

Abstract Abstract
Disruption of the endothelium leads to increased permeability, allowing extravasation of macromolecules and other solutes from blood vessels. Calcium entry through a calcium-selective, store-operated calcium (SOC) channel, Isoc, contributes to barrier disruption. An understanding of the mechanisms surrounding the regulation of Isoc is far from complete. We show that the calcium/calmodulin-activated phosphatase calcineurin (CN) plays a role in regulation of SOC entry, possibly through the dephosphorylation of stromal interaction molecule 1 (STIM1). Phosphorylation has been implicated as a regulatory mechanism of activity for a number of canonical transient receptor potential (TRPC) and SOC channels, including Isoc. Our results show that STIM1 phosphorylation increases in pulmonary artery endothelial cells (PAECs) upon activation of SOC entry. However, the phosphatases involved in STIM1 dephosphorylation are unknown. We found that a CN inhibitor (calcineurin inhibitory peptide [CIP]) increases the phosphorylation pattern of STIM1. Using a fura 2-acetoxymethyl ester approach to measure cytosolic calcium in PAECs, we found that CIP decreases SOC entry following thapsigargin treatment in PAECs. Luciferase assays indicate that thapsigargin induces activation of CN activity and confirm inhibition of CN activity by CIP in PAECs. Also, Isoc is significantly attenuated in whole-cell patch-clamp studies of PAECs treated with CIP. Finally, PAECs pretreated with CIP exhibit decreased interendothelial cell gap formation in response to thapsigargin-induced SOC entry, as compared to control cells. Taken together, our data show that CN contributes to the phosphorylation status of STIM1, which is important in regulation of endothelial SOC entry and Isoc activity.
Keywords: Isoc, transient receptor potential, TRPC, stromal interaction molecule 1, STIM1, phosphorylation, phosphatase
Introduction
Calcium signaling mediates a number of cellular events, and in many nonexcitable cells, store-operated calcium (SOC) entry represents the major calcium event for signaling. In the SOC entry process, depletion of endoplasmic reticulum (ER) calcium stores leads to calcium entry across the plasma membrane through SOC channels (as reviewed previously1,2). Calcium entry through these channels factors largely in calcium signaling cascades in both physiological and disease states.
The calcium-activated Ca2+ current, or ICRAC, was one of the first SOC currents studied. The ICRAC is a small, calcium-selective, inwardly rectifying current originally found in mast cells.1 Since its discovery, a number of SOC currents have been described in a variety of cell types. Pulmonary artery endothelial cells (PAECs) exhibit a calcium-selective SOC entry current termed ISOC, also found in other endothelial cells and a number of additional cell types, including epithelial cells and myocytes.1,3 Activation of ISOC in PAECs leads to formation of interendothelial cell gaps and disruption of the endothelial cell barrier.3,4
Insight into the regulation of the ISOC channel remains elusive, as the complete molecular makeup of the ISOC channel is still under investigation. The current paradigm is that the channel is made up of mammalian homologs of the canonical transient receptor potential (TRPC) protein subfamily (reviewed by Cioffi et al.4). In particular, TRPC family members 1, 3, 4, and 5 have been implicated as potential SOC channel subunits, and at least TRPC1 and TRPC4 are part of the ISOC channel structure,5,6 with the TRPC4 C-terminus interacting with protein 4.1 close to the putative channel pore.7 As well, Orai1, which makes up the ICRAC channel,8-10 constitutively interacts with TRPC4 (and potentially TRPC1, after SOC activation) of the ISOC channel.11 Here, Orai1 is responsible for the calcium selectivity of the channel.11
Activation of the ISOC channel allows for influx of calcium, whereas inactivation restricts calcium entry via channel closure. It is now known that the region encompassing the protein 4.1 binding site and the adjacent proline-rich region on the TRPC4 subunit is essential for ISOC channel activation.7 Conversely, while it is known that ISOC channel inactivation is both phosphorylation and calcium dependent,12-14 the molecular details of that process, including the identity of kinases and phosphatases and their targets, have not been determined. An understanding not only of the targets but also of the enzymes and signaling pathways involved is imperative for the larger picture of ISOC regulation.
Calcineurin (CN), also known as protein phosphatase 2B (PP2B), is a calcium/calmodulin-activated serine-threonine phosphatase. In this heterodimeric phosphatase, the A subunit contains the catalytic activity and 3 regulatory domains: the CN B subunit binding site, the calmodulin-binding domain, and the autoinhibitory domain (reviewed by Rusnak and Mertz15). The CN B subunit is responsible for calcium binding via 4 calcium-binding EF-hand motifs.16 The autoinhibitory domain binds in the active site in the absence of calcium and calmodulin. CN is implicated in the regulation of some calcium channels. CN is shown to be associated with the inositol triphosphate (InsP3) and ryanodine receptors and is suggested to modulate calcium flux via its phosphatase activity.17 In addition, CN is found as a subunit of a multiprotein complex on TRPC6 channels of neuronal PC12 cells.18 Traditionally, CN is thought to be activated by influxes of extracellular calcium into the cell. However, recently it was shown that the calcium released from intracellular stores via thapsigargin treatment is sufficient to activate CN in cardiomyocyes.19 As well, Kar et al.20 showed that CN-dependent signaling is activated by calcium microdomains in HEK293 cells.
While it is known that ISOC channel inactivation is phosphorylation dependent, the phosphorylation target or targets of ISOC have yet to be elucidated. A potential target in PAECs is stromal interaction molecule 1 (STIM1), an 85-kDa calcium sensor. STIM1 is phosphorylated at several serine and threonine residues,21-24 and its phosphorylation is linked to regulation of SOC entry, including inactivation of SOC entry.21,23,25 STIM1 initiates SOC entry by oligomerizing into puncta at the ER and joining the plasma membrane-bound SOC channels through interaction with Orai1 following calcium depletion (reviewed by Wu et al.26). That STIM1 may be a functionally important target is supported by studies showing that STIM1 interacts with TRPC1/4 to regulate SOC entry.27 Further, Orai1 interacts with TRPC4 in the endogenous ISOC channel to control activation and calcium selectivity.11 In the CRAC channel, STIM1 interacts directly with Orai1 to allow calcium entry (reviewed by Hewavitharana et al.28).
Here, we sought to determine whether CN plays a role in SOC entry and ISOC regulation in PAECs. Using a cell-permeable calcineurin inhibitory peptide (CIP) that mimics the autoinhibitory domain of CN,29-32 we found that CN inhibition reduces global SOC entry and attenuates ISOC. In addition, using small interfering RNA (siRNA) knockdown, we show that STIM1 is necessary for SOC entry in PAECs. Finally, we found that STIM1 is phosphorylated upon thapsigargin-induced store depletion and that CN inhibition increases the phosphorylation profile of STIM1 in thapsigargin-stimulated PAECs.
