

Abstract
Parenteral nutrition–associated liver disease (PNALD) is a serious complication of PN in infants who do not tolerate enteral feedings, especially those with acquired or congenital intestinal diseases. Yet, the mechanisms underlying PNALD are poorly understood. It has been suggested that a component of soy oil (SO) lipid emulsions in PN solutions, such as plant sterols (phytosterols), may be responsible for PNALD, and that use of fish oil (FO)–based lipid emulsions may be protective. We used a mouse model of PNALD combining PN infusion with intestinal injury to demonstrate that SO-based PN solution causes liver damage and hepatic macrophage activation and that PN solutions that are FO-based or devoid of all lipids prevent these processes. We have furthermore demonstrated that a factor in the SO lipid emulsions, stigmasterol, promotes cholestasis, liver injury, and liver macrophage activation in this model and that this effect may be mediated through suppression of canalicular bile transporter expression (Abcb11/BSEP, Abcc2/MRP2) via antagonism of the nuclear receptors Fxr and Lxr, and failure of up-regulation of the hepatic sterol exporters (Abcg5/g8/ABCG5/8). This study provides experimental evidence that plant sterols in lipid emulsions are a major factor responsible for PNALD and that the absence or reduction of plant sterols is one of the mechanisms for hepatic protection in infants receiving FO-based PN or lipid minimization PN treatment. Modification of lipid constituents in PN solutions is thus a promising strategy to reduce incidence and severity of PNALD.
INTRODUCTION
Administration of parenteral nutrition (PN) is critical for survival and growth in infants who cannot tolerate enteral feedings. However, in many of these infants, PN is associated with rapid development of cholestatic liver injury, referred to as PN-associated liver disease (PNALD; also known as intestinal failure–associated liver disease), which can progress to cirrhotic liver failure. Chronicity and severity of PNALD are highest in preterm infants and in infants with intestinal failure secondary to short bowel syndrome, congenital absorption defects, necrotizing enterocolitis, and intestinal malformations (1–3). PNALD is the leading indication for multivisceral (liver/intestine) transplantation in infants (4) because current therapies are not effective (5). The pathogenesis and etiology of PNALD remain poorly understood. Recent studies have focused on the possible contributing roles of lipid emulsions, which are obligate components of the PN solutions (6–13), and the absorption of bacterial products such as lipopolysaccharides (LPSs) from the compromised, inflamed intestinal mucosa (14, 15).
The intravenous lipid emulsion most commonly used in PN in the United States (Intralipid, Fresenius Kabi) is derived from soy oil (SO). Recent reports have shown that infusion with lipid emulsions derived from fish oil (FO) rather than SO improves established PNALD and that reduction of the SO lipid dose in PN solutions attenuates PNALD (6–12). These data suggest that a lipid component in the SO emulsion may be responsible for promoting PNALD. One of the components of SO emulsions implicated in PNALD is the presence of plant sterols (phytosterols) (14, 16–19). On the basis of in vitro studies, the phytosterol stigmasterol, which is one of three major phytosterols present in SO lipid emulsions, has been suggested as promoting cholestasis through inhibition of the nuclear receptor FXR, which, in turn, would result in reduced hepatocyte expression of a wide variety of FXR-dependent genes, including the principal determinant of bile secretion, the bile salt export pump (BSEP) (Abcb11) (14). In this in vitro model focusing on phytosterol-mediated alterations in nuclear receptor–mediated gene expression, stigmasterol antagonized FXR signaling, but the most prevalent phytosterol in soy lipid, β-sitosterol, did not (14). Proinflammatory cytokine-induced cell signaling in hepatocytes, primarily derived from LPS-activated Kupffer cells residing in hepatic sinusoids, also suppresses nuclear receptor–mediated gene expression in liver, including FXR-dependent pathways (referred to as cytokine-mediated cholestasis) (20–22). Thus, the combination of cytokine signaling and plant sterols could potentially exert synergistic potent inhibitory effects on hepatocyte expression of canalicular transport systems and thus promote intracellular bile salt retention or cholestasis.
To explore the contributions from inflammation and phytosterols in the etiology of PNALD, we have recently developed a PNALD mouse model in which intestinal injury in conjunction with PN infusion for 7 days promoted hepatic macrophage (Kupffer cell) activation and cytokine production, leading to PNALD, whereas neither intestinal injury nor PN alone was sufficient to induce liver injury (15). These findings suggested that both a component within the PN solution and the activation of Kupffer cells by intestinally derived Toll-like receptor 4 (TLR4) agonists were mechanistically involved in this liver injury (15).
Here, we have used this mouse model to test the hypothesis that select plant sterols in SO-derived PN solutions were critical factors leading to PNALD. We experimentally addressed four questions: (i) Does infusion with FO-PN prevent PNALD in mice? (ii) Can PNALD in mice be prevented by infusion of PN devoid of SO lipids? (iii) Does stigmasterol in PN solution promote liver injury? (iv) Does infusion of phytosterol-containing PN solutions reduce expression of hepatic bile acid transporter genes? Our results demonstrate that infusion of PN solutions devoid of plant sterols (FO-PN or no lipid PN) prevented cholestasis and liver injury, thus replicating the experience in human infants. Moreover, infusion with FO-based emulsion to which one phytosterol, stigmasterol, was added produced cholestasis and liver injury, despite the absence of other SO-derived lipids and the presence of the FO lipids. In addition, gene expression of the canalicular bile acid transporter Abcb11 and conjugated bilirubin transporter Abcc2 was reduced in mice infused with plant sterol–containing emulsions but not in mice receiving plant sterol–free PN, and serum stigmasterol concentrations correlated with the severity of cholestasis. Finally, pro-inflammatory activation of liver macrophages was limited to those mice administered plant sterol–containing emulsions.
These results provide direct experimental evidence that phytosterols play a role in the pathogenesis of PNALD and that the absence of phytosterols in FO lipid emulsions and in lipid-free emulsions is the likely mechanism of protection against PNALD. Our study thus provides a rationale for improving the design of lipid emulsions for PN solutions to prevent or treat PNALD while maintaining essential fatty acid homeostasis.
RESULTS
FO-based emulsion prevents PNALD in mice
We first determined if infusion with FO-based PN solution compared to SO-based PN solution would prevent or attenuate PNALD in mice, as reported in human infants (6, 7, 10). In these experiments, we used the recently described PNALD mouse model (15). Eight-week-old male C57BL/6 mice with intestinal injury produced by low-dose oral dextran sulfate sodium (DSS) pretreatment were randomized into four groups. Group 1 was continued on a regular chow diet for 7 days while receiving infusion with normal saline through a central venous catheter (CVC) (DSS/NS; n = 11); group 2 received a chow diet but did not undergo CVC placement (DSS; n = 10); group 3 was infused with SO-PN for 7 days (DSS/SO-PN; n = 19); and group 4 was infused with FO-PN for 7 days (DSS/FO-PN; n = 11). PN-infused mice had no access to chow but were given free access to water. A final control group underwent no treatment and had free access to water and chow (Chow; n = 10). Both PN solutions were identical with regard to concentration of total lipids, amino acids, and dextrose; both groups of mice received an equal dose of lipids per day relative to body weight (1.4 g/kg per day), and both PN solutions were isocaloric. Caloric intake of PN-infused mice was adjusted to 8.4 kcal/day to match the caloric intake of chow-fed mice (23). A summary of the treatment groups of mice and experimental design is depicted in fig. S1. The composition of the PN solutions and lipid emulsions is summarized in Tables 1 and 2.
