

Abstract
Biometals such as zinc, iron, copper and calcium play key roles in diverse physiological processes in the brain, but can be toxic in excess. A hallmark of neurodegeneration is a failure of homeostatic mechanisms controlling the concentration and distribution of these elements, resulting in overload, deficiency or mislocalization. A major roadblock to understanding the impact of altered biometal homeostasis in neurodegenerative disease is the lack of rapid, specific and sensitive techniques capable of providing quantitative subcellular information on biometal homeostasis in situ. Recent advances in X-ray fluorescence detectors have provided an opportunity to rapidly measure biometal content at subcellular resolution in cell populations using X-ray Fluorescence Microscopy (XFM). We applied this approach to investigate subcellular biometal homeostasis in a cerebellar cell line isolated from a natural mouse model of a childhood neurodegenerative disorder, the CLN6 form of neuronal ceroid lipofuscinosis, commonly known as Batten disease. Despite no global changes to whole cell concentrations of zinc or calcium, XFM revealed significant subcellular mislocalization of these important biological second messengers in cerebellar Cln6nclf (CbCln6nclf) cells. XFM revealed that nuclear-to-cytoplasmic trafficking of zinc was severely perturbed in diseased cells and the subcellular distribution of calcium was drastically altered in CbCln6nclf cells. Subtle differences in the zinc K-edge X-ray Absorption Near Edge Structure (XANES) spectra of control and CbCln6nclf cells suggested that impaired zinc homeostasis may be associated with an altered ligand set in CbCln6nclf cells. Importantly, a zinc-complex, ZnII(atsm), restored the nuclear-to-cytoplasmic zinc ratios in CbCln6nclf cells via nuclear zinc delivery, and restored the relationship between subcellular zinc and calcium levels to that observed in healthy control cells. ZnII(atsm) treatment also resulted in a reduction in the number of calcium-rich puncta observed in CbCln6nclf cells. This study highlights the complementarities of bulk and single cell analysis of metal content for understanding disease states. We demonstrate the utility and broad applicability of XFM for subcellular analysis of perturbed biometal metabolism and mechanism of action studies for novel therapeutics to target neurodegeneration.
Introduction
Tight regulation of biologically active metal homeostasis is critical to proper functioning of the brain. Metals such as copper, iron and zinc are essential cofactors to over 30% of all mammalian enzymes, and are increasingly recognized as important second messengers in neuronal signaling. Hence, it is not surprising that deregulation of biometal metabolism is reported to significantly contribute to pathology in a wide spectrum of neurological conditions. The precise metal disturbances are specific for each neurological disorder and may be related to toxic mislocalization as observed in Alzheimer’s disease (AD) 1 and Friedrich’s Ataxia 2, altered metal transporter function as in Menkes and Wilson’s diseases 3, 4 or altered metal-protein interactions as in amyotrophic lateral sclerosis (ALS) 5. Due to the sophisticated physiological control of cellular biometal homeostasis exerted by scores of metal transport, buffering, muffling and chaperone proteins, even subtle mislocalization of biometals can have a critical impact on cell functions in a non-obvious manner 6. AD presents a pertinent example - although the widely accepted metal-amyloid hypothesis implicates increased extracellular zinc and copper in amyloid β aggregation 7, reports on bulk metal levels in whole tissue homogenates are rarely indicative of subtle subcellular changes involving intracellular metal depletion in AD 1, 8, 9. Hence while it is evident that metal dyshomeostasis is a key hallmark of impaired neurological function, it is not widely appreciated that disease-associated changes in metal metabolism involve complex and dynamic intracellular trafficking of multiple metal pools (both protein bound or readily-exchangeable) between different organelles.
Similarly, our recent work demonstrates subtle accumulation of zinc, copper, manganese and cobalt in animal models of the CLN6 form of an inherited childhood neurodegenerative disorder, the neuronal ceroid lipofuscinoses (NCLs) 10, 11. This disease is characterized by progressive blindness and motor impairment leading invariably to premature death (reviewed in 12). CLN6 encodes an ER protein of unknown function; recessive mutations in CLN6 exert pathological consequences on many subcellular compartments including the ER, lysosomes and mitochondria 13. We determined that the disrupted biometal functions involved in progression of NCLs are driven by loss of an intracellular metal transporter, Zip7 14, which is predicted to result in subcellular zinc mislocalization. Consistent with this, we demonstrated redistribution of brain zinc in diseased mice and sheep into ER and lysosome/Golgi compartments using subcellular fractionation techniques 10, although multiple subcellular organelles are known to co-localize within individual fractions 15. Importantly, a metal complex, ZnII(atsm), restored cellular zinc-dependent functions in Cln6nclf cells, however the mechanism is not clearly defined. Hence, complementary methods of accurate visualization and quantitation of subcellular metal distributions in cells and tissues are critical to understanding metal dysregulation in neurological disease and for defining novel therapeutic targets for treatment.
It remains the case that there is a lack of effective tools to probe the quantitative biometal distribution at the single cell level. Current bulk analysis techniques such as Inductively Coupled Plasma Mass Spectrometry (ICP-MS) are extremely sensitive but cannot routinely assess inter-cell variation and require destruction of the sample. Advances in Laser Ablation-(LA-) ICP-MS have successfully extended this approach to providing quantitative spatially-resolved information on elemental distribution but currently cannot interrogate the subcellular organization of elements within intact cells and of course the measurement process remains entirely destructive 16. Metalloproteomic techniques, ie. size exclusion chromatography (SEC) coupled to ICP-MS, which has revealed critical information on protein-bound metal pools 17, are incapable of probing loosely bound or labile metals and are further limited by the inability to measure single cells or provide information on the subcellular compartmentalization of metals. Cell permeable metal-specific optical fluorophores provide valuable information (i.e. cell to cell variability) on pools of “exchangeable” ions, but the extent to which these compounds may influence the native cellular metalloarchitecture is less clear (for a comprehensive review see 18). Moreover, care must be taken to avoid spurious interpretations, stemming from changed spectral characteristics due to organelle-specific differences in uptake, or local changes to the chemical environment (pH or redox potential) 19, 20. X-ray fluorescence (XRF) microscopy (XFM; for a recent review see 21) can provide probe free, quantitative, spatially resolved information of the nanoscale organization of elements within single cells with minimal measurement artifacts 21. In addition to mapping the distribution of elements, more specialized XFM experiments allow complementary X-ray Absorption Near Edge Structure (XANES) studies to be performed on the same sample 22. XANES can reveal information about elemental oxidation state and allow interrogation of the chemical environment for the element under investigation. XFM is also compatible with optical and ion beam microscopies, making powerful correlative studies possible. XFM has been used to assess elemental content and distribution at the single cell level for over thirty years 23 but due to the typically extended periods of time required (many hours of data collection per cell) XFM has not yet found routine use. However, advances in XRF detectors have increased the speed with which data can be collected, thus making the mapping of statistically relevant cell numbers achievable 24. Here we use XFM to demonstrate disturbed bio-metal homeostasis at the subcellular level in CbCln6nclf cells and show that treatment with ZnII(atsm) is able to correct altered cellular physiology.
Results
Loss of Zip7 metal transporter expression in cerebellar cells from Cln6nclf mice
We previously reported accumulation of zinc, copper, cobalt, manganese and iron in brain tissue homogenates from natural sheep and mouse models of NCLs, a group of inherited childhood neurodegenerative and lysosomal storage disorders 10, 11. We observed early and progressive loss of the ER/Golgi resident biometal transporter Zip7, in the brains of sheep and primary cortical neurons from embryonic mice containing natural mutations in the Cln6 gene 14. Mutations in the gene, including the identical mutation to that observed in the Cln6nclf mouse, are associated with variant late infantile NCL in humans 25, 26. These observed changes are indicative of deregulated subcellular biometal homeostasis in NCL, and we have since identified analogous loss of biometal regulation in additional sub-types of NCL (CLN1, 3 and 5; 27). Biometal accumulation in the cerebellum occurred with concomitant reduction in cerebellar Cln6 mRNA in Cln6nclf mice 10. The cerebellum controls motor functioning, an early impairment in the CLN6 sub-type of NCL disease, and is a region of initial pathology both in human disease and associated animal models 28–30. A cerebellar cell line established from Cln6nclf mice (CbCln6nclf cells) recapitulated aberrant lysosomal protein accumulation as well as mitochondrial and ER dysfunction evident in human CLN6 disease 31, providing a highly relevant secondary cell line to investigate how subcellular biometal homeostasis is affected in CbCln6nclf cells. Western blotting revealed a significant loss of Zip7 expression in homozygous CbCln6nclf cells compared to control cells (Fig. 1A–B), supporting previous observations that Slc39a7 (encoding Zip7) expression was −1.6 fold downregulated in 3 different CbCln6nclf cell lines31. We examined whether these changes altered global cellular metal levels. Bulk analysis via ICP-MS revealed no differences in the cellular concentrations of zinc, copper, iron, calcium, manganese or cobalt (Fig. 1C–F and data not shown).