Methods
Reagents
Reagents were obtained from Sigma unless noted below. Cell culture medium, penicillin, and streptomycin were obtained from Life Technologies (Grand Island, NY). Heat-inactivated fetal bovine serum (FBS) was purchased from Cell Generation (Fort Collins, CO). Pan-CN A antibody was obtained from Cell Signaling (Boston, MA). Monoclonal antibody to β-actin was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and STIM1 monoclonal antibody was obtained from BD Biosciences (San Jose, CA). Goat anti-rabbit-HRP (horseradish peroxidase) and goat anti-mouse-HRP secondary antibodies were obtained from Thermo Scientific (Pittsburgh, PA). Cell-permeable CIP was purchased from EMD Millipore (Billerica, MA), and Superfect transfection reagent was purchased from QIAGEN (Valencia, CA). Monolight luciferase reagents were from BD Biosciences. The IL-2/NFAT (nuclear factor of activated T cells) luciferase reporter has been previously described.33 The bicinchonic acid (BCA) protein assay kit was obtained from Thermo Scientific (Rockford, IL).
Cell culture
PAECs were isolated and cultured with a method described by Creighton et al.34 Cells were grown in DMEM (Dulbecco’s Modified Eagle Medium) high glucose, supplemented with 10% FBS, 50 U/mL penicillin G, and 0.05 mg/mL streptomycin.
Immunocytochemistry
PAEC monolayers were fixed and permeabilized with 90% ice-cold methanol in phosphate-buffered saline, and cells were blocked with 10% normal goat serum, 0.1% BSA, and 20 mM glycine in Hanks’s buffered salt solution (HBSS) for 1 hour. Primary antibody to pan-CN A was added at a 1∶100 dilution at 4°C overnight. >FITC (fluorescein isothiocyanate)-conjugated secondary antibody was used at a 1∶100 dilution for 1 hour at room temperature. Fluorescent images were obtained with a Leica TCS SP2 confocal laser scanning microscope fitted with a 63× water immersion objective.
Western blot analysis
Whole-cell lysates were made with radio immunoprecipitation assay (RIPA) lysis buffer (Boston Bioproducts, Ashland, MA) plus 1% of 10× protease inhibitor cocktail. Cell membrane/cytoskeleton extractions were performed as described by Cioffi et al.,7 modified from the methods of Lockwich et al.35 Briefly, confluent PAEC monolayers were lysed in hypotonic buffer (0.1 M Tris chloride [pH 8.0], 1 mM MgCl2, 10% protease inhibitor cocktail [10×, Sigma]), and pelleted at 3000 g (24 minutes, 4°C). Cell pellets were resuspended in sucrose buffer (10 mM Tris base [pH 7.4], 250 mM sucrose, 0.9 mM dithiothreitol, 10% protease inhibitor cocktail) and subjected to Dounce homogenization. The homogenized samples were spun at 3000 g for 17 minutes at 4°C, and the resultant supernatant was collected and centrifuged at 50,000 g (30 minutes, 4°C). Octylglucoside/potassium iodide (KI) extraction buffer (50 mM Tris base [pH 7.5], 150 mM NaCl, 5.2 mM EDTA (ethylenediaminetetraacetic acid), 50 mM octylglucoside, 100 mM KI, 10% protease inhibitor cocktail) was then used to extract the pellet. The final centrifugation (145,000 g, 50 minutes, 4°C) resolved two fractions: a pellet fraction, consisting of the cytoskeletal proteins and any protein strongly attached to them, and the supernatant fraction, containing the dissolved phospholipids, plasma membrane proteins, and any protein strongly attached to them. Whole-cell lysates and detergent extractions were subjected to electrophoresis on 4%–12% bis-tris gels (Invitrogen) and transferred to nitrocellulose membranes at 350 mA. Membranes were incubated with primary antibodies overnight at 4°C and then with appropriate HRP-conjugated secondary antibodies for 1 hour. Chemiluminescence detection of proteins was visualized using either Supersignal West Pico or West Femto chemiluminescent substrates (Thermo Scientific, Waltham, MA).
Ca2+ measurements and patch-clamp electrophysiology
PAECs were seeded onto 35-mm glass coverslips and grown to confluence. Changes in intracellular calcium levels were determined as previously described36 using the fluorophore fura 2-acetoxymethyl ester (Fura 2/AM; Life Technologies, Grand Island, NY). Transmembrane currents were measured using conventional whole-cell voltage-clamp configuration as previously described.3,5,7,11,36 Briefly, transmembrane currents in single, electrically isolated cells were measured with an EPC-9 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). Pulse+PulseFit software (ver. 8.5, HEKA Elektronik) was used to generate the voltage stimuli and to acquire the current signals, which were filtered at 2.9 kHz. The currents were evoked by step depolarization (i.e., 200-millisecond testing pulses ranging from −100 to +60 mV in 20-mV increments) from a holding potential of 0 mV; currents were measured as the mean value of the current amplitude during the last 20 milliseconds of each step. The standard pipette solution contained (in mmol/L) 130 N-methyl-D-glucamine, 10 Hepes, 2 Mg2+-ATP (adenosine triphosphate), 1 N-phenylanthranilicacid, 0.1 5-nitro-2-(3-phenylpropylamino)benzoic acid, 2 EGTA (ethylene glycol tetraacetic acid), and 1 Ca(OH)2 (pH 7.2, adjusted with methane sulfonic acid); the free [Ca2+] was estimated as 100 nmol/L, as calculated by the CaBuf program (G. Droogmans, ftp://ftp.cc.kuleuven.ac.be/pub/droogmans/cabuf.zip). To examine Ca2+ currents, the bath (external) solution contained (in mmol/L) 120 aspartic acid, 5 Ca(OH)2, 5 CaCl2, 10 Hepes, and 0.5 3,4-diaminopyridine (pH 7.4, adjusted with tetraethylammonium hydroxide). All solutions were adjusted to 290–300 mOsm with sucrose. Recording pipettes were made of hemo capillaries (Warner Instrument, Hamden, CT), pulled by a 2-stage puller (PC-10; Narishige, Tokyo), and heat-polished with a microforge (MF-200; World Precision Instruments, Sarasota, FL). Pipette resistance was in the range of 3–5 MΩ when filled with the standard pipette solution. All of the experiments were performed at room temperature (22°–25°C). Data are expressed as mean ± SEM for the number of cells (n) in which whole-cell patch-clamp recordings were obtained.
Co-immunoprecipitation
Immunoprecipitation experiments were performed using the Pierce Co-Immunoprecipitation Kit (Thermo Fisher Scientific, Rockford, IL) following manufacturer’s instructions. In brief, TRPC4 antibody was covalently attached to activated agarose beads using sodium cyanoborohydride. Whole-cell lysate was rotated with antibody-coupled beads overnight at 4°C. Input pass-through was collected, and beads were washed. Proteins were eluted from beads with a 1% sodium dodecylsulfate (SDS) treatment for 5 minutes, repeated twice. Protein eluates were then subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblot analysis for phosphoserine immunoreactivity.
NFAT luciferase reporter gene assays
PAECs were plated at a density of 1 × 105 cells/well in 6-well dishes. Approximately 24 hours later, cells were transfected with 2 μg DNA/well of the IL-2 NFAT reporter gene33 via Superfect transfection reagent in complete medium for 3 hours. Cells were washed, fresh medium was added, and cells were incubated for an additional 18–24 hours. Cells were then pretreated with 50 μM CIP or vehicle for 1 hour, followed by treatment with 1 μM thapsigargin for 7 hours with or without CIP, and harvested in 300 μL ice-cold Monolight lysis buffer (BD Biosciences). Luciferase activity was measured with a Monolight 2010 luminometer (Analytical Luminescence Laboratory, San Diego, CA). Results were normalized to total protein concentration of lysates, as measured with the Thermo Scientific BCA protein assay.