Table 1.
PN solution components used in experiments (per 100 ml).
| Aminosyn 15% (Abbott) | 26.7 ml; 4 g |
| Dextrose* | 36 ml; 25.5 g |
| Lipid† | 10 ml; 2 g |
| Sodium phosphate | 1.34 mmol |
| Potassium chloride | 1.6 mEq |
| Sodium chloride | 3.2 mEq |
| Potassium acetate | 12 mEq |
| Magnesium sulfate | 0.8 mEq |
| Calcium gluconate | 1.32 mEq |
| Multitrace-5 concentrate (MTE-5)‡ | 0.1 ml |
| Heparin | 500 U |
| Nitrogen content | 0.61 g |
| Nonprotein calories | 105.68 kcal |
| Protein calories | 16.02 kcal |
| Dextrose calories | 85.68 kcal |
| Lipid calories | 20.00 kcal |
| Total calories | 121.7 kcal |
| Multivitamins | 2 ml |
Dextrose was increased to 31.45 g (to provide 105.68 kcal) in the PN solution to which no lipids were added (NoL-PN) to make the solution isocaloric to PN with lipids.
Lipid source was Intralipid (20% emulsion) in SO-based PN (SO-PN) or Omegaven (10% emulsion) in FO-based PN (FO-PN). The composition of the lipid emulsions is detailed in Table 2.
MTE-5 contains zinc, copper, manganese, chromium, and selenium.
Table 2.
Lipid composition of SO-based lipid emulsion (Intralipid) and FO-based lipid emulsion (Omegaven).
| Intralipid | Omegaven | |
|---|---|---|
| Soy bean oil | 20 g/100 ml | 0 |
| Fish oil | 0 | 10 g/100 ml |
| Egg phospholipids | 1.2 g/100 ml | 1.2 g/100 ml |
| Glycerin | 2.25 g/100 ml | 2.5g/100 ml |
| Stigmasterol (45) | 6.0 mg/100 ml | 0 |
| Fatty acids in g/10 g of lipid | ||
| Palmitic | 1 | 0.25–1 |
| Stearic | 0.4 | 0.05–0.2 |
| Oleic | 2.6 | 0.6–1.3 |
| Linoleic | 5 | 0.1–0.7 |
| α-Linoleic | 0.9 | 0.2 |
| Eicosapentaenoic acid* | 0 | 1.28–2.82 |
| Docosahexaenoic acid* | 0 | 1.44–3.09 |
ω-3 fatty acids.
As reported previously (15), neither DSS nor DSS/NS treatment resulted in significant (P > 0.6) increases in serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, or bile acids, nor in liver histologic changes, demonstrating that intestinal injury alone was not associated with liver injury or cholestasis (Fig. 1, A to D, and table S1). Infusion of SO-PN in DSS-pretreated mice resulted in markedly increased serum concentrations of AST, ALT, total bilirubin, and total serum bile acids (TSBAs) compared to all control groups. In contrast, infusion of FO-PN in DSS-pretreated mice was associated with markedly lower serum AST, ALT, bilirubin, and bile acids that were no different from control mice [P < 0.0004, FO-PN versus SO-PN, one-way analysis of variance (ANOVA)] (Fig. 1, A to D, and table S1). These data demonstrated that FO-based PN solutions prevented both hepatocyte injury and cholestasis in mice pretreated with DSS.
Fig. 1. PN solutions devoid of plant sterols prevent PNALD in mice.

(A to D) Hepatocyte injury is indicated by elevated serum AST (A) and ALT (B) concentrations, and cholestasis is indicated by increased total bilirubin (C) and TSBAs (D) in response to infusion with SO-based PN (n = 19) in DSS-pretreated mice. DSS-pretreated mice were infused for 7 days with PN solutions containing either SO-PN or FO-PN or completely devoid of all lipids (NoL-PN). Hepatocyte injury and cholestasis were attenuated after infusion with FO-PN (n = 11) or no lipid PN (NoL-PN; n = 9), both devoid of phytosterols. Control groups consisted of DSS-pretreated mice that were infused with NS for 7 days (n = 11) or not infused at all (n = 10), in addition to completely untreated mice (no DSS, no infusion; n = 10). Means and SEM are shown. Adjusted P values were calculated with one-way ANOVA and Tukey correction for multiple comparisons. (A and D) *P < 0.0001, DSS/SO-PN versus all other groups. (B) *P < 0.0002, DSS/SO-PN versus control groups and DSS/FO-PN; P = 0.0136 versus DSS/NoL-PN. (C) *P < 0.0001, DSS/SO-PN versus control groups; P = 0.0003 versus DSS/FO-PN and DSS/NoL-PN. i.v., intravenous.
Removal of lipids from PN solution attenuates PNALD in mice
To further determine the role of lipids in promoting PNALD in mice, we infused DSS-pretreated mice with a PN solution completely devoid of all lipids (DSS/NoL-PN; n = 9) that was made isocaloric by increasing dextrose content (Table 1). NoL-PN–infused mice underwent DSS pretreatment and 7 days of PN infusion treatment identical to SO-PN– and FO-PN–infused mice (fig. S1). Compared to SO-PN mice, infusion with NoL-PN resulted in marked attenuation of hepatocyte injury (reduced serum AST and ALT) and cholestasis (reduced serum total bilirubin and total bile acids) to values that were similar to those in FO-PN mice and controls (Fig. 1, A to D, and table S1).
These data suggested that the protective effect of FO-PN infusion was likely due to the absence of component(s) present in SO-PN, and not the presence of ω-3 fatty acids, the predominant lipid species in FO-PN (Table 2). The main phytosterols present in SO-PN (Intralipid 20%) are stigmasterol, β-sitosterol, and campesterol. Among these three phytosterols, stigmasterol has the greatest potential to promote cholestasis through antagonism of FXR function and specifically reduce expression of Abcb11 in primary mouse hepatocytes (14), and was therefore further investigated.
Stigmasterol promotes PNALD in mice
We next determined if stigmasterol promoted cholestasis and liver injury in mice by adding stigmasterol in two different concentrations to FO-PN solutions. One group of DSS-pretreated mice was infused for 7 days with FO-PN in which 1 mg of stigmasterol was added to 100 ml of PN solution (DSS/FO-PN-Stig1; n = 12). The final concentration of stigmasterol in this PN solution, measured by mass spectrometry, was 1.085 mg/100 ml, which was in the range of the stigmasterol concentration measured in the SO-PN solution (0.671 mg/100 ml). A second group of DSS-pretreated mice was infused for 7 days with FO-PN containing 3 mg/100 ml of stigmasterol (DSS/FO-PN-Stig3; n = 9) to replicate the stigmasterol concentration present in SO-PN infused to human infants, who receive lipid (up to 3.5 g/kg per day) in typical PN (24). The final concentration of stigmasterol in this PN solution, measured by mass spectrometry, was 2.79 mg/100 ml.