Fig 1. Zip7 loss in CbCln6nclf cells is not associated with altered global biometal levels.

A. Representative immunoblot of Zip7 and total ERK expression in cellular homogenates (5μg) harvested from non-treated control and CbCln6nclf mutant cells. B. Densitometry analysis of immunoblots (N=3 biological replicates), normalized to expression of total ERK protein was performed using ImageJ. Values are mean + SEM. ** p<0.01 by Student’s t-test. C–F Total cellular zinc (C), copper (D), iron (E) and calcium (F) concentrations were analysed using ICP-MS. Metal concentrations are normalized to total cellular protein content, as measured by BCA assay, and are expressed as mean + SEM.
Use of XFM to interrogate subcellular metal levels
Though no changes to total biometal levels were detected by bulk analysis, previous reports have shown that biometal deregulation associated with disease states can be more nuanced than simple gross changes in abundance 8, 32. Additionally, recent reports have highlighted the value of performing “per-cell” analysis of biometal abundance and distribution 24. To test our hypothesis that reduced Zip7 expression may impair zinc metabolism, we investigated whether biometal trafficking rather than total metal concentrations were altered in CbCln6nclf cells. XFM provided an ideal technique to collect detailed information on the sub-cellular distribution of zinc, in situ.
Fixation of samples for elemental analysis is a critical consideration, as use of traditional chemical fixatives can induce a variety of artifacts 33. Thus, to ensure preservation of native biometal distribution and with organelle integrity in an “as near to native” state as possible, chemical fixation was eschewed as it is known to induce leakage and reorganization of metal ions 34. In contrast, cryoprotection followed by dehydration preserves biometal levels and distributions while rendering specimens suitable for XFM analysis. Previous work comparing biometal distribution between hydrated anesthetized C. elegans and cryopreserved and desiccated animals, provided confidence that XFM of cryopreserved specimens is appropriate for the preparation of biological samples 35. Advances in low-latency XRF detectors (such as the 384-channel Maia detector installed at the Australian Synchrotron) provide significantly faster data acquisition rates and allow high-definition subcellular elemental mapping of ~10–25 cells in ~30 minutes (a typical scan area of 0.015 mm2 area containing 94,000 pixels). The increased sensitivity is concurrent with a reduction in dose, enabling the imaging of optical fluorescence organelle markers both before and after elemental mapping. These served a dual role, providing reference points for correlating the elemental maps while also acting as sensitive probes for any distortions to cellular ultrastructure that exposure to ionizing radiation may induce (Fig. S1).
The Compton scatter of incident photons (Fig. 2A–B) provides an ideal guide to delineate cellular boundaries (Fig. S2). To ensure that treatment of cells with the organelle markers ER-Tracker™ Green and MitoTracker® Deep Red did not alter elemental abundance or distribution, elemental mapping was performed on parallel preparations of cells that had not been treated with these organelle markers. The presence of organelle markers did not have an impact on the concentrations of cellular zinc or calcium (Fig. S3, S4). However, treatment with organelle markers resulted in increased concentrations of copper and iron detected by XFM (Fig. S4). We expect that the treatment with the markers is likely to exert a corresponding effect in both control and CbCln6nclf cells. Thus, relative comparisons of copper and iron data between these groups still remain biologically meaningful. For the purpose of the present study, we chose to focus on the distributions of calcium and zinc in these cells.
Fig 2. Identification of subcellular metal distribution in cerebellar cells using XFM.

Cerebellar granule cells from control and Cln6nclf mice were grown on X-ray transparent SiN windows (500nm thick) and fixed by plunge freezing in liquid ethane (−180°C). Fixed cells were exposed to an incident beam of 12.7 keV X-rays. A–B. Grayscale-colored images of inelastic scatter (Compton) of incident photons was used to define cell boundaries (Figure S2). C–F. Heat maps of elemental concentrations for zinc (C–D) and calcium (E–F). The maximum concentration of each element is shown at the top left of the relevant panel. Red arrows indicate cells with high nuclear:cytoplasmic zinc ratios, while the green arrow indicates a cell with a low nuclear:cytoplasmic zinc ratio. G–H. Heat maps of calcium concentrations divided by zinc concentrations for each pixel. I–L. Cerebellar cells were stained with MitoTracker® Deep Red (500nM; I–J) and ER-Tracker™ Green (500nM; K–L) for 30min prior to fixation. Confocal images depicting subcellular localization of mitochondria and endoplasmic reticulum in cerebellar cells. Scale bar, 50μm
Identification of deregulated subcellular metal distribution in CbCln6nclf cells
Consistent with previous reports, zinc was concentrated primarily in the cell nucleus, particularly evident in the control cells (Fig. 2C–D) 36, 37. This is presumably a reflection of the importance of zinc as a cofactor for over 400 predicted zinc-finger transcription factors 38. We observed dramatic alterations in subcellular calcium distributions between control and homozygous CbCln6nclf mutant cells (Fig. 2E–F), which is consistent with recent work showing impaired Ca2+ metabolism in several NCL models. In the Cln8mnd mouse model of NCL, an age related inability to clear cellular Ca2+ was due to a mitochondrial calcium accumulation 39, while CLN3 is reported to protect cells from calcium-dependent cytotoxicity via interactions with the calcium sensor calsenilin 40. In control cells, calcium was evenly distributed throughout the entire cell (Fig. 2E), while in CbCln6nclf mutant cells, despite the total amount of cellular calcium being similar to controls, we observed small puncta (−1σ to 1σ: area = 1.94 μm2 to 9.48 μm2) highly enriched in calcium with average calcium concentrations 5.6 fold above controls (1.8 μg cm−2; σ=0.67 μg cm−2; Fig. 2F). Conversely, these data reveal that the majority of the CbCln6nclf mutant cell interior is calcium deficient, despite total calcium content being appropriate (Fig. 3B). Though calcium puncta were observed in multiple cultures of CbCln6nclf and rarely in the healthy preparations we harbored concerns that these structures may be artifacts of insoluble calcium settling out of solution and decorating the cell surface. If true, the projected area of the cell would be strongly (and positively) correlated with the number of calcium deposits. To test this hypothesis, the relationship between a cell’s size (projected area) and the number of calcium deposits was determined. We observed that the cell area accounted for less than 5% of the observed variation in the number of calcium deposits per cell. This analysis provided strong evidence that accumulation of calcium-enriched deposits in CbCln6nclf cells is not due to passive calcium precipitation from the media (Fig. S5) but reflects deeply altered calcium homeostasis. The cellular distribution of calcium and zinc within control cells provided a useful means of determining the position of the nucleus based on the Ca:Zn ratio (Fig. 2G). However, the extensive mislocalization of calcium in CbCln6nclf cells precluded such an analysis for these cells (Fig. 2H).
Fig 3. Subcellular biometal homeostasis is deregulated in CbCln6nclf cells.

Inelastic scatter was used as a guide for the generation of cell masks in ImageJ. As zinc demonstrates primarily nuclear distribution in all cells, high zinc intensity was used to delineate nuclei, and cytoplasm was defined as any cellular area not encompassing the nucleus. The mean concentration of zinc (A) and calcium (B) in each individual control (black bars) and CbCln6nclf (grey bars) cell, nucleus and cytoplasm was calculated using the masks (Figure S2). Data are presented as mean + SEM. * p<0.05, *** p<0.0001 by two-way ANOVA and Bonferroni multiple comparison posttests. (C–D) The nuclear concentration of zinc (C) and calcium (D), in each individual control (black circles) and CbCln6nclf (grey triangles) cell was plotted against the cytoplasmic concentration of each respective metal in the same cell. Linear regression analysis was performed in GraphPad Prism. r2 values for the strength of each correlation are presented in each panel. Red triangles indicate cells with high nuclear:cytoplasmic zinc ratios (corresponding to Fig. 2), while the green triangle indicates a cell with a low nuclear:cytoplasmic zinc ratio (corresponding to Fig. 2).
CbCln6nclf cerebellar cells possess lysosomal, mitochondrial and ER abnormalities 31. Though we attempted to include the lysosomal marker, LysoTracker® Blue DND-22, as part of our suit of optical fluorophores, we were unable to successfully capture meaningful images of this tag. These technical difficulties could be due to a number of issues, potentially including fluorophore quenching due to cryopreservation and desiccation of the specimen (loss of aqueous environment or increased optical density of the sample)41. Though in this investigation we were unable to examine lysosomal localization of metal content, we continue to develop protocols to enable this work to be undertaken in the future. However, ER-Tracker™ Green and MitoTracker® Deep Red retained fluorescence post our sample preparation protocol and we made use of these markers in our search for disrupted metal homeostasis in subcellular compartments. Prior to XFM analysis, cryopreserved and dehydrated cells were imaged using confocal immunofluorescence (Fig. 2I–L, for higher magnification images, see Fig. S6). Consistent with previous observations 31, CbCln6nclf cells displayed altered mitochondrial and ER staining compared to control cells. However, co-registration of organelle markers and the calcium deposits present in the CbCln6nclf cells was unconvincing, suggesting that the calcium accumulation observed in these cells was not occurring exclusively in either the mitochondria or ER. Usefully, fluorescence was maintained even after XFM (Fig. S1), presenting a technical advance with ready application to future high resolution XFM studies of bioinorganic chemistry.