STIM1 siRNA knockdown
For knockdown of STIM1 in PAECs, siRNA to the target sequence 5′-TTCGGTTGTCGAAGGAGGTAA-3′ of the rat STIM1 gene (NM_001108496) was purchased from QIAGEN. PAECs were grown to ∼30–40% confluency, and siRNA to STIM1 or scrambled siRNA was introduced to cells via HiPerFect transfection reagent (QIAGEN) in serum- and antibiotic-free medium. After 3 hours, complete medium was added, cells were incubated with siRNA at a final concentration of 20 nM for 72 hours, and intracellular [Ca2+] was measured. Knockdown efficiency was validated by Western blot.
Time-lapse microscopy
PAECs were grown on 35-mm glass coverslips to confluence and imaged in 2 mM Ca2+ HBSS every 5 seconds for 30 minutes with a Zeiss Axiovert D-1 microscope fitted with a 40× oil immersion objective. Thapsigargin was added at approximately frame 12 (1 minute), and time-lapse photography continued for an additional ∼348 frames (29 additional minutes). Representative photographs are shown at frame 0 and frame 300 (Fig. 7).
Figure 7.
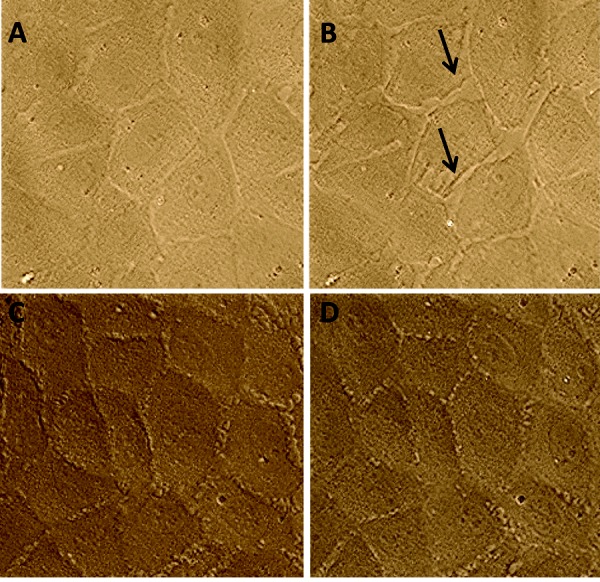
Calcineurin inhibitory peptide (CIP) reduces thapsigargin-induced intercellular gap formation in pulmonary artery endothelial cells (PAECs). A, PAECs grown on glass coverslips exhibit confluent monolayer at time 0. B, Thapsigargin treatment induces intercellular gap formation, as denoted by arrows. C, PAECs exhibit confluent monolayer after 1-hour pretreatment with 50 μM CIP. D, CIP-pretreated cells do not form intercellular gaps after thapsigargin treatment.
Statistical analysis
Statistical analysis was performed using GraphPad, version 5.0, software (San Diego, CA). Comparisons between multiple groups were performed with a one-way ANOVA and Newman-Keuls multiple-comparison post hoc test. Comparisons between two groups were analyzed with a Student’s T test. Values were considered significantly different when P < 0.05.
Results
CN is expressed in PAECs
To determine whether CN is present in PAECs, whole-cell lysates were made and Western blots were performed. By use of a pan-CN A antibody, CN was found to be expressed in PAECs (Fig. 1A). CN has been shown to be present both in the cytosol37-39 and at the membrane37,40 of a variety of cell types. For CN to play a significant role in the regulation of the ISOC channel in PAECs, it follows that at least part of the CN population localizes to the cell membrane. To test this, we prepared membrane-cytoskeletal fractions of PAECs.7 These fractions were then extracted with octylglucosidase in the presence of KI. Ocytlglucosidase is used to solubilize the plasma membrane including caveolae, and KI is used to cleave membrane proteins from the cytoskeleton such that the cytoskeleton resolves in the pellet fraction and membrane proteins are found in the supernatant fraction. Conversely, the supernatant fraction retains membrane and tightly associated proteins. CN resolved in the supernatant fraction of PAECs (Fig. 1B), indicating that CN complexes with the membrane or membrane proteins. Finally, to confirm our fractionation results, we performed immunocytochemistry on PAECs. Cells were fixed and permeabilized before staining with CN antibody, and confocal images were acquired. We found that there is strong membrane staining for CN at cell-cell borders (Fig. 1C, 1D), consistent with our membrane fractionation data.
Figure 1.
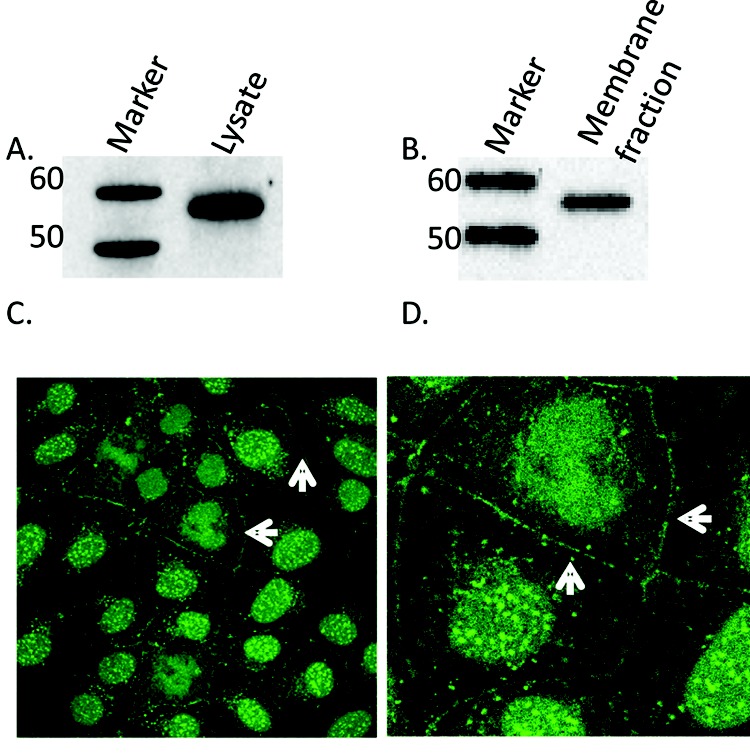
Localization of calcineurin (CN) in pulmonary artery endothelial cells (PAECs). A, Western blot analysis of CN in whole-cells lysates of PAECs. Whole-cell lysates of PAECs were run on 4%–12% bis-tris gels, transferred onto nitrocellulose, and subjected to Western blot analysis with antibody to pan-CN A. A ∼57-kDa band corresponding to CN A was observed. B, Immunoblot showing CN in supernatant detergent-extracted membrane fraction using octylglucoside/KI. CN A was found in the membrane fraction of PAECs. C, Immunocytochemical staining for CN in PAECs. Fluorescent images were obtained with a Leica TCS SP2 confocal laser scanning microscope fitted with a 63× water immersion objective. Arrows highlight membrane localization of positive CN staining. D, Enlarged detail of cell-cell border staining in C.