Infusion with FO-PN-Stig3 markedly increased AST, ALT, total bilirubin, and total bile acids, compared to infusion with FO-PN, to concentrations similar to those observed in DSS/SO-PN mice (Fig. 2, A to D, and table S1). Infusion with FO-PN-Stig1 increased these values to concentrations intermediate between FO-PN-Stig3 and FO-PN.
Fig. 2. Phytosterol-containing PN solutions promote PNALD in mice.

(A to D) Hepatocyte injury is indicated by elevated serum AST (A) and ALT (B) concentrations, and cholestasis is indicated by increased total bilirubin (C) and TSBAs (D) in response to infusion with FO-PN solution to which the plant sterol stigmasterol was added at 1 mg/100 ml (FO-PN-Stig1; n = 12) and 3 mg/100 ml (FO-PN-Stig3; n = 9), in comparison to infusion with FO-PN devoid of plant sterols. Control, SO-PN, and FO-PN groups are reproduced from Fig. 1. *P < 0.05. Adjusted P values were calculated by one-way ANOVA with Tukey correction for multiple comparisons, and exact P values for each comparison are displayed in table S2. Means and SEM are shown.
Total serum bilirubin was elevated in both DSS/FO-PN-Stig1– and DSS/FO-PN-Stig3–infused mice and did not differ significantly (P > 0.7) from that detected in DSS/SO-PN mice (Fig. 2C and table S1). These data demonstrated that a 7-day infusion of PN solution containing stigmasterol in this model caused both hepatocellular injury and cholestasis.
Phytosterol-containing PN solutions promote activation of macrophages
We have previously reported that the combination of intestinal injury and PN infusion in this mouse model was associated with proinflammatory activation of hepatic macrophages as reflected by increased transcription of Il6 mRNA in purified hepatic macrophages, whereas either treatment alone did not induce macrophage activation (15). To determine the role of plant sterols in PN in this activation of macrophages, we isolated and purified liver macrophages from mice in the current experiments, and measured mRNA expression of Il6 by quantitative reverse transcription polymerase chain reaction (qRT-PCR). Consistent with our previous report, transcription of Il6 was increased in liver macrophages from DSS-pretreated mice infused with SO-PN compared to liver macrophages from chow-fed and DSS-pretreated chow or NS control mice (Fig. 3A and table S1). In contrast, transcription of Il6 was not increased in liver macrophages from DSS-pretreated mice infused with either FO-PN or NoL-PN (Fig. 3A and table S1). However, DSS-pretreated mice infused with FO-PN-Stig3 showed enhanced Il6 transcription in liver macrophages to the extent observed in the SO-PN group (Fig. 3A and table S1).
Fig. 3. Phytosterol-containing PN solutions promote activation of liver macrophages and BMDMs.

(A) qRT-PCR analysis depicting relative amounts of Il6 mRNA (canonical indicator of macrophage activation) in purified hepatic CD11b+ cells (presented as hepatic macrophages) derived from the various groups of mice. Hepatic macrophage activation is increased in SO-PN–infused mice. DSS treatment alone with infusion of NS does not induce macrophage activation. Macrophage activation was attenuated in livers from mice infused with FO-PN and NoL-PN. Macrophage activation was also promoted in livers from mice infused with stigmasterol-spiked PN-SO solution (PN-FO-Stig3). Gene expression was determined after normalization to hypoxanthine-guanine phosphoribosyltransferase 1 (Hprt1) as an endogenous control gene. mRNA amounts are expressed relative to results obtained from untreated chow-fed control mice. Livers were pooled from three to five mice per group, and means and SEM from technical triplicates are shown. One representative of two independent CD11b+ cell isolations is shown. (B to D) StigAc induces transcription of the canonical macrophage proinflammatory genes Il6 (B), Il1β (C), and Tnfα (D) in naïve BMDMs. mRNA amounts are expressed relative to results obtained from untreated BMDMs. Shown are means and SEM from a technical triplicate from one representative experiment of a series of three independent experiments (BMDMs obtained from three different mice). *P < 0.05 versus all groups without *; *#P < 0.05 versus all other groups by one-way ANOVA with Tukey correction for multiple comparisons; exact P values for each comparison are depicted in table S2.
We next determined if stigmasterol could activate cytokine transcription in primary naïve macrophages in vitro. We exposed naïve wild-type mouse bone marrow–derived macrophages (BMDMs) for 4 hours to 10 μM stigmasterol acetate (StigAc). StigAc was chosen because of its reported superior solubility and stability compared with its parent compound (14); this concentration was shown to inhibit bile acid activation of Fxr and Abcb11 expression in primary mouse hepatocytes, and is seen in the plasma of young children receiving SO-based PN (16–18). We found that, compared to untreated and vehicle (ethanol in transfection reagent)–treated BMDMs, StigAc induced significantly (P < 0.0002) increased transcription of canonical macrophage proinflammatory cytokines, including Il6, Il1β, and Tnfα (Fig. 3, B to D, and table S1), which was between 30 and 60% of that observed with the canonical macrophage activator LPS (10 ng/ml) (Fig. 3, B to D, and table S1).
Serum and liver concentrations of stigmasterol correlate with PNALD in mice
Serum concentrations of stigmasterol measured by mass spectrometry were increased about sixfold in DSS-pretreated mice that had been infused with SO-PN compared to chow-fed controls (Fig. 4A and table S1), but not in DSS-pretreated mice infused with FO-PN. Serum stigmasterol concentrations were increased further in DSS/FO-PN-Stig1 mice (>20-fold). Liver stigmasterol was increased in DSS-pretreated mice that had been infused with SO-PN compared to chow-fed controls, but not in those that had been infused with FO-PN. Addition of stigmasterol to FO-PN led to increased liver stigmasterol concentrations (Fig. 4B and table S1). Together, these data show that stigmasterol accumulated in the serum and liver of mice administered phytosterol-containing PN solutions, which were associated with hepatic injury and cholestasis.
Fig. 4. Serum and liver concentrations of stigmasterol correlate with PNALD in mice.

(A and B) The bar graphs show serum (A) and liver (B) concentrations of stigmasterol in response to infusion with stigmasterol-containing PN solutions. Serum and liver stigmasterol is increased after infusion with stigmasterol-containing PN solutions [SO-PN (n = 7) and FO-PN-Stig1 (n = 7)] compared to DSS-pretreated mice that did not receive any PN (chow/DSS, n = 7) and DSS-pretreated mice infused with FO-PN (n = 7). Completely untreated mice (chow, n = 4) are shown for comparison. Means and SEM are shown. Unpaired t test was performed for statistical analysis. (A) *P = 0.0351, DSS/SO-PN versus DSS; *P < 0.0001, DSS/FO-PNStig1 versus DSS/FO-PN. (B) *P = 0.0055, DSS/SO-PN versus DSS; *P < 0.0001, DSS/FO-PNStig1 versus DSS/FO-PN.