In order to extract per-cell elemental content, the elastic scatter (Compton Scatter) of incident X-rays was used to generate Boolean masks to separate the “cell” area from “non-cell” area. as a guide. Nuclear masks were generated using a combination of cellular zinc concentrations, Ca:Zn ratios and absence of ER and/or mitochondrial staining as a guide (Fig. S2). Cytoplasmic regions were defined as the remaining cellular area that was not the nucleus. Mean cellular, nuclear and cytoplasmic zinc concentrations were unchanged between control and CbCln6nclf cells (Fig. 3A). Similarly, despite the dramatically perturbed calcium distributions between control and CbCln6nclf cells (Fig. 2E–F), analysis of mean cellular, nuclear and cytoplasmic calcium concentrations revealed no differences (Fig. 3B). Although a loss of total cellular zinc and calcium content is detectable, such an analysis would be misleading due to the inherent size differences between control and CbCln6nclf cells (mean cell area is 605 μm2 for control cells, 271 μm2 for CbCln6nclf cells).
It is important to recognize the limitations of bulk measurements, which provide no information on the variability in metal content between single cells within a population, or the intracellular distribution of different metal pools within cells. To examine the metal handling dynamics within single cells, we measured the relationship between the mean nuclear and cytoplasmic metal concentration in individual cells. Despite substantial inter-cell variation in total metal concentrations (ranging from 5.4 to 13.2 ng cm−2 for zinc and 199.3 to 484.2 ng cm−2 for calcium), we observed a strong positive correlation between the nuclear and cytoplasmic concentration of zinc and calcium in control cells (r2=0.85, and 0.92, respectively; Fig. 3C–D), reflective of stringent control of inter-organelle metal concentrations as maintained by membrane bound transporters, metal chaperones, storage proteins and the suit of cytoplasmic buffers and mufflers 42. Importantly, there was a complete loss of relationship between the nuclear to cytoplasmic zinc ratios in CbCln6nclf cells (r2=0.03, p=0.00012; Fig. 3C), indicating severely impaired subcellular zinc homeostasis. The correlations between nuclear and cytoplasmic calcium concentrations were also reduced, but to a lesser extent than for zinc (r2=0.32, p=0.0131; Fig. 3D). Together, these data demonstrate the utility of high-definition elemental mapping using XFM over traditional analytical techniques such as ICP-MS for understanding subcellular metal handling dynamics in health and disease.
XFM identifies restoration of subcellular metal regulation after zinc delivery with the zinc-complex, ZnII(atsm)
We previously demonstrated that a zinc-delivering complex, ZnII(atsm)43, induced upregulation of Zip7 and improved activity of a zinc-dependent enzyme in primary Cln6nclf astrocytes 14. To directly investigate whether the therapeutic effect of ZnII(atsm) involved subcellular biometal redistribution in CbCln6nclf cells, we treated cultures for 2h with ZnII(atsm) (1μM) and performed elemental mapping using XFM (Fig. 4). ZnII(atsm) treatment of control cells resulted in primarily nuclear zinc uptake into some, but not all, cells (Fig. 4C). Following ZnII(atsm) treatment, zinc accumulated in CbCln6nclf cells, although the distribution was more clearly nuclear than in untreated cells (Fig. 4D). Indeed, ZnII(atsm) substantially restored the relationship between the nuclear and cytoplasmic zinc concentrations in individual cells (from r2=0.03 to r2=0.67; Fig. 5A).
Fig 4. Identification of subcellular metal distribution in cerebellar cells treated with ZnII(atsm).
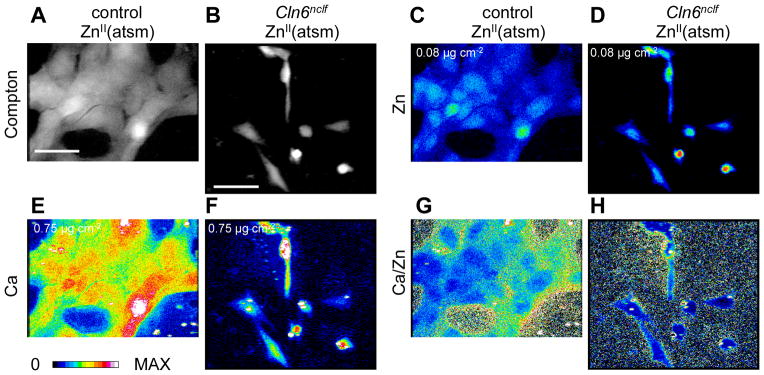
Cerebellar granule cells from control and Cln6nclf mice were grown on X-ray transparent SiN windows (500nm thick), treated with 1μM ZnII(atsm) for 2h and fixed by plunge freezing in liquid ethane (−180°C). Fixed cells were exposed to an incident beam of 12.7 keV X-rays. A–B. Grayscale-colored images of inelastic scatter (Compton) of incident photons was used to define cell boundaries. C–F. Heat maps of elemental concentrations for zinc (C–D) and calcium (E–F). The maximum concentration of each element is shown at the top left of the relevant panel. G–H. Heat maps of calcium concentrations divided by zinc concentrations for each pixel. Scale bar, 50μm.
Fig 5. ZnII(atsm) treatment restores subcellular biometal homeostasis in CbCln6nclf cells.

A. The nuclear concentration of zinc in each individual cerebellar control cell (black circles), CbCln6nclf cell (grey triangles) and CbCln6nclf cell treated for 2h with 1μM ZnII(atsm) (red squares) was plotted against the cytoplasmic concentration of zinc in the same cell. Linear regression analysis was performed in GraphPad Prism. r2 values for the strength of each correlation are presented. B. The concentration of zinc in each individual control cell (black circles), CbCln6nclf cell (grey triangles) and CbCln6nclf cell treated for 2h with 1μM ZnII(atsm) (red squares) was plotted against the concentration of calcium in the same cell. C. Binary masks were generated to examine the number of regions of high calcium intensity (above 2μg cm−2) per cell area for each treatment group. ** p<0.01, *** p<0.0001 by one-way ANOVA and Bonferroni multiple comparison posttests. Error bars represent SEM. D. The histograms (tabulated frequency, bin width=0.0385 ng cm−2) of the log-transformed calcium concentrations in cells were modeled by Gaussian curves (solid black, grey or red lines). The residual plots are presented above the histograms. E. Zinc K-edge XANES of control cell, CbCln6nclf cells and CbCln6nclf cells treated for 2h with 1μM ZnII(atsm) was performed using X-rays at 58 energies ranging from 9.604 to 9.76 keV. Pre-edge absorbance values were linearized and normalized XANES is expressed as a percentage of the absorbance maxima. XANES spectra for ZnCl2 and ZnO nanoparticles are presented as controls.
In light of the well described promiscuity of many calcium channels to zinc (reviewed in 44), and the reported role of zinc in the regulation of cellular calcium-binding proteins 45–47, we hypothesized that cellular zinc and calcium concentrations may be, at least partially, co-regulated. Indeed, a very strong correlation between per-cell zinc and calcium concentrations in individual control cells was observed (r2=0.90, Fig. 5B), whereas this correlation was lost in CbCln6nclf cells (r2=0.26). Importantly, the relationship between cellular zinc and calcium was partially corrected upon ZnII(atsm) treatment (r2=0.46). This correction may be due, in part, to ZnII(atsm)-dependent calcium redistribution throughout CbCln6nclf cells (Fig. 4F). As shown in Fig. 3B, the mean calcium concentration does not reflect the clear visual differences in calcium distributions between control and CbCln6nclf cells. Therefore the distribution of calcium was analyzed by defining and counting ROIs in ImageJ that were thresholded above a calcium concentration of 2 μg cm−2 (the maximum calcium concentration for 99.95% of pixels in control cells). The number of calcium-rich deposits per μm2 in each cell was significantly higher in CbCln6nclf cells, and was substantially reduced by ZnII(atsm) treatment (Fig. 5C). The distribution of per-pixel calcium concentrations for cerebellar cells was well modeled by a log-Gaussian curve (Fig. 5D), consistent with previous reports for biological systems where an analyte’s concentration is the product of multiple dependent reactions that fluctuate between individual cells 48. In control cells, a single log-Gaussian with a log-mean of 2.503 (calcium concentration = 318 ng cm−2) was modeled to the data. The distribution in CbCln6nclf cells was accurately modeled by a log-Gaussian with a log-mean of 2.297 (calcium concentration = 198 ng cm−2). This represents the majority of calcium within the cells, and is calcium deficient, as compared to control cells. ZnII(atsm) treatment induced a shift in the major calcium peak in CbCln6nclf cells towards that of control cells (log-mean = 2.365, [Ca]= 232 ng cm−2) further supporting a protective effect of ZnII(atsm) on subcellular metal homeostasis.
