Google Scholar]
- 10.Zhang TZ, Pan JJ. Genetic analysis of a fuzzless-lintless mutant in Gossypium hirsutum L. Jiangsu J Agr Sci. 1991;7:13–16. [Google Scholar]
- 11.Wang QQ, Liu F, Chen XS, Ma XJ, Zeng HQ, Yang ZM. Transcriptome profiling of early developing cotton fiber by deep-sequencing reveals significantly differential expression of genes in a fuzzless/lintless mutant. Genomics. 2010;96(6):369–376. doi: 10.1016/j.ygeno.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Liu K, Han M, Zhang C, Yao L, Sun J, Zhang T. Comparative proteomic analysis reveals the mechanisms governing cotton fiber differentiation and initiation. J Proteomics. 2012;75(3):845–856. doi: 10.1016/j.jprot.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 13.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143(7):1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H, Sadygov RG, Yates JR. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 15.Wang K, Wang Z, Li F, Ye W, Wang J, Song G, Yue Z, Cong L, Shang H, Zhu S. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012;44(10):1098–1103. doi: 10.1038/ng.2371. [DOI] [PubMed] [Google Scholar]
- 16.Salih E. Phosphoproteomics by mass spectrometry and classical protein chemistry approaches. Mass Spectrom Rev. 2005;24(6):828–846. doi: 10.1002/mas.20042. [DOI] [PubMed] [Google Scholar]
- 17.Wicks SJ, Lui S, Abdel-Wahab N, Mason RM, Chantry A. Inactivation of smad-transforming growth factor β signaling by Ca2+-calmodulin-dependent protein kinase II. Mol Cell Biol. 2000;20(21):8103–8111. doi: 10.1128/MCB.20.21.8103-8111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cieśla J, Frączyk T, Rode W. Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim Pol. 2011;58:137–148. [PubMed] [Google Scholar]
- 19.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20(3):301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 20.Villén J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc. 2008;3(10):1630–1638. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng J, Liu L, Wang J, Jin Q. Urinary proteomic and non-prefractionation quantitative phosphoproteomic analysis during pregnancy and non-pregnancy. BMC Genomics. 2013;14(1):777. doi: 10.1186/1471-2164-14-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugiyama N, Nakagami H, Mochida K, Daudi A, Tomita M, Shirasu K, Ishihama Y. Large-scale phosphorylation mapping reveals the extent of tyrosine phosphorylation in Arabidopsis. Mol Syst Biol. 2008;4(1):1–7. doi: 10.1038/msb.2008.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whiteman SA, Nühse TS, Ashford DA, Sanders D, Maathuis FJ. A proteomic and phosphoproteomic analysis of Oryza sativa plasma membrane and vacuolar membrane. Plant J. 2008;56(1):146–156. doi: 10.1111/j.1365-313X.2008.03578.x. [DOI] [PubMed] [Google Scholar]
- 24.Fu Q, Liu PC, Wang JX, Song QS, Zhao XF. Proteomic identification of differentially expressed and phosphorylated proteins in epidermis involved in larval-pupal metamorphosis of Helicoverpa armigera. BMC Genomics. 2009;10(1):600. doi: 10.1186/1471-2164-10-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heazlewood JL, Durek P, Hummel J, Selbig J, Weckwerth W, Walther D, Schulze WX. PhosPhAt: a database of phosphorylation sites in Arabidopsis thaliana and a plant-specific phosphorylation site predictor. Nucleic Acids Res. 2008;36(Suppl 1):1015–1021. doi: 10.1093/nar/gkm812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao J, Agrawal GK, Thelen JJ, Xu D. P3DB: a plant protein phosphorylation database. Nucleic Acids Res. 2009;37(Suppl 1):960–962. doi: 10.1093/nar/gkn733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao Q, Bollinger C, Gao J, Xu D, Thelen JJ. P3DB: an integrated database for plant protein phosphorylation. Front Plant Sci. 2012;3:1–8. doi: 10.3389/fpls.2012.00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rose CM, Venkateshwaran M, Grimsrud PA, Westphall MS, Sussman MR, Coon JJ, Ané J-M. Medicago PhosphoProtein Database: a repository for Medicago truncatula phosphoprotein data. Front Plant Sci. 2012;3:1–6. doi: 10.3389/fpls.2012.