Phytosterol-containing PN solutions suppress expression of canalicular genes critical for bile secretion
We next sought to determine whether the cholestatic effects of stigmasterol were related to expression of hepatocyte bile acid transporters. We determined liver mRNA expression of Abcb11 and the canalicular multispecific organic anion transporter involved in conjugated bilirubin excretion (MRP2; encoded by Abcc2). Compared to expression in untreated mice, liver mRNA expression was mildly reduced in DSS-pretreated control mice for both Abcb11 and Abcc2, but was markedly suppressed in livers from DSS-pretreated mice infused with SO-PN (Fig. 5, A and B, and table S1). In contrast, Abcb11 and Abcc2 expression in livers from mice infused with NoL-PN or FO-PN was similar to that observed in DSS-pretreated control mice and markedly higher than in the livers from SO-PN–infused mice (Fig. 5, A and B, and table S1). Moreover, expression of Abcb11 and Abcc2 in livers from FO-PN-Stig1 and FO-PN-Stig3 infused mice was markedly reduced to amounts that were comparable to those detected in livers from mice infused with SO-PN (Fig. 5, A and B, and table S1). Because expression of Abcb11 and Abcc2 is regulated by bile acid–stimulated FXR, we measured hepatic expression of Fxr and found that Fxr mRNA was markedly reduced in livers from DSS-pretreated mice infused with SO-PN, FO-PN-Stig1, and FO-PN-Stig3 compared to FO-PN and NoL-PN (Fig. 5C and table S1), mirroring expression of Abcb11 and Abcc2. Thus, stigmasterol reduces both expression and function of Fxr/FXR.
Fig. 5. Phytosterol-containing PN solutions suppress hepatic bile salt exporters.

qRT-PCR analysis depicting relative amounts of liver transcripts for hepatic canalicular exporters Abcb11 (encoding BSEP) and Abcc2 [encoding multidrug resistance–associated protein 2 (MDR2)] in livers from wild-type (WT) mice infused with various PN solutions (A and B), in TLR4 signaling–deficient mice (Tlr4), and in WT mice after antibiotic suppression of intestinal microbiota (Abx) (D and E). (C) Transcript amounts for Fxr are shown in the same groups of mice. Gene expression was determined after normalization to Hprt1 as an endogenous control gene. mRNA amounts are expressed relative to results obtained from untreated chow-fed controls. Groups consisted of 5 to 15 mice. Individual mice were tested in triplicate. Mean values from triplicates were analyzed and are displayed as means and SEM for each group. Exact adjusted P values obtained by one-way ANOVA and Tukey correction for multiple comparisons are depicted in table S2. (A) *P < 0.0001, DSS/SO-PN versus controls; P = 0.0006, DSS/SO-PN versus DSS/FO-PN; *P < 0.02, DSS/FO-PN-Stig1 and DSS/FO-PN-Stig3 versus DSS/FO-PN. (B) *P < 0.0014, DSS/SO-PN versus chow, DSS/NS, and DSS/FO-PN; P < 0.0165, DSS/FO-PN-Stig1 and DSS/FO-PN-Stig3 versus DSS/FO-PN. (C) *P < 0.0001, DSS/SO-PN versus controls and DSS/FO-PN; *P < 0.02, DSS/FO-PN-Stig1 and DSS/FO-PN-Stig3 versus DSS/FO-PN. (D and E) *P < 0.0004, DSS-SO-PN versus DSS-SO-PN-Abx and DSS-SO-PN-Tlr4; #P > 0.05 versus controls.
Gut microbiota and TLR4 signaling are involved in suppression of liver bile salt export systems
We have previously reported that antibiotic suppression of the intestinal microbiota or global abrogation of TLR4 signaling attenuated PNALD in mice pretreated with DSS and infused with SO-PN for 7 days (15). We therefore examined the role of the intestinal microbiota and TLR4 signaling pathways in regulating expression of Abcb11 and Abcc2 in livers of PN-infused mice with intestinal injury. For this purpose, we measured mRNA amounts in livers obtained from previously studied and reported wild-type C57BL/6 mice that had undergone DSS pretreatment and SO-PN infusion for 7 days with concomitant broad-spectrum oral antibiotic treatment (DSS/SO-PN-Abx; n = 5) (15). We also measured mRNA amounts in livers from previously reported C57BL/6 mice with a naturally occurring mutation in the Tlr4 gene that renders the mice globally unresponsive to LPS (25); these were also subjected to DSS pretreatment and SO-PN infusion for 7 days (DSS/SO-PN-Tlr4; n = 5). Both of these groups of mice displayed attenuated PNALD (15). Expression of mRNA for Abcb11 and Abcc2 in livers from antibiotic-treated mice was markedly increased when compared to SO-PN mice that had not received oral antibiotic treatment and was similar to gene expression in DSS/chow mice (n = 6 to 7) (Fig. 5, D and E, and table S1). Similarly, hepatic expression of Abcb11 and Abcc2 from Tlr4 mutant mice that received DSS pretreatment and SO-PN was markedly increased compared to SO-PN wild-type mice and was similar to expression from DSS/chow mice (Fig. 5, D and E, and table S1). These results suggest that innate immune signaling pathways initiated by LPS absorbed from gut microbiota and mediated through host TLR4 signaling may contribute to the suppression of hepatic bile acid and bilirubin transporters.
Phytosterol-containing PN solutions suppress hepatic sterol transporters
Finally, we determined hepatic mRNA expression for the sterol transporters Abcg5 and Abcg8, the principal exporters of cholesterol and phytosterols from hepatocytes. Compared to expression in untreated mice (n = 5) and DSS-pretreated (n = 5) control mice, expression of both Abcg5 and Abcg8 was induced in DSS-pretreated NoL-PN–infused (n = 5) and FO-PN–infused (n = 5) mice (Fig. 6, A and B, and table S1). In contrast, this induction of mRNA expression was suppressed in livers from DSS-pretreated SO-PN–infused mice (n = 5) (Fig. 6, A and B, and table S1). Likewise, induction of hepatic expression for both Abcg5 and Abcg8 was also not observed in DSS-pretreated PN-FO-Stig1–infused (n = 5) and PN-FO-Stig3–infused (n = 5) mice (Fig. 6, A and B, and table S1). These data suggest that phytosterol-containing PN (SO-PN, FO-Stig1, or FO-Stig3) suppressed the normal induction of Abcg5 and Abcg8 necessary to transport the phytosterols out of the hepatocyte, possibly accounting, in part, for the elevated liver concentrations of stigmasterol observed (Fig. 4B and table S1). Because expression of Abcg5 and Abcg8 is regulated by the nuclear receptor liver X receptor (LXR, Lxr), we determined hepatic gene expression of Lxr. Lxr mRNA was markedly reduced in livers from DSS-pretreated SO-PN–, FO-PN-Stig1–, and FO-PN-Stig3–infused mice compared to FO-PN and NoL-PN mice (Fig. 6C and table S1). Thus, phytosterol-containing PN solutions suppressed Lxr expression and its downstream gene targets, Abcg5 and Abcg8.
Fig. 6. Phytosterol-containing PN solutions suppress hepatic sterol transporters.