We hypothesized that the distorted distribution of zinc observed in CbCln6nclf cells may be due to alterations in endogenous zinc ligands compared to healthy cells. To probe the nature of zinc binding partners in Battens disease, we performed Zn K-edge XANES to characterize the coordination environment of cellular zinc in both healthy and CbCln6nclf cells. The efficacy of data collection achievable with the Maia detector installed at the Australian Synchrotron49, allowed small groups of cells (3–6 individuals) to be identified and targeted for XANES mapping. In this instance Zn XANES-mapping entailed the region of interest (ROI) being re-scanned at 59 unique incident energies (see methods section for details) the resulting series of images provided spatially resolved spectroscopic information on the coordination environment of zinc within the targeted cells. While there was little difference between the white-line or edge position of the XANES spectra from control or CbCln6nclf cells, there was an additional feature, present at 9.683 keV, visible in the diseased group (Fig. 5E), indeed altered expression levels of Zn-proteins between mutant and wild type cells is the likely explanation rather than an entirely new class of ligand being present. Interestingly, this structure was no longer present in the spectra of CbCln6nclf cells following ZnII(atsm) treatment. Though we were unable to confidently assign any particular ligand(s), these data provide strong evidence that the cellular co-ordination environment of zinc in CbCln6nclf cells is altered and corrected by ZnII(atsm). At the same time, however, the similarities between the spectra limit the degree to which the “global “coordination sphere of zinc has changed (Fig. 5E). The spectra of ZnII(atsm)-treated cells is substantially more reminiscent of the cerebellar cell spectra than that of the ZnII(atsm) standard, consistent with zinc dissociation from ZnII(atsm) upon cell entry. This study highlights the utility of XFM for understanding subtle but critical alterations to discrete sub-cellular biometal pools in cells containing disease-associated protein mutations. Analogous studies can also be applied to cells subjected to diverse stresses associated with disease.
Discussion
Despite rapid improvements in the sensitivity of assessing biometal content of cells and tissues due to advancement of ICP-MS (and related techniques), there remains much to be understood about the inorganic physiology of the brain in health and disease. The use of XFM overcomes several analytical challenges associated with hyphenated-ICP-MS techniques by allowing investigation of individual cells at subcellular resolution with minimum disruption to native elemental abundance or distribution, and XFM is not prone to artifacts associated with optical metal-sensing fluorophores. The world-leading Maia XRF detector at the Australian Synchrotron has data acquisition rates orders of magnitude faster than conventional XFM setups, allowing the rapid and routine high-definition elemental mapping of many cells. This overcomes a major limitation of traditional bioXFM studies, where often only small numbers of cells (n = 3) were analyzed due to time constraints50, 51.
Additionally, the improvements in detector efficiency reduce the dose given to the sample and allowed the concomitant visualization of organelle makers within cells before and after XFM. This type of correlative microscopy maximizes the information content extracted from a given specimen and is preferred to the more typical approach of inferring elemental information with staining patterns worked up in parallel cultures 52 or within adjacent tissue sections53. To our knowledge, this work represents the most complete concurrent visualization of multiple organelle distributions within cells probed for metal content via XFM. As organelle structure is complex and often dynamic 54, 55, multiplexed imaging, as described here, of organelle markers and quantitative elemental mapping, provides a powerful opportunity to gather information on transient subcellular metal pools. However, what effects introducing organelle markers may have on metal ion distribution will need careful examination. Though work continues, the observed alteration in the cellular iron and copper load in stained cells may stem from sequestration of oxidized MitoTracker® within mitochondria. This will provide an opportunity for the dye to react with thiol moieties of Fe and Cu chaperone proteins linked to the biogenesis of Fe-S clusters (reviewed in 48) and delivery of Cu to components of the electron transport chain 49. Importantly, we determined that calcium and zinc content or distributions were not significantly altered by organelle marker treatment (Fig. S3, 4).
XFM is a scanning probe imaging technique and traditional setups suffer slow rates of data acquisition partly as a consequence of mechanical limitations in raster-scanning specimens through x-ray focus but until recently delays in detector read-out have been significant. Thus, while high resolution studies are possible 56, the length of time required to collect data can swiftly become unacceptably long, particularly if biological replicates are required. Often these practicalities result in collecting elemental maps that are somewhat under-sampled (i.e. the sampling rate fails to satisfy the Nyquist theorem). By employing a multiparameter (event-mode) approach to data acquisition, the Maia has reduced read-out overhead to zero, enabling faster scanning. Here samples were fly-scanned through x-ray focus and the resulting XRF was binned every 400 nm pixels (thus each pixel is 0.016 μm2). As the pixel size is 5-fold smaller than the probe size (Full Width Half Maximum of the incident beam intensity: horizontal: 2.3 μm; vertical 1.99 μm), oversampling in this manner provided significantly enhanced image contrast (over typical XFM setups) and facilitated ready demarcation of cell boundaries and internal structure(s), despite the relative large size of the probe and without a significant time penalty (Fig. 2I–L).
XFM provides an information-dense snapshot of subtle alterations to metal homeostasis in cells, which are unlikely to be detected in bulk analyses of global metal levels in cellular or tissue homogenates. XFM thereby enables novel avenues of investigation not afforded by bulk measurements. Firstly, the role of inter-cellular variability on metal homeostasis has seldom been examined. The striking variability of total elemental content between single cells even within a theoretically “homogeneous” secondary culture as observed here carries significant implications for the variability within the phenotypically distinct cellular subsets of the brain. For instance, copper sequestration by microglia in AD may limit the extracellular copper available to seed Aβ deposition57, while simultaneously rendering surrounding neurons copper-deficient58. Similar observations have been noted for zinc mislocalization in AD59. Examination of mean metal levels significantly underestimates the complexity of metal homeostasis due to the dilution of specific effects within population averages. This missing information on metal metabolism at the level of individual cells holds promise to shed light on selective neuronal vulnerability in neurodegeneration.
Secondly, visualization of subcellular elemental distribution is advantageous and reveals precise control of inter-organelle metal ratios despite immense inter-cell variability (Fig. 3C–D). Substantial deregulation of this metal homeostasis, especially in the case of zinc, was detected in CbCln6nclf cells, despite a lack of population-wide metal disturbances as measured by ICP-MS and XFM (Fig. 1, 3A–B). Optimal subcellular zinc levels are maintained by a network of over twenty specifically compartmentalized unidirectional membrane bound transporters belonging to the Zip and ZnT families (for a recent review, see 60). This potentially reflects the size and complexity of the zinc proteome, predicted to encompass approximately 2800 catalytic and regulatory proteins with an array of varied cellular functions 61. Altered expression of Zip and ZnT proteins has been reported for various neurodegenerative disorders, including AD 62, 63 and Friedrich’s Ataxia 64. We have also detected loss of Zip7 in 3 natural CLN6 disease models (Fig. 1A; 14), and Zip7 loss in zebrafish caused eye defects and reduced zinc concentrations in the eye 65, which may phenocopy CLN6 disease.
Subcellular biometal mislocalization can exert profound effects on pleotropic cellular functions. Zip7 is an ER/Golgi transmembrane zinc transporter in mammalian cells, controlling the influx of zinc, and potentially other metals 66, 67, into the cytoplasm. Therefore, loss of Zip7 would theoretically result in initial retention of labile or readily exchangeable zinc in the ER/Golgi. Due to the high relative affinity of Zn(II) for biomolecules (Irving-Williams series), increased concentrations of labile zinc in the ER or Golgi will displace other metals from protein co-ordination sites. The specific targets of aberrant excess zinc binding in the ER and Golgi are subject to further investigation in our laboratory. Cytoplasmic free zinc is low nanomolar 68, providing a reservoir of zinc for aberrant transport to the ER or Golgi, as labile zinc pools in these compartments are stringently maintained at less than 1pM 69. Additionally, Zip7 deficiency may prevent kinase-dependent “zinc wave” signals from being transduced throughout cells 70, 71. In the present study, we did not detect excess zinc in the ER (Fig. S7). However, given that total cellular metal levels are in the high micromolar range 72, even hundred-fold increases in labile zinc in the ER (from 1pM to 100pM) may not be directly visualized in the context of the million-fold excess of protein-bound zinc. It is also possible that we were unable to detect differences in ER zinc concentrations due to the resolution of the organelle staining or size of the x-ray beam. Nonetheless, we detected deregulation of subcellular zinc homeostasis in CbCln6nclf cells, that was rescued by ZnII(atsm) treatment. ZnII(atsm) induced primarily nuclear trafficking of zinc, restoring the ratios of nuclear:cytoplasmic zinc dynamics within two hours of treatment. Nuclear zinc is critical for the functioning of zinc finger transcription factors, however, the nuclear membrane is devoid of zinc transporters. Some zinc-finger transcription factors, including MTF-1 sense zinc in the cytoplasm, and translocation of the holo-protein is required for transcriptional activation of downstream processes 73. Nuclear localization of zinc-loaded metallothionein 74 has also been described and may represent an additional regulatory mechanism controlling nuclear:cytoplasmic zinc ratios. Whether ZnII(atsm) treatment results in direct nuclear zinc delivery, or delivered zinc is shuttled to the nucleus via alternative cellular processes remains to be determined.