Inhibition of CN attenuates SOC entry in PAECs
A CIP mimicking the autoinhibitory domain of CN has proven to be a useful tool for the study of CN interactions and signaling.29-32 To determine whether CN inhibition affects global SOC entry, we used a Fura 2/AM approach. PAECs were loaded with Fura 2/AM, washed, and then treated with 50 μM CIP or vehicle control in 2 mM Ca2+ buffer for 1 hour. After the incubation, buffer was replaced with low-Ca2+ buffer and 50 μM CIP or vehicle control. ER Ca2+ release was initiated by treatment with thapsigargin, and SOC entry was revealed by addition of 2 mM Ca2+. While calcium release was unchanged between treatments, PAECs treated with CIP exhibited an attenuated SOC entry, compared with untreated cells (Fig. 2A).
Figure 2.
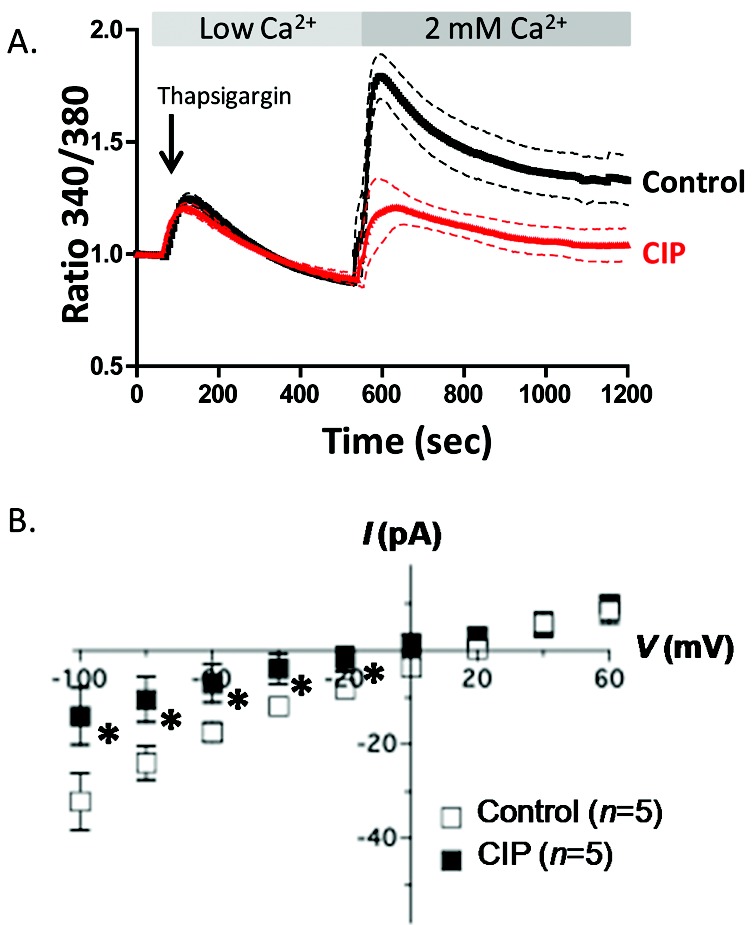
Calcineurin inhibitory peptide (CIP) attenuates store-operated calcium (SOC) entry and decreases ISOC in pulmonary artery endothelial cells (PAECs). A, PAECs were loaded with Fura 2/AM (fura 2-acetoxymethyl ester) and pretreated with 50 μM CIP or vehicle alone for 1 hour, and changes in intracellular [Ca2+] were measured. SOC entry was revealed by thapsigargin treatment in low-Ca2+ buffer. Upon 2 mM Ca2+ add-back, CIP-treated PAECs exhibited a decreased SOC (red tracing) compared to controls (black tracing). Each tracing represents mean ± SEM of at least 4 experiments with 2 regions of interest of 10–20 cells for each. B, Single cells were pretreated with CIP for 1–2 hours and examined in a whole-cell patch-clamp configuration with CIP added to the patch pipette. The ISOC channel was activated by thapsigargin in the patch pipette. Cells were held at 0 mV and stepped from −100 to +100 mV for 200 milliseconds. CIP treatment attenuated ISOC at all negative testing potentials, as compared to control cells. Asterisk indicates significant differences from control cells.
ISOC is decreased in cells treated with CIP
Because Fura 2/AM measures global calcium entry but does not differentiate between SOC entry channels, we used electrophysiology to measure the ISOC directly. Because global SOC entry is reduced in cells treated with CIP, we sought to determine whether CIP specifically affects ISOC in PAECs. Cells were pretreated with CIP for 1–2 hours, and CIP was added to the patch pipette in a whole-cell voltage-clamp configuration. Cells were held at 0 mV and stepped from −100 to +100 mV for 200 milliseconds. Compared with control cells, PAECs treated with CIP displayed a significantly attenuated ISOC at all negative testing potentials (Fig. 2B).
CN inhibition reduces thapsigargin-induced CN activity in PAECs
To determine whether CIP attenuates CN activity in PAECs, we used an interleukin-2/nuclear factor of activated T cells (IL-2/NFAT) luciferase reporter assay that measures CN-specific phosphatase activity.33 PAECs expressing the reporter gene were stimulated with thapsigargin, and luciferase activity was measured. Our results show that PAECs possess CN activity that increases upon thapsigargin treatment. This CN-specific phosphatase activity is significantly reduced in CIP-treated PAECs (Fig. 3).
Figure 3.
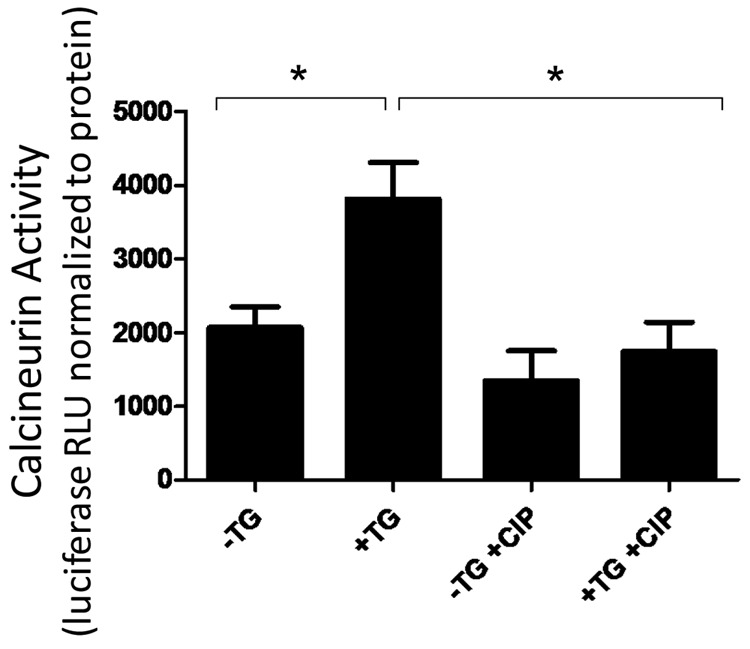
Calcineurin inhibitory peptide (CIP) reduces thapsigargin (TG)-induced calcineurin (CN) activity in pulmonary artery endothelial cells (PAECs). PAECs were transiently transfected with a nuclear factor of activated T cells (NFAT)–responsive luciferase reporter plasmid, an indicator of CN phosphatase activity. CN activity is shown as relative light units (RLU) of luciferase normalized to total protein. CN activity increased upon TG treatment, and this increase was attenuated in CIP-treated cells. Each bar represents the mean + SEM of at least 3 independent experiments. Asterisk indicates significant differences from TG-treated cells.