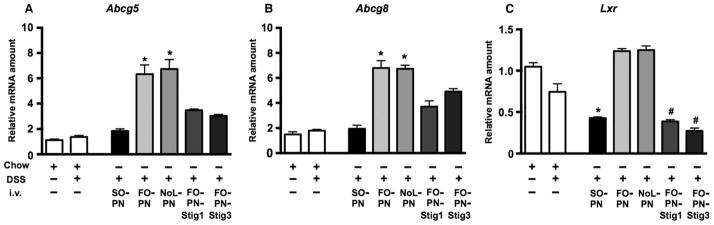
(A to C) qRT-PCR analysis depicting relative amounts of liver transcripts for hepatic sterol transporters Abcg5 (A), Abcg8 (B), and Lxr (C). Gene expression was determined after normalization to Hprt1 as an endogenous control gene. mRNA amounts are expressed relative to results obtained from untreated chow-fed controls. Groups consisted of five mice. Individual mice were tested in triplicate. Mean values from triplicates were analyzed and are displayed as means and SEM from groups of five mice. *P < 0.01 versus groups without * and #; #P < 0.0006 versus groups without *. Exact adjusted P values obtained by one-way ANOVA and Tukey correction for multiple comparisons are depicted in table S2.
DISCUSSION
Here, we have provided experimental evidence that implicates phytosterols, specifically stigmasterol, as a component of SO-based lipid emulsions that is responsible for cholestasis and hepatic macrophage activation in PNALD through a mechanism that involves phytosterol-mediated suppression of bile acid, bilirubin, and sterol transporters. We have demonstrated that substituting FO for SO-based lipid emulsions or elimination of all intravenous lipids in PN solutions prevents cholestasis, hepatocyte injury, and activation of liver macrophages in mice with intestinal injury receiving PN for 7 days, replicating reports in human infants in whom infusion with FO-based PN or reduction of intravenous lipid dose improved PNALD (6–13). These results indicate that the absence of a lipid component in FO-based PN, rather than the presence of ω-3 fatty acids as proposed by others (7, 10, 11, 13), is likely the major mechanism of protection from liver injury and cholestasis.
Phytosterols have been proposed as playing a potential role in the pathogenesis of PNALD for more than 20 years after it was demonstrated that phytosterols accumulate in livers and plasma from patients receiving SO-based PN (16, 17). More recent studies have shown that elevated serum phytosterol concentrations correlate with PNALD in human infants (18, 19). Mechanistic studies have demonstrated that among the phytosterols present in SO emulsions, stigmasterol was by far the most potent at inhibiting activity of FXR, which regulates transcription of bile acid transporters in cultured hepatocytes (14). On the basis of these observations, we evaluated the role of stigmasterol in promoting PNALD in our mouse model and took advantage of the fact that FO-based PN infusion, which was devoid of all phytosterols, prevented liver injury that was observed in mice infused with phytosterol-containing SO-PN. We also “spiked” FO-PN with stigmasterol to isolate its potential to induce liver injury and cholestasis. The two different concentrations of stigmasterol used in our study reflected the amount infused into mice receiving SO-PN, and the higher concentration reflected that infused into human infants receiving SO lipid emulsion (3 to 3.5 g/kg per day). The results provide experimental evidence that in an animal model of intestinal injury and PN infusion, plant sterols, specifically stigmasterol, contribute to cholestasis, liver injury, and macrophage activation. These findings support the emerging hypothesis that stigmasterol is the plant sterol in soy-based PN solutions that is likely to contribute to cholestasis and liver injury in patients with underlying intestinal diseases.
This hypothesis is further supported by clinical observations during PN administration in patients with short bowel syndrome (18, 19, 26). Transition from SO (Intralipid, Fresenius Kabi)–based PN to olive oil lipid (ClinOleic, Baxter/Clintec Parenteral SA) resulted in normalization of serum liver enzymes (26), indicating reduced liver injury. The major difference in plant sterol content between SO and olive oil lipid is the stigmasterol concentration, which is about three times higher in Intralipid than in ClinOleic, whereas the total plant sterol concentration is only 30% higher in Intralipid (27). On the basis of these and our observations, future studies should determine safe thresholds of plant sterols that can be administered in PN solutions to design improved lipid emulsions.
The mechanism by which phytosterol-containing PN solutions promote liver injury when infused in conjunction with intestinal injury/inflammation has not been fully evaluated. The prevailing hypothesis, based on in vitro studies using cell lines and primary hepatocytes, proposes that stigmasterol, through inhibition of activity of FXR, reduces expression of genes and proteins involved in canalicular excretion of bile acids and bilirubin, thus causing hepatocellular retention and toxicity from bile acids (14). On the basis of this hypothesis, we demonstrated that hepatic expression of Abcb11, Abcc2, and Fxr was markedly reduced in mice receiving PN solutions containing phytosterols, indicating marked perturbations of FXR and downstream signaling during PNALD. Although we did not directly investigate FXR binding activity to target promoter sequences, these data are consistent with a previous study implicating reduction of activity of BSEP and other transporters as a primary mechanism of stigmasterol-induced cholestasis (14).
We have previously reported that neither infusion with SO-PN solution for 7 days nor DSS-induced intestinal injury in mice is alone sufficient to induce liver injury, cholestasis, and macrophage activation (15). These findings in the mouse model are analogous to the experience in human infants, in whom PNALD is much more severe in infants with intestinal failure (3). We have thus hypothesized that components within the PN solution synergize with hepatic inflammatory pathways and cytokines that are induced by TLR agonists (such as LPS) absorbed through an injured intestine with bacterial overgrowth and reduced barrier function, and that activation of TLR4 signaling in hepatic macrophages leading to generation of proinflammatory cytokines is an important component of this hepatic injury (15). Indeed, in mice lacking TLR4 signaling or in which the intestinal microbiota had been suppressed by antibiotics, liver injury and cholestasis did not occur and Abcb11, Abcc2, and Fxr expression was restored to that in control mice. Moreover, we have demonstrated that the addition of stigmasterol to the PN solutions was associated with proinflammatory hepatic macrophage activation in vivo and that StigAc promoted cytokine expression in cultured macrophages in vitro at clinically relevant concentrations. These findings led us to speculate that LPS and plant sterols may synergize at the level of macrophage generation of proinflammatory cytokines in this liver injury model. Because stigmasterol inhibits FXR signaling in hepatocytes (14) and considering the emerging concept that FXR signaling may be anti-inflammatory in macrophages, inhibition of FXR activity in macrophages by stigmasterol may be a potential pathway involved in the observed ability of stigmasterol to activate macrophages (28–32). Our findings should prompt future studies to delineate the mechanism(s) through which plant sterols activate macrophages.
Our study also demonstrated that expression of the principal sterol exporters in the hepatocyte, encoded by Abcg5 and Abcg8, failed to be up-regulated in mice infused with phytosterol-containing PN solutions. Thus, hepatocytes of mice with PNALD are primed to accumulate phytosterols when relatively large amounts of phytosterols are infused in PN, potentially enhancing the antagonism of FXR signaling by higher hepatocyte concentrations of phytosterols. Because expression of Abcg5 and Abcg8 is regulated by LXR, we examined the possibility that altered LXR expression was responsible for failure to up-regulate Abcg5 and Abcg8. Indeed, hepatic Lxr transcription was markedly reduced in mice receiving intravenous stigmasterol. This may have particular importance because activation of LXR in macrophages has been shown to exert anti-inflammatory activity (33, 34); thus, the up-regulation of hepatic macrophage Il6 mRNA during PN may also be a result of inhibition of LXR activity by stigmasterol. A similar mechanism has recently been proposed in TLR-activated macrophages in which inhibition of LXR activity resulted in potentiated inhibition of cholesterol export (34).