XFM analysis also revealed striking differences in the subcellular distribution of calcium in control and CbCln6nclf cells. The presence of small, highly calcium-enriched deposits throughout CbCln6nclf cells is reminiscent of oxidative stress, as previously described 37. A limitation of the present generation of the Maia detector is the inability to measure elemental content of additional biologically relevant elements including P, S and Cl, as colocalization of these elements with calcium-enriched deposits may have provided clues regarding the composition of these structures. The subcellular distribution of the calcium-enriched deposits did not overlap with the distribution of the ER-Tracker™ Green and MitoTracker® Deep Red dyes, indicating that the typical cellular calcium storage compartments, the ER and the mitochondria, are not the sites of calcium storage or sequestration in CbCln6nclf cells. Alternatively, the calcium deposits may originate from lysosome-derived structures, as aberrant lysosomal calcium accumulation due to defects in the lysosomal Trpml1 channel has been described for the related LSDs, Mucolipidosis and Niemann-Pick C 75–77. This is an attractive possibility, as the lysosomes of CbCln6nclf cerebellar cells are known to accumulate the calcium-regulated 78 subunit c of mitochondrial ATP-synthase 31, providing a route for enhanced lysosomal calcium entry, whereas defects in Trpml1 may prevent lysosomal calcium extrusion. Although subunit c storage was not evident in subconfluent cultures 31, it is possible that low levels of increased lysosomal subunit c were below the detection limits in that study. Decreased ATP production, as reported in these cells 31, due to deregulation of mitochondrial ATP synthase, may also render cells more vulnerable to glutamate-mediated excitotoxicity, calcium influx and ROS production as described for CLN6 sheep models 79.
Lastly, bulk analysis does not enable investigation of metal co-regulation within cells. Correlations in the levels of calcium and zinc in neurons have been previously described 80, and may be indicative of co-regulation, co-trafficking or complexation. A number of calcium channels can transport zinc, both during synaptic activity through voltage-gated calcium channels, Nicotinic acetylcholine receptors, NMDA and AMPA receptors, as well as in resting cells at physiological calcium concentrations through plasma membrane and subcellular TRP channels (reviewed in 81). For example, the lysosomal Trmpl1 transporter, is reported to be zinc permeant 82. Not only can a vast array of calcium-permeable channels mediate zinc transport, but the physiology of these two metals in the brain is inextricably linked: calcium binding proteins that are critical for a wide range of cellular functions in signal transduction, cell communication, growth, including S100 family proteins and calmodulin, are reported to bind zinc in a structural or regulatory manner 45–47. Indeed, zinc is an allosteric modulator of calcium-bound calbindin 83. Moreover, Krebs et al (2013) found that calcium and zinc concentrations to significantly positively correlate in 11 post-mortem human brains in the absence of known brain pathology 84, suggestive of some degree of co-regulation or co-transport of these key second messengers. Although the subcellular distributions of calcium and zinc were not colocalized, we detected a correlation within individual control cells between the calcium and zinc concentrations (Fig. 5B), which was lost in CbCln6nclf mutant cells, and partially restored by ZnII(atsm).
We previously demonstrated the protective effect of ZnII(atsm), in primary Cln6nclf neurons and astrocytes, on numerous zinc-dependent functions, both structural and regulatory 14. These included improved activity of a zinc-containing enzyme, alkaline phosphatase, increased expression of the anti-oxidant zinc buffer metallothionein, and restored Zip7 expression. Consistent with this, we report here that ZnII(atsm) may exert its protective functions through subcellular redistribution of zinc and calcium, correcting the metal handling defect in CbCln6nclf cells. Recently, the efficacy of the natural turmeric-derived polyphenol, curcumin, on retinal function in Cln6nclf mice was described 85. In addition to its reported anti-inflammatory role, we hypothesize curcumin may act via complexation of mislocalized metal ions as proposed for its therapeutic action in AD 86.
The current major roadblock to dissecting physiological biometal homeostasis is that each analytical technique is capable of probing a discrete metal pool – proteomic techniques such as LC-ICP-MS can only detect protein-bound metals, whereas XFM can detect all metal species. Although the labile pool is negligible for metals with high affinity to biomolecules such as copper and zinc (as predicted by the Irving-Williams series), a readily exchangeable pool exists that is transiently sequestered by cytosolic ligands in metallothionein and glutathione. Exciting advances in the field of ratiometric, genetically encoded zinc sensors based on Fluorescence Resonance Energy Transfer (FRET) with specifically engineered subcellular targeting characteristics have been applied to measure resting and stress-dependent changes to mitochondrial and cytosolic zinc concentrations in hippocampal neurons 87 and readily exchangeable zinc in E. coli cells 88. The sensors are composed of transfected constructs containing a high-(K’d=1.7μM) or low-(K’d= 160μM) affinity zinc sensing domain, encoded by a Cys2His2 or His4 sequence, respectively, which change conformation upon zinc binding, enabling FRET between two fluorescent proteins fused to the domain 87. Other zinc-sensing domains, including various mutants of carbonic anhydrase have also been utilised, allowing modulation of zinc binding affinities from femto- to micromolar 89. Fusion of signal and export sequences within the construct enables subcellular targeting 87. These highly sophisticated systems are specific for zinc, display improved targeting properties compared to traditional small molecule sensors and provide precise measurements of labile (but not protein bound) zinc concentrations. A current limitation of the technique is the confounding effect on measured labile zinc concentrations of different transfection efficiencies within a population of cells, but these challenges will be addressed in the future by use of stably transfected cell lines or inducibly expressed genetic zinc-sensing constructs. Additionally, the technique is only currently feasible in cells amenable to transfection, and unlike XFM, cannot simultaneously measure the concentrations of multiple elements within cells.
A combination of techniques, including spatially-resolved XFM, highly-sensitive LC-ICP-MS, and novel ratiometric fluorescent metal sensors capable of probing total, protein-bound and readily exchangeable metal pools, respectively, will ultimately yield the greatest understanding of subcellular, intercellular and organ-level metal-handling dynamics in health and disease and will help to characterise the emerging signalling role that transient mobilization of labile metal ions from intracellular and protein stores play in both physiological and disease states.
Experimental
Cell culture conditions and treatments
Cerebellar granule neurons derived from control Cln6nclf mice (from both homozygous and wildtype littermates, on the C57BL6 genetic background) were cultured as previously described 31 up to 90% confluence in Dulbecco’s Modified Eagle Medium (DMEM, Life Technologies) supplemented with 10% (v/v) fetal bovine serum (FBS, Interpath, VIC, Australia), 24 mM KCl, 1X penicillin/streptomycin/glutamine (Life Technologies, VIC, Australia), and 200 μg/ml G418 (Life Technologies). Cells were routinely passaged by trypsinization, maintained at 33°C in 5% (v/v) CO2, and used up to passage 15.
For ICP-MS and Western Blotting analyses, cells were cultured to ~70% confluence in T75 flasks prior to collection by scraping and centrifugation. Pellets were extracted with Phosphosafe (Merck, Victoria, Australia) containing a protease inhibitor cocktail (Roche, NSW, Australia) and DNAse (Roche).
For XFM, cells were seeded at 30% confluence onto X-ray transparent silicon nitride membranes (membrane area 2 mm by 2 mm, membrane thickness 500 nm; Silson; pre-treated with 1mM HCl to remove trace metals) in 24 well plates 48 h prior to experiments. Cells were either left untreated or were exposed to 1μM ZnII(atsm)90, 91 for 2 h 14. Following treatment, cells were exposed to 500 nM ER-Tracker™ Green and 500 nM MitoTracker® Deep Red (both from Life Technologies) for 30 min in fresh media. Non-stained cells were included as controls to quantify any effect of the organelle marker staining on cellular metal content. Salts were removed by washing in 175 mM ammonium acetate for 40 sec, and excess liquid was removed using a tapered wick (Kimwipe). Cells were rapidly fixed by plunge freezing in liquid nitrogen chilled liquid ethane, lyophilized overnight and stored under desiccant prior to confocal immunofluorescence or XFM analysis.
Confocal immunofluorescence
Confocal microscopy was performed both prior to, and post, XFM. Silicon nitride windows were placed onto microscope slides for stability during optical microscopy analysis. Fluorescent images of 5-μm-thick confocal sections were acquired using the Zeiss Meta upright confocal microscope (Zeiss, NSW, Australia). A 20× objective (EC Plan Neofluar/0.5 M27) with 2× digital zoom was used to visualize cells, resulting in a pixel size of 400 nm, corresponding to pixel sizes obtained via XFM analysis (vide infra). Excitation of the ER-Tracker™ Green and MitoTracker® Deep Red dyes was achieved using the Argon (514nm) and HeNe (633nm) lasers, respectively. For optimal image acquisition, a pixel dwell time of 51.2 μsec was used and averaging was performed line by line (4×).