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi T, Serada S, Ako M, Fujimoto M, Miyazaki Y, Nakatsuka R, Ikezoe T, Yokoyama A, Taguchi T, Shimada K. New findings of kinase switching in gastrointestinal stromal tumor under imatinib using phosphoproteomic analysis. Int J Cancer. 2013;113(11):2737–2743. doi: 10.1002/ijc.28282. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen THN, Brechenmacher L, Aldrich JT, Clauss TR, Gritsenko MA, Hixson KK, Libault M, Tanaka K, Yang F, Yao Q. Quantitative phosphoproteomic analysis of soybean root hairs inoculated with Bradyrhizobium japonicum. Mol Cell Proteomics. 2012;11(11):1140–1155. doi: 10.1074/mcp.M112.018028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kosako H, Nagano K. Quantitative phosphoproteomics strategies for understanding protein kinase-mediated signal transduction pathways. Expert Rev Proteomic. 2011;8(1):81–94. doi: 10.1586/epr.10.104. [DOI] [PubMed] [Google Scholar]
- 32.Rudrabhatla P, Grant P, Jaffe H, Strong MJ, Pant HC. Quantitative phosphoproteomic analysis of neuronal intermediate filament proteins (NF-M/H) in Alzheimer's disease by iTRAQ. Faseb J. 2010;24(11):4396–4407. doi: 10.1096/fj.10-157859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwai LK, Benoist C, Mathis D, White FM. Quantitative phosphoproteomic analysis of T cell receptor signaling in diabetes prone and resistant mice. J Proteome Res. 2010;9(6):3135–3145. doi: 10.1021/pr100035b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Craig S, Beaton CD. A simple cryo‒SEM method for delicate plant tissues. J Microsc. 1996;182(2):102–105. doi: 10.1046/j.1365-2818.1996.128409.x. [DOI] [Google Scholar]
- 35.Wisacute JR. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 36.Zhao PM, Wang LL, Han LB, Wang J, Yao Y, Wang HY, Du XM, Luo YM, Xia GX. Proteomic identification of differentially expressed proteins in the Ligon lintless mutant of upland cotton (Gossypium hirsutum L.) J Proteome Res. 2009;9(2):1076–1087. doi: 10.1021/pr900975t. [DOI] [PubMed] [Google Scholar]
- 37.Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jørgensen TJ. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol Cell Proteomics. 2005;4(7):873–886. doi: 10.1074/mcp.T500007-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Sandberg A, Lindell G, Källström BN, Branca RM, Danielsson KG, Dahlberg M, Larson B, Forshed J, Lehtiö J. Tumor proteomics by multivariate analysis on individual pathway data for characterization of vulvar cancer phenotypes. Mol Cell Proteomics. 2012;11(7):1–14. doi: 10.1074/mcp.M112.016998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemeer S, Kunold E, Klaeger S, Raabe M, Towers MW, Claudes E, Arrey TN, Strupat K, Urlaub H, Kuster B. Phosphorylation site localization in peptides by MALDI MS/MS and the Mascot Delta Score. Anal Bioanal Chem. 2012;402(1):249–260. doi: 10.1007/s00216-011-5469-2. [DOI] [PubMed] [Google Scholar]
- 40.Colaert N, Barsnes H, Vaudel M, Helsens K, Timmerman E, Sickmann A, Gevaert K, Martens L. Thermo-msf-parser: an open source java library to parse and visualize thermo proteome discoverer msf files. J Proteome Res. 2011;10(8):3840–3843. doi: 10.1021/pr2005154. [DOI] [PubMed] [Google Scholar]
- 41.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.The cotton gene index databaseftp://occams.dfci.harvard.edu/pub/bio/tgi/data/Gossypium
- 43.Beausoleil S, Villn J, Gerber S, Rush J, Gygi S. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24(10):1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 44.Edwards D, Stajich JE, Hansen D. Bioinformatics : tools and applications. New York: Springer; 2009. [Google Scholar]
- 45.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18(20):6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwartz D, Gygi SP. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol. 2005;23(11):1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 48.CompariMotif @ Biowarehttp://bioware.ucd.ie/~compass/biowareweb/Server_pages/comparimotif.php
- 49.Edwards RJ, Davey NE, Shields DC. CompariMotif: quick and easy comparisons of sequence motifs. Bioinformatics. 2008;24(10):1307–1309. doi: 10.1093/bioinformatics/btn105. [DOI] [PubMed] [Google Scholar]