Phytosterols also accumulate to very high serum and hepatic concentrations in a rare genetic disease, sitosterolemia, in which mutations in ABCG5/G8 lead to impaired biliary secretion and increased intestinal absorption of phytosterols (35–37). Although these patients develop xanthomas and premature atherosclerosis, liver injury and cholestasis are not generally present in affected individuals or in the mouse model of this disease (35–38). Thus, our demonstration of the role of stigmasterol in PNALD appears to be contradicted by these observations. However, there are several important differences between PNALD and sitosterolemia. First, neonates, the group most affected by PNALD, have an increased susceptibility to cholestasis because of ongoing postnatal development of bile acid transport and detoxification/conjugating enzymatic systems, which improves with age (39, 40). Second, the amount of infused phytosterols in SO lipid emulsions greatly exceeds that normally encountered by neonates, potentially overwhelming normal physiologic homeostatic mechanisms. Third, PNALD is a multifactorial disease, as opposed to sitosterolemia, in which activated inflammatory pathways from intestinally absorbed bacterial products and systemic inflammation and infection are involved. LPS, cytokines, and, as we have shown, proinflammatory activation of macrophages play important roles in altering the expression of bile acid transport genes in PNALD, but have not been shown to be involved in sitosterolemia, to our knowledge. Thus, elevated hepatic concentrations of phytosterols in isolation may not lead to liver injury; however, in the setting of PNALD, there is accumulating clinical and experimental evidence that phytosterols are key pathogenic factors.
There are limitations of this study. Adult mice were used throughout the experiments rather than infant mice because of the technical issues of placing and maintaining CVCs in very small mice. Thus, the experimental model does not completely replicate the human most susceptible to PNALD, the infant with intestinal malformations or disease. However, we could expect the findings in the current study to be more severe if it were possible to adapt this model to neonatal mice because of the immaturity of hepatic bile acid transporter expression and the propensity toward cholestasis in both rodent and human infants (41, 42). Another limitation of this study is that only one phytosterol (stigmasterol) was investigated, so the roles of other phytosterols in this model are not known.
In summary, the present study and our previous report (15) provide experimental evidence in a mouse model that the pathogenesis of PNALD involves the gene-regulating effects of infused phytosterols combined with gut-derived LPS- and phytosterol-mediated activation of liver macrophages. The resulting deregulation of canalicular bile acid, bilirubin, and sterol transporters, likely through inhibition of nuclear receptor signaling by phytosterols and cytokines, leads to cholestasis and enhanced accumulation of hepatic phytosterols. Thus, modification of lipid constituents infused in PN solutions for infants with intestinal injury will be an important strategy to reduce the incidence and severity of PNALD.
MATERIALS AND METHODS
Study design
The PNALD mouse model was previously published (15). In brief, C57BL/6 wild-type and syngeneic TLR4 mutant (B6.B10ScN-Tlr4lps-del/JthJ) adult male mice (8 to 10 weeks old; Jackson Laboratories) were exposed ad lib to 2.5% DSS in the drinking water for 4 days. Mice then received regular drinking water for 24 hours (referred to as “DSS pre-treatment”) before surgical placement of a CVC into the right jugular vein. Mice were placed in a rubber harness (Instech Laboratories) connected by a swivel system. The rubber harness ensures secure placement of the catheter, whereas the swivel system connects the infusion line from the harness to the infusion pump (Harvard Apparatus) such that movement of the mouse within the cage is only minimally limited. Mice were recovered from surgery for 24 hours with NS infused in the CVC before continuous infusion for 7 days with PN at a rate of 0.29 ml/hour, providing a caloric intake of 8.4 kcal/24 hours. All PN-infused mice had access to water ad lib but not to chow during the PN infusion period. Several control groups of mice were also studied. DSS-pretreated mice were infused with NS for 7 days instead of PN (DSS/NS). DSS-pretreated mice that did not have a CVC placed were given free access to chow and water. Unmanipulated control mice had free access to chow and water without being connected to the harness-swivel system. Mice received PN that contained either SO lipid emulsion (10 ml of 20% Intralipid per 100 ml of PN solution), FO lipid emulsion (10 ml of 10% Omegaven per 100 ml of PN solution), no lipid emulsion, or FO lipid emulsion to which stigmasterol was added at concentration of 1 or 3 mg/100 ml of PN. All mice were individually housed in metabolic cages. A graphical representation of the experimental design is depicted in fig. S1. Blood was collected from the retro-orbital plexus under pentobarbital anesthesia. Serum was analyzed by the University of Colorado Hospital Clinical Chemistry Laboratory for AST, ALT, and total bilirubin concentrations. TSBAs were analyzed using a total bile acid detection kit (Diazyme Laboratories) according to the manufacturer’s instructions. PN solution, serum, and liver stigmasterol concentrations were determined by mass spectrometry as previously described (43).
All animals were treated humanely, and the protocol was approved by the Institutional Animal Care and Use Committee of the University of Colorado Denver.
Suppression of intestinal microbiota
A group of DSS-pretreated SO-PN mice was exposed ad lib to an oral cocktail of four broad-spectrum antibiotics provided in the drinking water during the entire course of the 7-day PN infusion as previously reported (15). The antibiotics, mixed fresh every 24 hours, included vancomycin (1 g/liter), streptomycin (2 g/liter), ampicillin (2 g/liter), and metronidazole (2 g/liter).
Hepatic macrophage purification
Hepatic macrophages were purified as previously described (15). In brief, pooled livers (three to six mice) were minced in cold Hanks’ buffer (Gibco/Invitrogen) and incubated in LiberaseR (Hoffmann–La Roche) and deoxyribonuclease (DNase) (Sigma-Aldrich) for 30 min at 37°C followed by low-speed centrifugation at 25g and further separation over Histodenz gradient (Sigma-Aldrich). CD11b+ cells (referred to as hepatic macrophages/Kupffer cells in this study) were then purified using positive selection with magnetic CD11b antibody beads (MACS, Miltenyi) as previously described (15).
In vitro activation of macrophages with StigAc
Mouse BMDMs were generated from 6- to 8-week-old male C57BL/6 mice (Jackson Laboratories) as previously described (44). BMDMs were treated with ultrapure LPS (10 ng/ml) (Escherichia coli K12 strain from Invivogen) and a final concentration of 10 μM StigAc (Steraloids). StigAc was solubilized as a 10,000 μM solution in 100% ethanol (EtOH). For BMDM treatment, StigAc was formulated in TransIT-LT1 Transfection Reagent (Mirus) at a concentration of 100 μM in serum- and antibiotic-free Dulbecco’s modified Eagle’s medium and incubated for 30 min before adding to BMDMs at a final concentration of 10 μM. Vehicle controls included equimolar preparations of transfection reagent alone (TR) and a mixture of transfection reagent with ethanol (TR-EtOH).
RNA isolation and quantitative gene expression analysis
RNA was extracted using TRIzol (Invitrogen), treated with DNase (Ambion), and reverse-transcribed with iScript (Bio-Rad) according to the manufacturer’s instructions. Gene expression was analyzed in triplicate by qRT-PCR analysis on an Applied Biosystems 7300 cycler using commercially available mouse TaqMan gene expression assays (Applied Biosystems). Data were analyzed by software provided by Applied Biosystems using the ΔΔCt method and expressed after normalization to Hprt1 and relative to untreated chow-fed control or DSS/NS-treated mice and untreated BMDMs.