An ∼85-kDa protein corresponding to STIM1 is phosphorylated upon thapsigargin treatment in PAECs
To uncover phosphorylation changes in response to ER store depletion, PAECs were treated with thapsigargin for 1 or 5 minutes or left untreated. Whole-cell lysates were immediately prepared following each thapsigargin treatment, and Western blot analysis was performed. Using a monoclonal phosphoserine antibody, a prominent band was observed at ∼85 kDa that increased following thapsigargin treatment (Fig. 4A). On a separate membrane, we also probed for STIM1 and observed a band at ∼85 kDa (Fig. 4A). Next, immunoprecipitation experiments were performed. PAECs were treated ± thapsigargin for 1 minute, whole-cell lysates were collected, and STIM1 was immunoprecipitated. SDS-PAGE and Western blots were run on immunoprecipitate elutions and probed with phosphoserine antibody. We resolved a phosphospecific band at 85 kDa (Fig. 4B), indicating that the immunoprecipitated STIM1 was phosphorylated upon thapsigargin treatment. Because thapsigargin is an irreversible inhibitor, we also examined phosphorylation in PAECs after thrombin treatment over time. Thrombin, a receptor-mediated inflammatory agonist, activates SOC entry via Ins(1,4,5)P3-dependent Ca2+ release and allows for reuptake of calcium into the ER.41 PAECs were treated with thrombin for 1, 5, 7, and 10 minutes, and whole-cell lysates were made. We found that activation of SOC entry in PAECs by thrombin induced phosphorylation of an ∼85-kDa protein that then decreased over time (Fig. 4C). Taken together, these data suggest that STIM1 is phosphorylated immediately following SOC entry and then dephosphorylated within minutes, likely to prepare the channel for reactivation.
Figure 4.

Stromal interaction molecule 1 (STIM1) is phosphorylated in pulmonary artery endothelial cell (PAECs). A, Western blot analysis of phosphoserine-immunoreactive proteins in whole-cell lysates of PAECs. Whole-cell lysates of PAECs treated with and without thapsigargin (+TG and −TG, respectively) for 1, 3, and 5 minutes were run on 4%–12% bis-tris gels, transferred onto nitrocellulose, and subjected to Western blot analysis with antibody to phosphoserine residues. An ∼85-kDa band was observed that was increased upon TG treatment. Actin is shown as loading control. Below, independent immunoblot of whole-cell lysates, showing STIM1 immunoreactivity at ∼85 kDa by use of a polyclonal antibody to STIM1. B, STIM1 was immunoprecipitated from PAEC whole-cell lysates, subjected to Western blot analysis, and probed for phosphoserine immunoreactivity. A band corresponding to phospho-STIM1 at ∼85 kDa was observed. E1, elution, STIM1 immunoprecipitate. C, Western blot analysis of phosphoserine-immunoreactive proteins in whole-cell lysates of PAECs treated with or without thrombin (+Th and −Th, respectively) for 1, 3, 5, 7, and 10 minutes and run on 4%–12% bis-tris gels, transferred onto nitrocellulose, and subjected to Western blot analysis with antibody to phosphoserine residues. Actin is shown as loading control.
Because CIP reduced global CN activity of PAECs in response to thapsigargin, we next looked at CN-dependent phosphorylation changes of membrane proteins in PAECs. PAECs were treated with thapsigargin at the 0- and 5-minute time points and harvested for membrane/cytoskeleton preparation as described in “Methods,” and Western blot analysis was performed with a phosphoserine-specific antibody. The prominent phosphoserine-reactive band at ∼85 kDa increased in intensity upon thapsigargin treatment (Fig. 5A). Treatment of PAECs with CIP changed the phosphorylation profile of this band (Fig. 5A). In the presence of CIP, the ∼85-kDa band was more intense at 0 and 5 minutes in the thapsigargin treatment, as compared with that in the non-CIP-treated cells. An increase in phosphorylation after thapsigargin treatment was evident in the non-CIP-treated cells. However, this increase was not seen in the CIP-treated cells, as baseline phosphorylation was high, perhaps because phosphorylation sites were saturated because of the inhibitor. Indeed, densitometry analysis of independent membrane preparations showed that cells treated with CIP did not exhibit an increase in phosphorylation in response to thapsigargin at 85 kDa, as compared to control cells (Fig. 5B). Taken together, these data suggest a role for CN in the regulation of thapsigargin-induced SOC entry.
Figure 5.
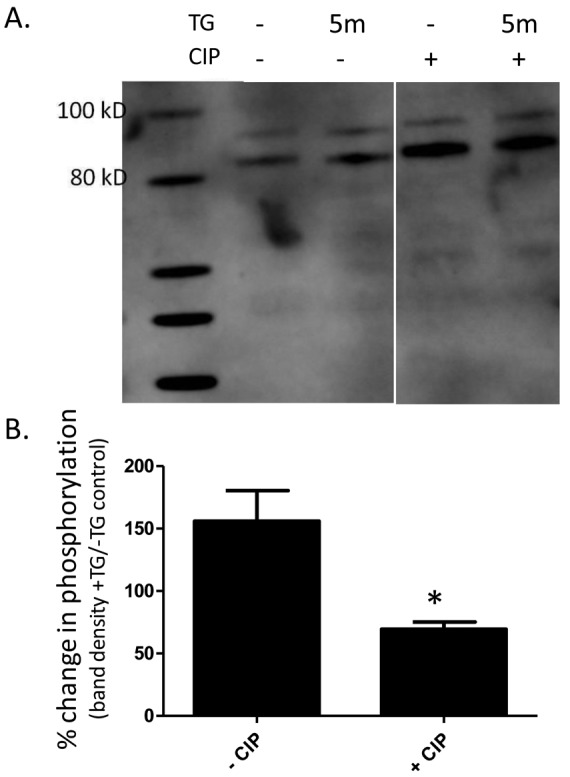
Calcineurin inhibitory peptide (CIP) increases phosphorylation of stromal interaction molecule 1 (STIM1) in pulmonary artery endothelial cells (PAECs). A, Western blot analysis of CIP-treated cells. Supernatant octylglucoside/KI-extracted membrane fractions of cells treated with or without 5-minute thapsigargin (TG) treatment and with or without CIP and subjected to Western blot analysis with antiphosphoserine antibody. A phosphospecific band at ∼85 kDa was observed that increased in intensity with TG and/or CIP treatment. B, Western blot densitometry results from A. ImageJ software was used to measure the densitometry of bands. Data are represented as percent change in phosphorylation, based on band density of TG-treated cells or untreated cells. Each bar represents the mean ± SEM of 2 independent membrane preparations. Asterisk indicates significant difference from non-CIP-treated controls.