ICP-MS
ICP-MS was performed on cell pellets as previously described 11. Cellular metal content was normalized to cellular protein content as measured by BCA assay (Thermo Fisher Scientific, VIC, Australia).
Western Blotting
Western blotting was performed on cell lysates (5μg) as before 10 using rabbit Zip7 (1:1200, Proteintech, IL, USA) and anti-rabbit IgG antibodies (1:5000, Cell Signaling, MA, USA). Zip7 band intensities were normalized to control bands (ImageJ, MD, USA) on blots probed with antibodies against total ERK.
X-ray Fluorescence Microscopy (XFM)
The distribution of elements was mapped at the XFM beamline at the Australian Synchrotron 92. An incident beam of 12.7 keV x-rays was chosen to induce K-shell ionization of elements with atomic numbers below 30 (Z ≤ Zn), while providing adequate separation of the Rayleigh and Compton peaks from the elemental fluorescence of interest, as described 24. The incident beam was focused to a ~2μm spot (full-width at half maximum) using a Kirkpatrick–Baez mirror pair and specimens were fly-scanned through x-ray focus. The resulting XRF was collected in event-mode using the low-latency, 384-channel Maia XRF detector (positioned in the backscatter geometry) and the full XRF spectra used to reconstruct elemental maps of the specimen using a virtual pixel size of 0.4 μm, giving an effective dwell time ~0.025 seconds. This meant elemental maps of each specimen (containing between 9 and 24 cells) were collected in well under 1h. Single element foils of Mn and Pt (Micromatter, Canada), were scanned in the same geometry and used as references to establish elemental quantitation. Deconvolution of the Maia data was performed using the GeoPIXE v6.6j (CSIRO, Australia) that incorporates a linear transformation matrix to perform spectral deconvolution 93. Spectra were calibrated using the metal foil measurements, and corrections made for self-absorption in the sample, absorption in air, and the efficiency response of the detector 94. The detected x-ray photons from each pixel were related to calculated-model fluorescence x-ray yields for an assumed specimen composition and thickness. The composition and thickness of the silicon nitride windows were known from manufactures specifications and the composition and average density typical of dried organic material (C22H10N2O4 and 1.42 g cm−3 respectively) 95 was used to model the bulk cell. Absorption effects for XRF from the lowest atomic number element relevant to this study (Ca Kα) are negligible for this specimen type.
XANES Imaging
Fluorescence-XANES imaging was achieved by repeat scanning of a region of interest at a series of different incident energies crossing the Zn K-edge (9.669 keV). This process generated a “stack” of images, each recorded at a unique incident energy. For the current study each fluorescence-XANES stack comprised a total of 59 individual energies (corresponding to 59 maps), spanning 9.604 keV – 9.764 keV and captured the energy location of major features of the Zn Kedge, whilst minimizing the measurement time. The parameters of each map within a fluorescence-XANES stack (pixel size, effective dwell time, etc) were kept constant and for technical reasons the incident energy was scanned progressively decreasing from the maximum to minimum values. The individual maps comprising a fluorescence-XANES stack were recorded in a manner largely analogous to the elemental mapping described above, but with some important differences. Briefly, in order to ensure maximum stability of the incident flux across the relevant range of energies during fluorescence-XANES imaging, a small taper was introduced to the undulators. Tapering the undulator gap (0.121 mm) broadens the spectral distribution at the expense of source brilliance and peak intensity. This setup helps minimize the introduction of artifacts into the spectra, although the decreased incident flux also lowers sensitivity. Thus in order to achieve counting statistics comparable to elemental maps acquired with an untapered undulator gap, samples were scanned through x-ray focus at a reduced velocity. A fluorescence-XANES stack of elemental maps could then be used extract Zn XANES spectra from individual cells. In total, 3 healthy, 3 diseased cells and 1 diseased cell treated with ZnII(atsm) were imaged in this manner and the resulting spectra analyzed using Demeter 0.9.18.2 96.
Statistical Analysis
Differences in global metal content and cellular Zip7 expression were assessed by Student’s t test in GraphPad Prism v5.0c (GraphPad, CA, USA). Changes to subcellular metal content were analysed using two-way ANOVA. For elements where significant main effects were observed, Bonferonni post-hoc tests were used to determine which groups were significantly different. Analysis of subcellular regulation of biometal homeostasis was performed using linear regression analysis. Effects of ZnII(atsm) treatment on subcellular metal homeostasis were determined using one-way ANOVA. Bin widths for analysis of histograms were calculated to be within the range given by the square-root choice (k√vn) and the Rice rule (k=2n1/3) for the following numbers of pixels for each dataset: control, 60595; CbCln6nclf, 19368; CbCln6nclf + ZnII(atsm), 11254. Non-linear curve fitting of log-transformed calcium histograms was performed using a Gaussian least squares fit. Comparison of models between data sets was performed using the exact sum of squares F-test. Deviation of data from the model was assessed using the runs and D’Agostino-Pearson normality tests. p values below 0.05 were considered significant.
Conclusions
Impaired biometal homeostasis is a key hallmark of neurodegenerative disorders, but limitations of current techniques prevent information-dense probing of subcellular biometal distributions in multiple cells. Advances in XFM detector efficiency have allowed unprecedented speed of data acquisition, which combined with low radiation dose enables rapid, high-resolution investigation of metal handling dynamics in populations of cells concomitantly with fluorescent labelling of transient subcellular organelles. Using this approach, we detected for the first time impairment of subcellular metal trafficking in a fatal childhood neurodegenerative disorder, despite a lack of global changes to biometal levels. We also demonstrated that these biometal imbalances can be partially corrected by a metallo-complex, ZnII(atsm). The techniques and methodology presented in this paper can be applied to offer molecular and elemental insight into the metal cell biology in other neurodegenerative diseases in single cells. Importantly, these approaches will prove invaluable for mechanism of action studies for novel therapeutics to target neurodegeneration.
Supplementary Material
Acknowledgments
Parts of this work were performed at the XFM beamline at the Australian Synchrotron, Victoria, Australia. This work was supported by the National Health and Medical Research Council of Australia (NHMRC) [628946] and the National Institutes of Health: National Institute of Neurological Disorders & Stroke [R01NS073813]. ARW is a recipient of an ARC Future Fellowship [FT100100674]. The funding sources had no influence in study design; collection, analysis, and interpretation of data; writing the report; or the decision to submit the report for publication. Patent protection has previously been sought by the University of Melbourne for the use of bis(thiosemicarbazones) for treatment of diseases. ARW and PSD are co-inventors on this patent application PCT/AU2007/001792, which is the subject of a commercialization contract between the University and a private company. The company has not funded nor contributed to research described in this manuscript.