Statistical analysis
GraphPad Prism software was used to determine statistical significance by one-way ANOVA with Tukey correction for multiple comparisons; unpaired t test was used for comparisons between two groups. Means and SEM are shown. A P value of <0.05 was considered statistically significant. All original data and exact adjusted P values, α, and 95% confidence intervals are depicted in tables S1 and S2.
Supplementary Material
Acknowledgments
Funding: This work was supported by NIH/National Center for Advancing Translational Science Colorado Clinical and Translational Sciences Institute (grant UL1 TR000154), the Colorado Clinical Nutrition Research Unit (grant P30DK048520), and the Academic Enrichment Fund of the University of Colorado School of Medicine. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
Omegaven was provided by Fresenius Kabi.
Footnotes
Author contributions: Study design, supervision, data analysis, data interpretation, and preparation of the manuscript: K.C.E.K., S.J.K., and R.J.S.; data collection: A.L.A., M.W.D., K.C.E.K., P.M.V., W.Z., and K.D.R.S.
Competing interests: The authors declare that they have no competing interests.
www.sciencetranslationalmedicine.org/cgi/content/full/5/206/206ra137/DC1
Fig. S1. Experimental design and experimental treatment groups.
Table S1. Original data for all figures.
Table S2. Statistical analyses for all figures.
REFERENCES AND NOTES
- 1.Squires RH, Duggan C, Teitelbaum DH, Wales PW, Balint J, Venick R, Rhee S, Sudan D, Mercer D, Martinez JA, Carter BA, Soden J, Horslen S, Rudolph JA, Kocoshis S, Superina R, Lawlor S, Haller T, Kurs-Lasky M, Belle SH. Pediatric Intestinal Failure Consortium, Natural history of pediatric intestinal failure: Initial report from the Pediatric Intestinal Failure Consortium. J Pediatr. 2012;161:723–728.e2. doi: 10.1016/j.jpeds.2012.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rangel SJ, Calkins CM, Cowles RA, Barnhart DC, Huang EY, Abdullah F, Arca MJ, Teitelbaum DH. 2011 American Pediatric Surgical Association Outcomes and Clinical Trials Committee, Parenteral nutrition–associated cholestasis: An American Pediatric Surgical Association Outcomes and Clinical Trials Committee systematic review. J Pediatr Surg. 2012;47:225–240. doi: 10.1016/j.jpedsurg.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Kaufman SS, Loseke CA, Lupo JV, Young RJ, Murray ND, Pinch LW, Vanderhoof JA. Influence of bacterial overgrowth and intestinal inflammation on duration of parenteral nutrition in children with short bowel syndrome. J Pediatr. 1997;131:356–361. doi: 10.1016/s0022-3476(97)80058-3. [DOI] [PubMed] [Google Scholar]
- 4.Duro D, Kamin D, Duggan C. Overview of pediatric short bowel syndrome. J Pediatr Gastroenterol Nutr. 2008;47(Suppl 1):S33–S36. doi: 10.1097/MPG.0b013e3181819007. [DOI] [PubMed] [Google Scholar]
- 5.Carter BA, Karpen SJ. Intestinal failure-associated liver disease: Management and treatment strategies past, present, and future. Semin Liver Dis. 2007;27:251–258. doi: 10.1055/s-2007-985070. [DOI] [PubMed] [Google Scholar]
- 6.Koletzko B, Goulet O. Fish oil containing intravenous lipid emulsions in parenteral nutrition-associated cholestatic liver disease. Curr Opin Clin Nutr Metab Care. 2010;13:321–326. doi: 10.1097/MCO.0b013e3283385407. [DOI] [PubMed] [Google Scholar]
- 7.Fallon EM, Le HD, Puder M. Prevention of parenteral nutrition-associated liver disease: Role of w-3 fish oil. Curr Opin Organ Transplant. 2010;15:334–340. doi: 10.1097/mot.0b013e3283394879. [DOI] [PubMed] [Google Scholar]
- 8.Cober MP, Teitelbaum DH. Prevention of parenteral nutrition-associated liver disease: Lipid minimization. Curr Opin Organ Transplant. 2010;15:330–333. doi: 10.1097/MOT.0b013e328338c2da. [DOI] [PubMed] [Google Scholar]
- 9.Cober MP, Killu G, Brattain A, Welch KB, Kunisaki SM, Teitelbaum DH. Intravenous fat emulsions reduction for patients with parenteral nutrition–associated liver disease. J Pediatr. 2012;160:421–427. doi: 10.1016/j.jpeds.2011.08.047. [DOI] [PubMed] [Google Scholar]
- 10.Puder M, Valim C, Meisel JA, Le HD, de Meijer VE, Robinson EM, Zhou J, Duggan C, Gura KM. Parenteral fish oil improves outcomes in patients with parenteral nutrition-associated liver injury. Ann Surg. 2009;250:395–402. doi: 10.1097/SLA.0b013e3181b36657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diamond IR, Sterescu A, Pencharz PB, Kim JH, Wales PW. Changing the paradigm: Omegaven for the treatment of liver failure in pediatric short bowel syndrome. J Pediatr Gastroenterol Nutr. 2009;48:209–215. doi: 10.1097/MPG.0b013e318182c8f6. [DOI] [PubMed] [Google Scholar]
- 12.Cheung HM, Lam HS, Tam YH, Lee KH, Ng PC. Rescue treatment of infants with intestinal failure and parenteral nutrition-associated cholestasis (PNAC) using a parenteral fish-oil-based lipid. Clin Nutr. 2009;28:209–212. doi: 10.1016/j.clnu.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Diamond IR, Pencharz PB, Feldman BM, Ling SC, Moore AM, Wales PW. Novel lipid-based approaches to pediatric intestinal failure–associated liver disease. Arch Pediatr Adolesc Med. 2012;166:473–478. doi: 10.1001/archpediatrics.2011.1896. [DOI] [PubMed] [Google Scholar]
- 14.Carter BA, Taylor OA, Prendergast DR, Zimmerman TL, Von Furstenberg R, Moore DD, Karpen SJ. Stigmasterol, a soy lipid–derived phytosterol, is an antagonist of the bile acid nuclear receptor FXR. Pediatr Res. 2007;62:301–306. doi: 10.1203/PDR.0b013e3181256492. [DOI] [PubMed] [Google Scholar]
- 15.El Kasmi KC, Anderson AL, Devereaux MW, Fillon SA, Harris JK, Lovell MA, Finegold MJ, Sokol RJ. Toll-like receptor 4–dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology. 2012;55:1518–1528. doi: 10.1002/hep.25500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clayton PT, Whitfield P, Iyer K. The role of phytosterols in the pathogenesis of liver complications of pediatric parenteral nutrition. Nutrition. 1998;14:158–164. doi: 10.1016/s0899-9007(97)00233-5. [DOI] [PubMed] [Google Scholar]
- 17.Clayton PT, Bowron A, Mills KA, Massoud A, Casteels M, Milla PJ. Phytosterolemia in children with parenteral nutrition-associated cholestatic liver disease. Gastroenterology. 1993;105:1806–1813. doi: 10.1016/0016-5085(93)91079-w. [DOI] [PubMed] [Google Scholar]
- 18.Kurvinen A, Nissinen MJ, Andersson S, Korhonen P, Ruuska T, Taimisto M, Kalliomäki M, Lehtonen L, Sankilampi U, Arikoski P, Saarela T, Miettinen TA, Gylling H, Pakarinen MP. Parenteral plant sterols and intestinal failure-associated liver disease in neonates. J Pediatr Gastroenterol Nutr. 2012;54:803–811. doi: 10.1097/MPG.0b013e3182474118. [DOI] [PubMed] [Google Scholar]