- 50.Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J. The Pfam protein families database. Nucleic Acids Res. 2012;40(1):290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.The plant transcription factor databasehttp://planttfdb.cbi.pku.edu.cn/
- 53.Jin J, Zhang H, Kong L, Gao G, Luo J. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014;42(1):1182–1187. doi: 10.1093/nar/gkt1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.The blast software packageftp://ftp.ncbi.nlm.nih.gov/blast/executables/release/
- 55.Larkin M, Blackshields G, Brown N, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 56.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(suppl 2):316–322. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reiland S, Messerli G, Baerenfaller K, Gerrits B, Endler A, Grossmann J, Gruissem W, Baginsky S. Large-scale Arabidopsis phosphoproteome profiling reveals novel chloroplast kinase substrates and phosphorylation networks. Plant Physiol. 2009;150(2):889–903. doi: 10.1104/pp.109.138677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Potikha TS, Collins CC, Johnson DI, Delmer DP, Levine A. The involvement of hydrogen peroxide in the differentiation of secondary walls in cotton fibers. Plant Physiol. 1999;119(3):849–858. doi: 10.1104/pp.119.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu CC, Liu CF, Wang HX, Shen ZY, Yang CP, Wei ZG. Identification and analysis of phosphorylation status of proteins in dormant terminal buds of poplar. BMC Plant Biol. 2011;11(1):158. doi: 10.1186/1471-2229-11-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nühse TS, Stensballe A, Jensen ON, Peck SC. Phosphoproteomics of the Arabidopsis plasma membrane and a new phosphorylation site database. Plant Cell. 2004;16(9):2394–2405. doi: 10.1105/tpc.104.023150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Facette MR, Shen Z, Björnsdóttir FR, Briggs SP, Smith LG. Parallel proteomic and phosphoproteomic analyses of successive stages of maize leaf development. Plant Cell. 2013;25(8):2798–2812. doi: 10.1105/tpc.113.112227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clevers H. Wnt/β-catenin signaling in development and disease. Cell. 2006;127(3):469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 64.Doré JJ, Edens M, Garamszegi N, Leof EB. Heteromeric and homomeric transforming growth factor-β receptors show distinct signaling and endocytic responses in epithelial cells. J Biol Chem. 1998;273(48):31770–31777. doi: 10.1074/jbc.273.48.31770. [DOI] [PubMed] [Google Scholar]
- 65.FreemanCook KD, Autry C, Borzillo G, Gordon D, Barbacci-Tobin E, Bernardo V, Briere D, Clark T, Corbett M, Jakubczak J. Design of selective, ATP-competitive inhibitors of Akt. J Med Chem. 2010;53(12):4615–4622. doi: 10.1021/jm1003842. [DOI] [PubMed] [Google Scholar]
- 66.Marshall CJ. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr Opin Genet Dev. 1994;4(1):82–89. doi: 10.1016/0959-437X(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 67.Stenmark H, Olkkonen VM. The Rab GTPase family. Genome Biol. 2001;2(5):30071–30077. doi: 10.1186/gb-2001-2-5-reviews3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.PereiraLeal JB, Seabra MC. The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J Mol Biol. 2000;301(4):1077–1087. doi: 10.1006/jmbi.2000.4010. [DOI] [PubMed] [Google Scholar]
- 69.Carol RJ, Takeda S, Linstead P, Durrant MC, Kakesova H, Derbyshire P, Drea S, Zarsky V, Dolan L. A RhoGDP dissociation inhibitor spatially regulates growth in root hair cells. Nature. 2005;438(7070):1013–1016. doi: 10.1038/nature04198. [DOI] [PubMed] [Google Scholar]
- 70.Dovas A, Couchman J. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J. 2005;390:1–9. doi: 10.1042/BJ20050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wymer CL, Bibikova TN, Gilroy S. Cytoplasmic free calcium distributions during the development of root hairs of Arabidopsis thaliana. Plant J. 1997;12(2):427–439. doi: 10.1046/j.1365-313X.1997.12020427.x. [DOI] [PubMed] [Google Scholar]
- 72.Huang QS, Wang HY, Gao P, Wang GY, Xia GX. Cloning and characterization of a calcium dependent protein kinase gene associated with cotton fiber development. Plant Cell Rep. 2008;27(12):1869–1875. doi: 10.1007/s00299-008-0603-0. [DOI] [PubMed] [Google Scholar]