STIM1 knockdown attenuates SOC entry in PAECs
Our data show that inhibition of CN attenuates SOC entry in PAECs, that STIM1 is phosphorylated upon SOC activation by thapsigargin, and that CN inhibition augments the phosphorylation of STIM1. To investigate whether CN is acting on STIM1 in SOC entry in PAECs, we used an siRNA approach to reduce the expression of STIM1 and treated cells with or without CIP. Cells were treated with siRNA to STIM1 or a scrambled-siRNA control for at least 72 hours, then treated with or without CIP for 1 hour, and SOC entry was measured. STIM1 expression was reduced by more than half (Fig. 6A, 6B). Treatment with thapsigargin revealed no difference in calcium release between groups, while STIM1 siRNA-treated cells exhibited attenuated SOC entry, compared to those treated with the scrambled control (Fig. 6C, 6D). Importantly, there was no difference in SOC entry between control or CIP-treated cells and STIM1 siRNA-and-CIP-treated cells. These data suggest that CN contributes to regulation of STIM1 endothelial SOC entry.
Figure 6.

Stromal interaction molecule 1 (STIM1) knockdown attenuates store-operated calcium (SOC) in pulmonary artery endothelial cells (PAECs). PAECs were transfected with 20 nM siRNA (small interfering RNA) to knockdown STIM1 using HiPerFect transfection reagent. Control cells received 20 nM scrambled (Scram) siRNA. A, Representative Western blots showing STIM1 levels in PAECs with and without siRNA treatment. Actin is shown as loading control. B, Densitometry analysis of siRNA-treated PAECs. An asterisk indicates significant differences from non-siRNA-treated control cells. C, Thapsigargin-induced SOC entry was decreased in PAECs treated with siRNA- and CIP-treated cells. PAECs received 20 nM of siRNA to knockdown STIM1 for >72 hours (or scrambled control). Cells were loaded with Fura 2/AM (fura 2-acetoxymethyl ester) and pretreated with 50 μM calcineurin inhibitory peptide (CIP) or vehicle alone for 1 hour, and changes in intracellular [Ca2+] were measured. SOC entry was initiated by thapsigargin treatment in low-Ca2+ buffer. Upon 2 mM Ca2+ add-back, cells treated with siRNA to STIM1 exhibited decreased SOC (red tracing) compared to controls (black tracing). CIP treatment had no effect on this decrease in SOC entry in STIM1 siRNA-treated cells (green tracing). Each tracing represents the average of at least 3 experiments (mean ± SEM) with 2 regions of interest of 10–20 cells for each. D, Graphical representation of calcium tracings in C at peak SOC entry (600 seconds).
CN inhibition decreases thapsigargin-induced gap formation in PAECs
Inhibition of CN by CIP decreased global calcium entry and reduced ISOC. In order to examine the physiological effects of CN inhibition on PAECs, we employed time-lapse microscopy of PAECs treated with thapsigargin and with or without CIP to determine whether thapsigargin induced interendothelial cell gap formation in PAECs. Figure 7A shows the PAEC monolayer before thapsigargin treatment in the absence of CIP. Thapsigargin treatment induced interendothelial cell gap formation in this cell layer (Fig. 7B). In cells pretreated with CIP, however, gap formation was reduced following thapsigargin stimulation. Thus, CN inhibition reduces interendothelial cell gap formation in PAECs.
Discussion
Results from this study reveal that the calcium/calmodulin-activated phosphatase CN is involved in the regulation of SOC entry in PAECs. CN, which was originally identified in bovine brain tissue,42 is a highly conserved phosphatase, and genes for both the CN A and CN B subunits are present in all eukaryotic organisms (reviewed by Rusnak and Mertz15). CN is found in a wide array of mammalian tissues and has a broad subcellular distribution, with a long list of substrates and cellular functions.15 Among these, CN phosphatase activity is known to regulate a number of ion channels in various cell types, for example, voltage-gated Ca2+ channels in the brain and L-type Ca2+ channels in smooth muscle cells.15
Calcium entry through SOC channels across the plasma membrane contributes significantly to endothelial barrier disruption, and specifically, activation of the calcium-selective ISOC channel leads to endothelial cell gap formation.3 An understanding of the mechanisms surrounding activation/inactivation of ISOC is far from complete. It is known that inactivation of ISOC appears to be phosphorylation dependent,12 yet the molecular details are unknown. The ISOC channel is made up of at least TRPC4 and TRPC1 subunits, and TRPC4 is necessary for proper function. The C-terminus of TRPC4 contains within it a number of sequences that may be involved in protein-protein interactions. A number of these regions are homologous to the C-termini of other TRPC proteins, including a proline-rich region residing near the channel pore43 and a more distal calmodulin/InsP3 receptor binding site.44 In addition, TRPC4 and TRPC1 contain 2 calmodulin binding sites, and calmodulin binding may be involved in inactivation of TRPC1.14,45 In the ISOC channel, these sites may dock calmodulin in a “ready” position for CN activation, which requires the binding of calcium and calmodulin (reviewed by Rusnak and Mertz15).
STIM1 can interact with TRPC1/4 to regulate endothelial SOC entry.27 In the CRAC channel, STIM1 initiates SOC entry via puncta formation at the ER membrane and subsequent union with the plasma membrane-bound SOC channels following calcium depletion (reviewed by Wu et al.26). Further, it was recently shown that Orai1 interacts with TRPC4 in the endogenous ISOC channel to control activation and calcium selectivity.46 In the CRAC channel, STIM1 interacts directly with Orai1 to allow calcium entry (reviewed by Hewavitharana et al.28). However, in the TRPC1/TRPC4 channel, Sundivakkam et al.27 found that while STIM1 is necessary for proper channel function, its interaction with Orai1 is not required. These observations lead to questions as to the ternary complex molecular arrangement of TRPC1/4-STIM1-Orai1.
Pozo-Guisado et al.23 very recently showed that STIM1 is phosphorylated at Ser575, Ser608, and Ser621 by ERK 1/2 (extracellular signal-regulated kinases 1/2) during calcium store depletion and suggested that the reversible phosphorylation of STIM1 controls SOC entry. This group found that phosphorylation at these sites actually promotes SOC entry, which opposes data revealing that phosphorylation of STIM1 is responsible for the inactivation of SOC entry.21,25 Sundivakkam et al.25 demonstrated that in pulmonary microvascular endothelial cells, however, phosphorylation of STIM1 suppresses SOC entry. This apparent discrepancy may be due to the phosphorylation of different residues on STIM1,21,23 suggesting that this is a very dynamic and complex regulation. Indeed, STIM1 has also been shown to be modified by O-linked N-acetylglucosamine (O-GlcNAc),47 a carbohydrate moiety attached at serine and/or threonine residues that can act as a nutrient and stress sensor.48 O-GlcNAcylation and phosphorylation can often occur on the same serine or threonine residues, requiring extensive crosstalk between the two processes. Increased STIM1 O-GlcNAcylation was shown to attenuate SOC entry in cardiomyocytes, and interestingly, inhibition of O-GlcNAcylation increased phosphorylation of STIM1 in these cells.47 This highlights the importance of identifying key signaling pathways, not only in the phosphorylation of STIM1 but also in its dephosphorylation, as regulators of SOC entry.