Footnotes
Electronic Supplementary Information (ESI) available: Supplementary Figures 1–5. See DOI: 10.1039/b000000x/
Notes and references
- 1.Adlard PA, Parncutt JM, Finkelstein DI, Bush AI. J Neurosci. 2010;30:1631–1636. doi: 10.1523/JNEUROSCI.5255-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Radisky DC, Babcock MC, Kaplan J. J Biol Chem. 1999;274:4497–4499. doi: 10.1074/jbc.274.8.4497. [DOI] [PubMed] [Google Scholar]
- 3.Chelly J, Monaco AP. Nat Genet. 1993;5:317–318. doi: 10.1038/ng1293-317. [DOI] [PubMed] [Google Scholar]
- 4.Chelly J, Tumer Z, Tonnesen T, Petterson A, Ishikawa-Brush Y, Tommerup N, Horn N, Monaco AP. Nat Genet. 1993;3:14–19. doi: 10.1038/ng0193-14. [DOI] [PubMed] [Google Scholar]
- 5.Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, Cabelli DE, Valentine JS, Brown RH., Jr J Biol Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- 6.Macomber L, Rensing C, Imlay JA. J Bacteriol. 2007;189:1616–1626. doi: 10.1128/JB.01357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnham KJ, Haeffner F, Ciccotosto GD, Curtain CC, Tew D, Mavros C, Beyreuther K, Carrington D, Masters CL, Cherny RA, Cappai R, Bush AI. FASEB J. 2004;18:1427–1429. doi: 10.1096/fj.04-1890fje. [DOI] [PubMed] [Google Scholar]
- 8.Rembach A, Hare DJ, Lind M, Fowler CJ, Cherny RA, McLean C, Bush AI, Masters CL, Roberts BR. Int J Alzheimers Dis. 2013 doi: 10.1155/2013/623241. Epub 2013 Oct 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrag M, Mueller C, Oyoyo U, Smith MA, Kirsch WM. Prog Neurobiol. 2011;94:296–306. doi: 10.1016/j.pneurobio.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanninen KM, Grubman A, Caragounis A, Duncan C, Parker SJ, Lidgerwood GE, Volitakis I, Ganio G, Crouch PJ, White AR. Biol Open. 2013;2:635–646. doi: 10.1242/bio.20134804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanninen KM, Grubman A, Meyerowitz J, Duncan C, Tan JL, Parker SJ, Crouch PJ, Paterson BM, Hickey JL, Donnelly PS, Volitakis I, Tammen I, Palmer DN, White AR. PLoS One. 2013;8:e58644. doi: 10.1371/journal.pone.0058644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mole WR, Goebel SH. The neuronal ceroid lipofuscinoses (batten disease) 2. Oxford University Press; 2011. [Google Scholar]
- 13.Palmer DN, Barry LA, Tyynela J, Cooper JD. Biochim Biophys Acta. 2013;1832:1882–1893. doi: 10.1016/j.bbadis.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 14.Grubman A, Lidgerwood GE, Duncan C, Bica L, Tan JL, Parker SJ, Caragounis A, Meyerowitz J, Volitakis I, Moujalled D, Liddell JR, Hickey JL, Horne M, Longmuir S, Koistinaho J, Donnelly PS, Crouch PJ, Tammen I, White AR, Kanninen KM. Acta Neuropathol Commun. 2014;2:25. doi: 10.1186/2051-5960-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber LA, Pfaller K, Vietor I. Circ Res. 2003;92:962–968. doi: 10.1161/01.RES.0000071748.48338.25. [DOI] [PubMed] [Google Scholar]
- 16.Hare DJ, George JL, Grimm R, Wilkins S, Adlard PA, Cherny RA, Bush AI, Finkelstein DI, Doble P. Metallomics. 2010;2:745–753. doi: 10.1039/c0mt00039f. [DOI] [PubMed] [Google Scholar]
- 17.Hare DJ, Grubman A, Ryan TM, Lothian A, Liddell JR, Grimm R, Matsuda T, Doble PA, Cherny RA, Bush AI, White AR, Masters CL, Roberts BR. Metallomics. 2013;5:1656–1662. doi: 10.1039/c3mt00227f. [DOI] [PubMed] [Google Scholar]
- 18.Penner-Hahn JE. Technologies for Detecting Metals in Single Cells. 2013;12:15–40. doi: 10.1007/978-94-007-5561-1_2. [DOI] [PubMed] [Google Scholar]
- 19.New EJ. Dalton Trans. 2013;42:3210–3219. doi: 10.1039/c2dt31933k. [DOI] [PubMed] [Google Scholar]
- 20.Price KA, Hickey JL, Xiao ZG, Wedd AG, James SA, Liddell JR, Crouch PJ, White AR, Donnelly PS. Chemical Science. 2012;3:2748–2759. [Google Scholar]
- 21.Vogt S, Lanzirotti A. Synchrotron Radiation News. 2013;26:32–38. [Google Scholar]
- 22.Zhang X, Balhorn R, Mazrimas J, Kirz J. J Struct Biol. 1996;116:335–344. doi: 10.1006/jsbi.1996.0051. [DOI] [PubMed] [Google Scholar]
- 23.Bacaner M, Broadhurst J, Hutchinson T, Lilley J. Proc Natl Acad Sci U S A. 1973;70:3423–3427. doi: 10.1073/pnas.70.12.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.James SA, Feltis BN, de Jonge MD, Sridhar M, Kimpton JA, Altissimo M, Mayo S, Zheng C, Hastings A, Howard DL, Paterson DJ, Wright PF, Moorhead GF, Turney TW, Fu J. ACS Nano. 2013;7:10621–10635. doi: 10.1021/nn403118u. [DOI] [PubMed] [Google Scholar]
- 25.Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L, Antonellis KA, Gillis T, Qin X, Liu S, Donahue LR, Bronson RT, Faust JR, Stout D, Haines JL, Lerner TJ, MacDonald ME. Am J Hum Genet. 2002;70:324–335. doi: 10.1086/338190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wheeler RB, Sharp JD, Schultz RA, Joslin JM, Williams RE, Mole SE. Am J Hum Genet. 2002;70:537–542. doi: 10.1086/338708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grubman A, Pollari E, Duncan C, Caragounis A, Blom T, Volitakis I, Wong A, Cooper J, Crouch PJ, Koistinaho J, Jalanko A, White AR, Kanninen KM. Metallomics. 2014 doi: 10.1039/c4mt00032c. [DOI] [PubMed] [Google Scholar]
- 28.Al-Maawali A, Blaser S, Yoon G. J Child Neurol. 2012;27:1121–1132. doi: 10.1177/0883073812448680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thelen M, Damme M, Schweizer M, Hagel C, Wong AM, Cooper JD, Braulke T, Galliciotti G. PLoS One. 2012;7:e35493. doi: 10.1371/journal.pone.0035493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thelen M, Fehr S, Schweizer M, Braulke T, Galliciotti G. J Neurosci Res. 2012;90:568–574. doi: 10.1002/jnr.22773. [DOI] [PubMed] [Google Scholar]
- 31.Cao Y, Staropoli JF, Biswas S, Espinola JA, MacDonald ME, Lee JM, Cotman SL. PLoS One. 2011;6:e17118. doi: 10.1371/journal.pone.0017118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James SA, Volitakis I, Adlard PA, Duce JA, Masters CL, Cherny RA, Bush AI. Free Radic Biol Med. 2012;52:298–302. doi: 10.1016/j.freeradbiomed.2011.10.446. [DOI] [PubMed] [Google Scholar]
- 33.Hackett MJ, McQuillan JA, El-Assaad F, Aitken JB, Levina A, Cohen DD, Siegele R, Carter EA, Grau GE, Hunt NH, Lay PA. Analyst. 2011;136:2941–2952. doi: 10.1039/c0an00269k. [DOI] [PubMed] [Google Scholar]
- 34.Matsuyama S, Shimura M, Fujii M, Maeshima K, Yumoto H, Mimura H, Sano Y, Yabashi M, Nishino Y, Tamasaku K, Ishizaka Y, Ishikawa T, Yamauchi K. X-Ray Spectrometry. 2010;39:260–266. [Google Scholar]
- 35.James SA, de Jonge MD, Howard DL, Bush AI, Paterson D, McColl G. Metallomics. 2013;5:627–635. doi: 10.1039/c3mt00010a. [DOI] [PubMed] [Google Scholar]
- 36.McColl G, James SA, Mayo S, Howard DL, Ryan CG, Kirkham R, Moorhead GF, Paterson D, de Jonge MD, Bush AI. PLoS One. 2012;7:e32685. doi: 10.1371/journal.pone.0032685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Witting PK, Harris HH, Rayner BS, Aitken JB, Dillon CT, Stocker R, Lai B, Cai Z, Lay PA. Biochemistry. 2006;45:12500–12509. doi: 10.1021/bi0604375. [DOI] [PubMed] [Google Scholar]
- 38.Maret W. Adv Nutr. 2013;4:82–91. doi: 10.3945/an.112.003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolikova J, Afzalov R, Surin A, Lehesjoki AE, Khiroug L. Cell Calcium. 2011;50:491–501. doi: 10.1016/j.ceca.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 40.Chang JW, Choi H, Kim HJ, Jo DG, Jeon YJ, Noh JY, Park WJ, Jung YK. Hum Mol Genet. 2007;16:317–326. doi: 10.1093/hmg/ddl466. [DOI] [PubMed] [Google Scholar]
- 41.Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer; US: 2006. [Google Scholar]
- 42.Sekler I, Sensi SL, Hershfinkel M, Silverman WF. Mol Med. 2007;13:337–343. doi: 10.2119/2007-00037.Sekler. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donnelly PS, Caragounis A, Du T, Laughton KM, Volitakis I, Cherny RA, Sharples RA, Hill AF, Li QX, Masters CL, Barnham KJ, White AR. J Biol Chem. 2008;283:4568–4577. doi: 10.1074/jbc.M705957200. [DOI] [PubMed] [Google Scholar]
- 44.Bouron A, Oberwinkler J. Pflugers Arch. 2013 [Google Scholar]
- 45.Baudier J, Glasser N, Gerard D. J Biol Chem. 1986;261:8192–8203. [PubMed] [Google Scholar]
- 46.Baudier J, Haglid K, Haiech J, Gerard D. Biochem Biophys Res Commun. 1983;114:1138–1146. doi: 10.1016/0006-291x(83)90681-2. [DOI] [PubMed] [Google Scholar]