- 73.Hirose Y, Manley JL. RNA polymerase II and the integration of nuclear events. Gene Dev. 2000;14(12):1415–1429. [PubMed] [Google Scholar]
- 74.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138(1):129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jung JH, Seo PJ, Park CM. The E3 ubiquitin ligase HOS1 regulates Arabidopsis flowering by mediating CONSTANS degradation under cold stress. J Biol Chem. 2012;287(52):43277–43287. doi: 10.1074/jbc.M112.394338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hennig L, Christner C, Kipping M, Schelbert B, Rücknagel KP, Grabley S, Küllertz G, Fischer G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry. 1998;37(17):5953–5960. doi: 10.1021/bi973162p. [DOI] [PubMed] [Google Scholar]
- 77.Iwasaki S, Takeda A, Motose H, Watanabe Y. Characterization of Arabidopsis decapping proteins AtDCP1 and AtDCP2, which are essential for post-embryonic development. Febs Lett. 2007;581(13):2455–2459. doi: 10.1016/j.febslet.2007.04.051. [DOI] [PubMed] [Google Scholar]
- 78.Alahari SK, Schmidt H, Kaufer NF. The fission yeast prp4+ gene involved in pre-mRNA splicing codes for a predicted serine/threonine kinase and is essential for growth. Nucleic Acids Res. 1993;21(17):4079–4083. doi: 10.1093/nar/21.17.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu C, Li LC, Chen WQ, Chen X, Xu ZH, Bai SN. HDA18 affects cell fate in Arabidopsis root epidermis via histone acetylation at four kinase genes. Plant Cell. 2013;25(1):257–269. doi: 10.1105/tpc.112.107045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schmalstig JG, Hitz WD. Contributions of sucrose synthase and invertase to the metabolism of sucrose in developing leaves estimation by alternate substrate utilization. Plant Physiol. 1987;85(2):407–412. doi: 10.1104/pp.85.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sheen J, Zhou L, Jang JC. Sugars as signaling molecules. Curr Opin Plant Biol. 1999;2(5):410–418. doi: 10.1016/S1369-5266(99)00014-X. [DOI] [PubMed] [Google Scholar]
- 82.Rolland F, Moore B, Sheen J. Sugar sensing and signaling in plants. Plant Cell. 2002;14(Suppl 1):185–205. doi: 10.1105/tpc.010455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Albrecht G, Mustroph A. Localization of sucrose synthase in wheat roots: increased in situ activity of sucrose synthase correlates with cell wall thickening by cellulose deposition under hypoxia. Planta. 2003;217(2):252–260. doi: 10.1007/s00425-003-0995-6. [DOI] [PubMed] [Google Scholar]
- 84.Salnikov VV, Grimson MJ, Seagull RW, Haigler CH. Localization of sucrose synthase and callose in freeze-substituted secondary-wall-stage cotton fibers. Protoplasma. 2003;221(4):175–184. doi: 10.1007/s00709-002-0079-7. [DOI] [PubMed] [Google Scholar]
- 85.Nolte KD, Hendrix DL, Radin JW, Koch KE. Sucrose synthase localization during initiation of seed development and trichome differentiation in cotton ovules. Plant Physiol. 1995;109(4):1285–1293. doi: 10.1104/pp.109.4.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shi YH, Zhu SW, Mao XZ, Feng JX, Qin YM, Zhang L, Cheng J, Wei LP, Wang ZY, Zhu YX. Transcriptome profiling, molecular biological, and physiological studies reveal a major role for ethylene in cotton fiber cell elongation. Plant Cell. 2006;18(3):651–664. doi: 10.1105/tpc.105.040303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruan YL, Llewellyn DJ, Furbank RT. Suppression of sucrose synthase gene expression represses cotton fiber cell initiation, elongation, and seed development. Plant Cell. 2003;15(4):952–964. doi: 10.1105/tpc.010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pang CY, Wang H, Pang Y, Xu C, Jiao Y, Qin YM, Western TL, Yu SX, Zhu YX. Comparative proteomics indicates that biosynthesis of pectic precursors is important for cotton fiber and Arabidopsis root hair elongation. Mol Cell Proteomics. 2010;9(9):2019–2033. doi: 10.1074/mcp.M110.000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prigge MJ, Wagner DR. The Arabidopsis SERRATE gene encodes a zinc-finger protein required for normal shoot development. Plant Cell. 2001;13(6):1263–1280. doi: 10.1105/tpc.13.6.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoke SM, Guzzo J, Andrews B, Brandl CJ. Systematic genetic array analysis links the Saccharomyces cerevisiae SAGA/SLIK and NuA4 component Tra1 to multiple cellular processes. BMC Genet. 