Data suggest that inactivation of ISOC is phosphorylation dependent. In HSG (human submandibular gland) cells, treatment with the potent protein kinase C inhibitor staurosporine delayed inactivation of ISOC.12 Here, we showed that STIM1, resolving at ∼85 kDa, is phosphorylated upon thapsigargin treatment and that inhibition of CN increases the phosphorylation of an ∼85-kDa protein, suggesting that STIM1 phosphorylation is regulated at least in part by CN. We found that CN is expressed in PAECs and can be found at the cell membrane, as evidenced by Western blot analysis of membrane fraction preparations and immunocytochemistry. That CN plays a role in the regulation of endothelial SOC entry was supported by decreased global SOC entry and decreased ISOC in CIP-treated PAECs. Finally, thapsigargin-induced gap formation was decreased in PAECs pretreated with CIP. These data have important clinical relevance in understanding mechanisms underlying endothelial health, especially in light of recent discoveries. Spiekerkoetter and colleagues49 found that inhibition of CN activity, at least in part, reversed aberrant bone morphogenic protein receptor-2 (BMPR-2) signaling in PAECs from patients with idiopathic pulmonary arterial hypertension (PAH) and reversed severe PAH in a rat model of PAH.
In conclusion, our studies introduce CN as a regulator of endothelial SOC entry in PAECs. While it is known that the phosphorylation status of ISOC plays a role in its inactivation, the signaling molecules important for this dynamic phosphorylation have not been determined. Here we show that CN is a phosphatase involved in the regulation of ISOC. Further, we suggest that STIM1 is a principal target of CN. Future studies will investigate how STIM1 interacts with TRPC4/TRPC1/Orai1 in PAECs and whether STIM1 dephosphorylation by CN is regulated by O-GlcNAcylation.
Acknowledgments
We would like to thank Drs. Jonathan Scammell and Troy Stevens for invaluable scientific input and manuscript review, as well as Linn Ayers and Anna Buford for endothelial cell isolation and culture.
Source of Support: This work was supported by National Institutes of Health grants 1F32HL112565–01, R00HL089361, and R01HL107778–01A1.
Conflict of Interest: None declared.
References
- 1.Parekh AB, Putney JW Jr. Store-operated calcium channels. Physiol Rev 2005;85:757–810. [DOI] [PubMed]
- 2.Cioffi DL, Wu S, Stevens T. Transient receptor potential cation channels and store-operated Ca2+ channels. In: Yuan JX-J, ed. Ion channels in pulmonary vasculature. Lung Biology in Health and Disease, no. 197. Boca Raton, FL: Taylor & Francis, 2005:125–146.
- 3.Wu S, Cioffi EA, Alvarez D, Sayner SL, Chen H, Cioffi DL, King J, et al. Essential role of a Ca2+-selective, store-operated current (ISOC) in endothelial cell permeability: determinants of the vascular leak site. Circ Res 2005;96:856–863. [DOI] [PubMed]
- 4.Cioffi DL, Lowe K, Alvarez DF, Barry C, Stevens T. TRPing on the lung endothelium: calcium channels that regulate barrier function. Antioxid Redox Signal 2009;11:765–776. [DOI] [PMC free article] [PubMed]
- 5.Brough GH, Wu S, Cioffi D, Moore TM, Li M, Dean N, Stevens T. Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway. FASEB J 2001;15:1727–1738. [PubMed]
- 6.Freichel M, Suh SH, Pfeifer A, Schweig U, Trost C, Weissgerber P, Biel M, et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat Cell Biol 2001;3:121–127. [DOI] [PubMed]
- 7.Cioffi DL, Wu S, Alexeyev M, Goodman SR, Zhu MX, Stevens T. Activation of the endothelial store-operated ISOC Ca2+ channel requires interaction of protein 4.1 with TRPC4. Circ Res 2005;97:1164–1172. [DOI] [PubMed]
- 8.Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol 2008;586:419–425. [DOI] [PMC free article] [PubMed]
- 9.Penna A, Demuro A, Yeromin AV, Zhang SL, Safrina O, Parker I, Cahalan MD. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 2008;456:116–120. [DOI] [PMC free article] [PubMed]
- 10.Madl J, Weghuber J, Fritsch R, Derler I, Fahrner M, Frischauf I, Lackner B, Romanin C, Schutz GJ. Resting state Orai1 diffuses as homotetramer in the plasma membrane of live mammalian cells. J Biol Chem 2010;285:41135–41142. [DOI] [PMC free article] [PubMed]
- 11.Cioffi DL, Wu S, Chen H, Alexeyev M, St Croix CM, Pitt BR, Uhlig S, Stevens T. Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ Res 2012;110:1435–1444. [DOI] [PMC free article] [PubMed]
- 12.Liu X, O’Connell A, Ambudkar IS. Ca2+-dependent inactivation of a store-operated Ca2+ current in human submandibular gland cells: role of a staurosporine-sensitive protein kinase and the intracellular Ca2+ pump. J Biol Chem 1998;273:33295–33304. [DOI] [PubMed]
- 13.Litjens T, Harland ML, Roberts ML, Barritt GJ, Rychkov GY. Fast Ca2+-dependent inactivation of the store-operated Ca2+ current (ISOC) in liver cells: a role for calmodulin. J Physiol 2004;558:85–97. [DOI] [PMC free article] [PubMed]
- 14.Singh BB, Liu X, Tang J, Zhu MX, Ambudkar IS. Calmodulin regulates Ca2+-dependent feedback inhibition of store-operated Ca2+ influx by interaction with a site in the C terminus of TrpC1. Mol Cell 2002;9:739–750. [DOI] [PubMed]
- 15.Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev 2000;80:1483–1521. [DOI] [PubMed]
- 16.Aitken A, Klee CB, Cohen P. The structure of the B subunit of calcineurin. Eur J Biochem 1984;139:663–671. [DOI] [PubMed]
- 17.Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell 1995;83:463–472. [DOI] [PubMed]
- 18.Kim JY, Saffen D. Activation of M1 muscarinic acetylcholine receptors stimulates the formation of a multiprotein complex centered on TRPC6 channels. J Biol Chem 2005;280:32035–32047. [DOI] [PubMed]
- 19.Turner JD, Thomas AP, Reeves JP, Hantash BM. Calcineurin activation by slow calcium release from intracellular stores suppresses protein kinase C regulation of L-type calcium channels in L6 cells. Cell Calcium 2009;46:242–247. [DOI] [PubMed]
- 20.Kar P, Nelson C, Parekh AB. Selective activation of the transcription factor NFAT1 by calcium microdomains near Ca2+ release-activated Ca2+ (CRAC) channels. J Biol Chem 2011;286:14795–14803. [DOI] [PMC free article] [PubMed]
- 21.Smyth JT, Petranka JG, Boyles RR, DeHaven WI, Fukushima M, Johnson KL, Williams JG, Putney JW Jr. Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat Cell Biol 2009;11:1465–1472. [DOI] [PMC free article] [PubMed]