- 47.Schafer BW, Fritschy JM, Murmann P, Troxler H, Durussel I, Heizmann CW, Cox JA. J Biol Chem. 2000;275:30623–30630. doi: 10.1074/jbc.M002260200. [DOI] [PubMed] [Google Scholar]
- 48.Furusawa C, Suzuki T, Kashiwagi A, Yomo T, Kaneko K. Biophysics. 2005;1:25–31. doi: 10.2142/biophysics.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryan CG, Kirkham R, Hough RM, Moorhead G, Siddons DP, de Jonge MD, Paterson DJ, De Geronimo G, Howard DL, Cleverley JS. Nuclear Instruments & Methods in Physics Research Section A. 2010;619:37–43. [Google Scholar]
- 50.Aitken JB, Lay PA, Duong TT, Aran R, Witting PK, Harris HH, Lai B, Vogt S, Giles GI. J Biol Inorg Chem. 2012;17:589–598. doi: 10.1007/s00775-012-0879-y. [DOI] [PubMed] [Google Scholar]
- 51.Kosior E, Bohic S, Suhonen H, Ortega R, Deves G, Carmona A, Marchi F, Guillet JF, Cloetens P. J Struct Biol. 2012;177:239–247. doi: 10.1016/j.jsb.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 52.Carmona A, Deves G, Roudeau S, Cloetens P, Bohic S, Ortega R. ACS Chem Neurosci. 2010;1:194–203. doi: 10.1021/cn900021z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bervoets AR, Behets GJ, Schryvers D, Roels F, Yang Z, Verberckmoes SC, Damment SJ, Dauwe S, Mubiana VK, Blust R, De Broe ME, D’Haese PC. Kidney Int. 2009;75:389–398. doi: 10.1038/ki.2008.571. [DOI] [PubMed] [Google Scholar]
- 54.Chan DC. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 55.Sitia R, Meldolesi J. Mol Biol Cell. 1992;3:1067–1072. doi: 10.1091/mbc.3.10.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ortega R, Carmona A. Journal of Environmental and Analytical Toxicology. 2012:S4. [Google Scholar]
- 57.Zheng Z, White C, Lee J, Peterson TS, Bush AI, Sun GY, Weisman GA, Petris MJ. J Neurochem. 2010;114:1630–1638. doi: 10.1111/j.1471-4159.2010.06888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schrag M, Crofton A, Zabel M, Jiffry A, Kirsch D, Dickson A, Mao XW, Vinters HV, Domaille DW, Chang CJ, Kirsch W. J Alzheimers Dis. 2011;24:137–149. doi: 10.3233/JAD-2010-101503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bush AI. Alzheimer’s Disease: Advances for a New Century. 2013;3:277–281. [Google Scholar]
- 60.Lichten LA, Cousins RJ. Annu Rev Nutr. 2009;29:153–176. doi: 10.1146/annurev-nutr-033009-083312. [DOI] [PubMed] [Google Scholar]
- 61.Andreini C, Banci L, Bertini I, Rosato A. J Proteome Res. 2006;5:196–201. doi: 10.1021/pr050361j. [DOI] [PubMed] [Google Scholar]
- 62.Beyer N, Coulson DT, Heggarty S, Ravid R, Hellemans J, Irvine GB, Johnston JA. J Alzheimers Dis. 2012;29:863–873. doi: 10.3233/JAD-2012-112105. [DOI] [PubMed] [Google Scholar]
- 63.Lyubartseva G, Smith JL, Markesbery WR, Lovell MA. Brain Pathol. 2010;20:343–350. doi: 10.1111/j.1750-3639.2009.00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koeppen AH, Kuntzsch EC, Bjork ST, Ramirez RL, Mazurkiewicz JE, Feustel PJ. Acta Neuropathol Commun. 2013;1:26. doi: 10.1186/2051-5960-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yan G, Zhang Y, Yu J, Yu Y, Zhang F, Zhang Z, Wu A, Yan X, Zhou Y, Wang F. PLoS One. 2012;7:e42939. doi: 10.1371/journal.pone.0042939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dempski RE. Curr Top Membr. 2012;69:221–245. doi: 10.1016/B978-0-12-394390-3.00009-4. [DOI] [PubMed] [Google Scholar]
- 67.Nam H, Knutson MD. Biometals. 2012;25:115–124. doi: 10.1007/s10534-011-9487-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Colvin RA, Bush AI, Volitakis I, Fontaine CP, Thomas D, Kikuchi K, Holmes WR. Am J Physiol Cell Physiol. 2008;294:C726–742. doi: 10.1152/ajpcell.00541.2007. [DOI] [PubMed] [Google Scholar]
- 69.Qin Y, Dittmer PJ, Park JG, Jansen KB, Palmer AE. Proc Natl Acad Sci U S A. 2011;108:7351–7356. doi: 10.1073/pnas.1015686108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hogstrand C, Kille P, Nicholson RI, Taylor KM. Trends Mol Med. 2009;15:101–111. doi: 10.1016/j.molmed.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 71.Taylor KM, Hiscox S, Nicholson RI, Hogstrand C, Kille P. Sci Signal. 2012;5:ra11. doi: 10.1126/scisignal.2002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palmiter RD, Findley SD. EMBO J. 1995;14:639–649. doi: 10.1002/j.1460-2075.1995.tb07042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saydam N, Georgiev O, Nakano MY, Greber UF, Schaffner W. J Biol Chem. 2001;276:25487–25495. doi: 10.1074/jbc.M009154200. [DOI] [PubMed] [Google Scholar]
- 74.Nagano T, Itoh N, Ebisutani C, Takatani T, Miyoshi T, Nakanishi T, Tanaka K. J Cell Physiol. 2000;185:440–446. doi: 10.1002/1097-4652(200012)185:3<440::AID-JCP15>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 75.Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W, Michiels C, Munck S, Baert V, Sugita S, Wuytack F, Hiesinger PR, Grinstein S, Annaert W. J Cell Biol. 2012;198:23–35. doi: 10.1083/jcb.201201076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kiselyov K, Colletti GA, Terwilliger A, Ketchum K, Lyons CW, Quinn J, Muallem S. Cell Calcium. 2011;50:288–294. doi: 10.1016/j.ceca.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, Xu H. Nat Commun. 2012;3:731. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McGeoch JE, McGeoch MW, Mao R, Guidotti G. Biochem Biophys Res Commun. 2000;274:835–840. doi: 10.1006/bbrc.2000.3231. [DOI] [PubMed] [Google Scholar]
- 79.Jolly RD, Walkley SU. Mol Genet Metab. 1999;66:376–380. doi: 10.1006/mgme.1999.2821. [DOI] [PubMed] [Google Scholar]
- 80.Ishihara R, Ide-Ektessabi A, Ikeda K, Mizuno Y, Fujisawa S, Takeuchi T, Ohta T. Neuroreport. 2002;13:1817–1820. doi: 10.1097/00001756-200210070-00026. [DOI] [PubMed] [Google Scholar]
- 81.Bouron A, Oberwinkler J. Pflugers Arch. 2014;466:381–387. doi: 10.1007/s00424-013-1295-z. [DOI] [PubMed] [Google Scholar]
- 82.Eichelsdoerfer JL, Evans JA, Slaugenhaupt SA, Cuajungco MP. J Biol Chem. 2010;285:34304–34308. doi: 10.1074/jbc.C110.165480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bauer MC, Nilsson H, Thulin E, Frohm B, Malm J, Linse S. Protein Sci. 2008;17:760–767. doi: 10.1110/ps.073381108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krebs N, Langkammer C, Goessler W, Ropele S, Fazekas F, Yen K, Scheurer E. J Trace Elem Med Biol. 2014;28:1–7. doi: 10.1016/j.jtemb.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 85.Mirza M, Volz C, Karlstetter M, Langiu M, Somogyi A, Ruonala MO, Tamm ER, Jagle H, Langmann T. PLoS One. 2013;8:e75963. doi: 10.1371/journal.pone.0075963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Baum L, Ng A. J Alzheimers Dis. 2004;6:367–377. doi: 10.3233/jad-2004-6403. discussion 443–369. [DOI] [PubMed] [Google Scholar]
- 87.Dittmer PJ, Miranda JG, Gorski JA, Palmer AE. J Biol Chem. 2009;284:16289–16297. doi: 10.1074/jbc.M900501200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang D, Hurst TK, Thompson RB, Fierke CA. J Biomed Opt. 2011;16:087011. doi: 10.1117/1.3613926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fierke CA, Thompson RB. Biometals. 2001;14:205–222. doi: 10.1023/a:1012980628412. [DOI] [PubMed] [Google Scholar]
- 90.Gingras BA, Suprunchuk T, Bayley CH. Canadian Journal of Chemistry. 1962;40:1053–1059. [Google Scholar]
- 91.Cowley AR, Dilworth JR, Donnelly PS, Labisbal E, Sousa A. Journal of American Chemical Society. 2002;124:5270–5271. doi: 10.1021/ja012668z. [DOI] [PubMed] [Google Scholar]
- 92.Paterson D, de Jonge MD, Howard DL, Lewis W, McKinlay J, Starritt A, Kusel M, Ryan CG, Kirkham R, Moorhead G. AIP Conf Proc. 2011;1365:219–222. [Google Scholar]
- 93.Ryan CG. International Journal of Imaging Systems and Technology. 2000;11:219–230. [Google Scholar]
- 94.Ryan CG. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2001;181:170–179. doi: 10.1016/s0168-583x(01)00693-0. [DOI] [PubMed] [Google Scholar]
- 95.McMaster WH, Kerr del Grande N, Mallett JH, Hubbell JH, editors. U S D o Commerce. 1969. [Google Scholar]
- 96.Ravel B, Newville M. J Synchrotron Radiat. 2005;12:537–541. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.