2008;9(1):46. doi: 10.1186/1471-2156-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jin Y, Rodriguez AM, Stanton JD, Kitazono AA, Wyrick JJ. Simultaneous mutation of methylated lysine residues in histone H3 causes enhanced gene silencing, cell cycle defects, and cell lethality in Saccharomyces cerevisiae. Mol Cell Biol. 2007;27(19):6832–6841. doi: 10.1128/MCB.00745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nawy T, Lee JY, Colinas J, Wang JY, Thongrod SC, Malamy JE, Birnbaum K, Benfey PN. Transcriptional profile of the Arabidopsis root quiescent center. Plant Cell. 2005;17(7):1908–1925. doi: 10.1105/tpc.105.031724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y, Zhang WZ, Song LF, Zou JJ, Su Z, Wu WH. Transcriptome analyses show changes in gene expression to accompany pollen germination and tube growth in Arabidopsis. Plant Physiol. 2008;148(3):1201–1211. doi: 10.1104/pp.108.126375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.The Arabidopsis information resourcehttp://www.arabidopsis.org/servlets/TairObject?type=locus&name=AT3G01160
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1: Cotton quantitative phosphoproteomic analysis workflow. WT -3 DPA and WT 0 DPA: Ovules from -3 and 0 DPA developmental stages of Xuzhou 142 WT. fl -3 DPA and fl 0 DPA: Ovules from -3 and 0 DPA developmental stages of Xuzhou 142 fl mutant. A: Peptide sequence identification from peptide backbone fargment ions. B: Quantification from iTRAQ reporter ions. Scale bars: WT -3 DPA and fl -3 DPA, 200 μm; WT 0 DPA and fl 0 DPA, 20 μm. (TIFF 757 KB)
Additional file 2: The file contains all original MS/MS spectra of the 830 phosphopeptides identified in this research. (ZIP 15 MB)
Additional file 3: Table S1: Two sheets were included. Sheet 1: Detected phosphorylated sites. Sheet 2: Phosphoprotein annotations. (XLSX 367 KB)
Additional file 4: Table S2: Comparison of phosphosites conserved between cotton and species in the P3DB database. (XLSX 233 KB)
Additional file 5: Table S3: Phosphoprotein Pfam domain information (sheet 1) and phosphosites location in characterized protein domains (sheet 2). (XLSX 232 KB)
Additional file 6: Table S4: Three sheets were included. Distribution of identified phosphoproteins in cellular component (sheet 1), biological process (sheet 2), and molecular function (sheet 3) categories. (XLSX 46 KB)
Additional file 7: Table S5: Novel phosphoproteins (sheet 1) and phosphoproteins with homologs in other species in P3DB (sheet 2). (XLSX 9 MB)
Additional file 8: Table S6: Number of transcription factors among the identified phosphoproteins. (XLSX 17 KB)
Additional file 9: Table S7: Sequence alignment of phosphorylation sites and extraction of significantly enriched phosphorylation motifs. (XLSX 61 KB)
Additional file 10: Table S8: Six sheets were included. Motifs identified via Motif-X surrounding localized pSer in nine plant species (sheet 1); pThr residues in nine species (sheet 2); pSer in cotton (sheet 3); pThr residues in cotton (sheet 4); prealigned “phosphor-13-mers” in cotton and nine other species (sheet 5); random protein sequences derived from the genomes of cotton and the nine other species (sheet 6). (XLSX 12 MB)
Additional file 11: Table S9: Five sheets were included. Sheet 1: quantitative analysis of 830 phosphopeptides. Sheet 2: Differentially phosphorylated proteins between WT and fl -3 DPA ovules. Sheet 3: Differentially phosphorylated proteins between WT and fl 0 DPA ovules. Sheet 4: Differentially phosphorylated proteins between WT 0 DPA and fl -3 DPA ovules. Sheet 5: Differentially phosphorylated proteins between fl 0 DPA and fl -3 DPA ovules. (XLSX 257 KB)
Additional file 12: Figure S2: Molecular functional classification of identified differentially phosphorylated proteins (-3 DPA WT vs. -3 DPA fl, and 0 DPA WT vs. 0 DPA fl). (TIFF 190 KB)
Additional file 13: Table S10: Molecular functional classification of the 69 differentially phosphorylated proteins identified in this study. (XLSX 13 KB)
Additional file 14: Table S11: The significantly enriched pathways were identified by KOBAS. (XLSX 14 KB)