- 19.Kurvinen A, Nissinen MJ, Gylling H, Miettinen TA, Lampela H, Koivusalo AI, Rintala RJ, Pakarinen MP. Effects of long-term parenteral nutrition on serum lipids, plant sterols, cholesterol metabolism, and liver histology in pediatric intestinal failure. J Pediatr Gastroenterol Nutr. 2011;53:440–446. doi: 10.1097/MPG.0b013e3182212130. [DOI] [PubMed] [Google Scholar]
- 20.Bhogal HK, Sanyal AJ. The molecular pathogenesis of cholestasis in sepsis. Front Biosci. 2013;5:87–96. doi: 10.2741/e598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kosters A, Karpen SJ. The role of inflammation in cholestasis: Clinical and basic aspects. Semin Liver Dis. 2010;30:186–194. doi: 10.1055/s-0030-1253227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geier A, Fickert P, Trauner M. Mechanisms of disease: Mechanisms and clinical implications of cholestasis in sepsis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:574–585. doi: 10.1038/ncpgasthep0602. [DOI] [PubMed] [Google Scholar]
- 23.Yang H, Finaly R, Teitelbaum DH. Alteration in epithelial permeability and ion transport in a mouse model of total parenteral nutrition. Crit Care Med. 2003;31:1118–1125. doi: 10.1097/01.CCM.0000053523.73064.8A. [DOI] [PubMed] [Google Scholar]
- 24.Lapillonne A, Fellous L, Mokthari M, Kermorvant-Duchemin E. Parenteral nutrition objectives for very low birth weight infants: Results of a national survey. J Pediatr Gastroenterol Nutr. 2009;48:618–626. doi: 10.1097/MPG.0b013e31818c52bc. [DOI] [PubMed] [Google Scholar]
- 25.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 26.Hallikainen M, Huikko L, Kontra K, Nissinen M, Piironen V, Miettinen T, Gylling H. Effect of parenteral serum plant sterols on liver enzymes and cholesterol metabolism in a patient with short bowel syndrome. Nutr Clin Pract. 2008;23:429–435. doi: 10.1177/0884533608321138. [DOI] [PubMed] [Google Scholar]
- 27.Forchielli ML, Bersani G, Tala S, Grossi G, Puggioli C, Masi M. The spectrum of plant and animal sterols in different oil-derived intravenous emulsions. Lipids. 2010;45:63–71. doi: 10.1007/s11745-009-3371-x. [DOI] [PubMed] [Google Scholar]
- 28.Renga B, Mencarelli A, Cipriani S, D’Amore C, Carino A, Bruno A, Francisci D, Zampella A, Distrutti E, Fiorucci S. The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PLoS One. 2013;8:e54472. doi: 10.1371/journal.pone.0054472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S, Klomp LW, Siersema PD, Schipper ME, Danese S, Penna G, Laverny G, Adorini L, Moschetta A, van Mil SW. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–472. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 30.McMahan R, Wang XX, Cheng LL, Krisko T, Smith M, El Kasmi K, Pruzanski M, Adorini L, Golden-Mason L, Levi M, Rosen HR. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem. 2013;288:11761–11770. doi: 10.1074/jbc.M112.446575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology. 2008;48:1632–1643. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, Rizzo G, Gioiello A, Adorini L, Pellicciari R, Auwerx J, Schoonjans K. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14:747–757. doi: 10.1016/j.cmet.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong C, Walczak R, Dhamko H, Bradley MN, Marathe C, Boyadjian R, Salazar JV, Tontonoz P. Constitutive activation of LXR in macrophages regulates metabolic and inflammatory gene expression: Identification of ARL7 as a direct target. J Lipid Res. 2011;52:531–539. doi: 10.1194/jlr.M010686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P. Crosstalk between LXR and Toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. 2003;12:805–816. doi: 10.1016/s1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- 35.Bhattacharyya AK, Connor WE. β-Sitosterolemia and xanthomatosis. A newly described lipid storage disease in two sisters. J Clin Invest. 1974;53:1033–1043. doi: 10.1172/JCI107640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee MH, Lu K, Patel SB. Genetic basis of sitosterolemia. Curr Opin Lipidol. 2001;12:141–149. doi: 10.1097/00041433-200104000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salen G, Shefer S, Nguyen L, Ness GC, Tint GS, Shore V. Sitosterolemia. J Lipid Res. 1992;33:945–955. [PubMed] [Google Scholar]
- 38.Yu L, Hammer RE, Li-Hawkins J, Von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci USA. 2002;99:16237–16242. doi: 10.1073/pnas.252582399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomer G, Ananthanarayanan M, Weymann A, Balasubramanian N, Suchy FJ. Differential developmental regulation of rat liver canalicular membrane transporters Bsep and Mrp2. Pediatr Res. 2003;53:288–294. doi: 10.1203/01.PDR.0000047509.54253.01. [DOI] [PubMed] [Google Scholar]
- 40.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology—Drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349:1157–1167. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- 41.Balasubramaniyan N, Shahid M, Suchy FJ, Ananthanarayanan M. Multiple mechanisms of ontogenic regulation of nuclear receptors during rat liver development. Am J Physiol Gastrointest Liver Physiol. 2005;288:G251–G260. doi: 10.1152/ajpgi.00351.2004. [DOI] [PubMed] [Google Scholar]
- 42.Suchy FJ, Bucuvalas JC, Novak DA. Determinants of bile formation during development: Ontogeny of hepatic bile acid metabolism and transport. Semin Liver Dis. 1987;7:77–84. doi: 10.1055/s-2008-1040567. [DOI] [PubMed] [Google Scholar]
- 43.Kritchevsky D, Tepper SA, Czarnecki SK, Wolfe B, Setchell KD. Serum and aortic levels of phytosterols in rabbits fed sitosterol or sitostanol ester preparations. Lipids. 2003;38:1115–1118. doi: 10.1007/s11745-003-1168-8. [DOI] [PubMed] [Google Scholar]
- 44.El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, Basaraba RJ, König T, Schleicher U, Koo MS, Kaplan G, Fitzgerald KA, Tuomanen EI, Orme IM, Kanneganti TD, Bogdan C, Wynn TA, Murray PJ. Toll-like receptor–induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol. 2008;9:1399–1406. doi: 10.1038/ni.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Z, Harvey KA, Pavlina T, Dutot G, Hise M, Zaloga GP, Siddiqui RA. Steroidal compounds in commercial parenteral lipid emulsions. Nutrients. 2012;4:904–921. doi: 10.3390/nu4080904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.