- 22.Yu F, Sun L, Machaca K. Orai1 internalization and STIM1 clustering inhibition modulate SOCE inactivation during meiosis. Proc Natl Acad Sci USA 2009;106:17401–17406. [DOI] [PMC free article] [PubMed]
- 23.Pozo-Guisado E, Casas-Rua V, Tomas-Martin P, Lopez-Guerrero AM, Alvarez-Barrientos A, Martin-Romero FJ. Phosphorylation of STIM1 at ERK1/2 target sites regulates interaction with the microtubule plus-end binding protein EB1. J Cell Sci 2013;126:3170–3180. [DOI] [PubMed]
- 24.Lopez E, Jardin I, Berna-Erro A, Bermejo N, Salido GM, Sage SO, Rosado JA, Redondo PC. STIM1 tyrosine-phosphorylation is required for STIM1-Orai1 association in human platelets. Cell Signal 2012;24:1315–1322. [DOI] [PubMed]
- 25.Sundivakkam PC, Natarajan V, Malik AB, Tiruppathi C. Store-operated Ca2+ entry (SOCE) induced by protease-activated receptor-1 mediates STIM1 protein phosphorylation to inhibit SOCE in endothelial cells through AMP-activated protein kinase and p38β mitogen-activated protein kinase. J Biol Chem 2013;288:17030–17041. [DOI] [PMC free article] [PubMed]
- 26.Wu MM, Luik RM, Lewis RS. Some assembly required: constructing the elementary units of store-operated Ca2+ entry. Cell Calcium 2007;42:163–172. [DOI] [PMC free article] [PubMed]
- 27.Sundivakkam PC, Freichel M, Singh V, Yuan JP, Vogel SM, Flockerzi V, Malik AB, Tiruppathi C. The Ca2+ sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca2+ entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol Pharmacol 2012;81:510–526. [DOI] [PMC free article] [PubMed]
- 28.Hewavitharana T, Deng X, Soboloff J, Gill DL. Role of STIM and Orai proteins in the store-operated calcium signaling pathway. Cell Calcium 2007;42:173–182. [DOI] [PubMed]
- 29.Terada H, Matsushita M, Lu YF, Shirai T, Li ST, Tomizawa K, Moriwaki A, et al. Inhibition of excitatory neuronal cell death by cell-permeable calcineurin autoinhibitory peptide. J Neurochem 2003;87:1145–1151. [DOI] [PubMed]
- 30.Nolasco LH, Gushiken FC, Turner NA, Khatlani TS, Pradhan S, Dong JF, Moake JL, Vijayan KV. Protein phosphatase 2B inhibition promotes the secretion of von Willebrand factor from endothelial cells. J Thromb Haemost 2009;7:1009–1018. [DOI] [PMC free article] [PubMed]
- 31.Pandey D, Goyal P, Dwivedi S, Siess W. Unraveling a novel Rac1-mediated signaling pathway that regulates cofilin dephosphorylation and secretion in thrombin-stimulated platelets. Blood 2009;114:415–424. [DOI] [PubMed]
- 32.Henesy MB, Britain AL, Zhu B, Amable L, Honkanen RE, Corbin JD, Francis SH, Rich TC. Calcineurin regulates homologous desensitization of natriuretic peptide receptor-a and inhibits ANP-induced testosterone production in MA-10 cells. PLoS ONE 2012;7:e41711. [DOI] [PMC free article] [PubMed]
- 33.Gross KL, Cioffi EA, Scammell JG. Increased activity of the calcineurin-nuclear factor of activated T cells pathway in squirrel monkey B-lymphoblasts identified by PowerBlot. In Vitro Cell Dev Biol Anim 2004;40:57–63. [DOI] [PubMed]
- 34.Creighton JR, Masada N, Cooper DM, Stevens T. Coordinate regulation of membrane cAMP by Ca2+-inhibited adenylyl cyclase and phosphodiesterase activities. Am J Physiol Lung Cell Mol Physiol 2003;284:L100–L107. [DOI] [PubMed]
- 35.Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS. Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J Biol Chem 2000;275:11934–11942. [DOI] [PubMed]
- 36.Wu S, Sangerman J, Li M, Brough GH, Goodman SR, Stevens T. Essential control of an endothelial cell ISOC by the spectrin membrane skeleton. J Cell Biol 2001;154:1225–1233. [DOI] [PMC free article] [PubMed]
- 37.Kosiorek M, Podszywalow-Bartnicka P, Zylinska L, Zablocki K, Pikula S. Interaction of plasma membrane Ca2+-ATPase isoform 4 with calcineurin A: implications for catecholamine secretion by PC12 cells. Biochem Biophys Res Commun 2011;411:235–240. [DOI] [PubMed]
- 38.Sun L, Blair HC, Peng Y, Zaidi N, Adebanjo OA, Wu XB, Wu XY, et al. Calcineurin regulates bone formation by the osteoblast. Proc Natl Acad Sci USA 2005;102:17130–17135. [DOI] [PMC free article] [PubMed]
- 39.Usuda N, Arai H, Sasaki H, Hanai T, Nagata T, Muramatsu T, Kincaid RL, Higuchi S. Differential subcellular localization of neural isoforms of the catalytic subunit of calmodulin-dependent protein phosphatase (calcineurin) in central nervous system neurons: immunohistochemistry on formalin-fixed paraffin sections employing antigen retrieval by microwave irradiation. J Histochem Cytochem 1996;44:13–18. [DOI] [PubMed]
- 40.Lukyanetz EA. Evidence for colocalization of calcineurin and calcium channels in dorsal root ganglion neurons. Neuroscience 1997;78:625–628. [DOI] [PubMed]
- 41.Goligorsky MS, Menton DN, Laszlo A, Lum H. Nature of thrombin-induced sustained increase in cytosolic calcium concentration in cultured endothelial cells. J Biol Chem 1989;264:16771–16775. [PubMed]
- 42.Klee CB, Crouch TH, Krinks MH. Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc Natl Acad Sci USA 1979;76:6270–6273. [DOI] [PMC free article] [PubMed]
- 43.Cioffi DL, Wu S, Stevens T. On the endothelial cell ISOC. Cell Calcium 2003;33:323–336. [DOI] [PubMed]
- 44.Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of Trp channels. J Biol Chem 2001;276:21303–21310. [DOI] [PMC free article] [PubMed]
- 45.Trost C, Bergs C, Himmerkus N, Flockerzi V. The transient receptor potential, TRP4, cation channel is a novel member of the family of calmodulin binding proteins. Biochem J 2001;355:663–670. [DOI] [PMC free article] [PubMed]
- 46.Cioffi DL, Wu S, Chen H, Alexeyev C, St Croix CM, Pitt BR, Uhlig S, Stevens T. Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ Res 2012;110:1435–1444. [DOI] [PMC free article] [PubMed]
- 47.Zhu-Mauldin X, Marsh SA, Zou L, Marchase RB, Chatham JC. Modification of STIM1 by O-linked N-acetylglucosamine (O-GlcNAc) attenuates store-operated calcium entry in neonatal cardiomyocytes. J Biol Chem 2012;287:39094–39106. [DOI] [PMC free article] [PubMed]
- 48.Hu P, Shimoji S, Hart GW. Site-specific interplay between O-GlcNAcylation and phosphorylation in cellular regulation. FEBS Lett 2010;584:2526–2538. [DOI] [PubMed]
- 49.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 2013;123:3600–3613. [DOI] [PMC free article] [PubMed]