

Abstract Abstract
Phosphodiesterase-5 (PDE5) is the primary phosphodiesterase in the pulmonary vasculature. It degrades cyclic guanosine monophosphate (cGMP) and inhibits cGMP-mediated vasorelaxation. We previously reported that hydrocortisone treatment decreased hyperoxia-induced PDE5 activity and markers of oxidative stress in lambs with persistent pulmonary hypertension of the newborn (PPHN) ventilated with 100% O2. The objective of our study was to determine the molecular mechanism by which hydrocortisone downregulates PDE5 and oxidative stress in fetal pulmonary artery smooth muscle cells (FPASMCs) from PPHN lambs. PPHN FPASMC were incubated for 24 hours in either 21% or 95% O2. Some cells were treated with 100 nM hydrocortisone and/or ±1 μM helenalin, an inhibitor of nuclear factor κ B (NFκB), a redox-sensitive transcription factor. Exposure to hyperoxia led to increased PDE5 activity, oxidative stress, and NFκB activity. Pretreatment of PPHN FPASMC with hydrocortisone normalized PDE5 activity, decreased cytosolic oxidative stress, increased expression of extracellular superoxide dismutase and NFκB inhibitory protein, and decreased NFκB activity. Similarly, treatment with NFκB inhibitor, helenalin, decreased PDE5 activity. These data suggest that hyperoxia activates NFκB, which in turn induces PDE5 activity in PPHN FPASMC, whereas treatment with hydrocortisone attenuates these changes by blocking reactive oxygen species–induced NFκB activity.
Keywords: hydrocortisone, PDE5, PPHN, oxidative stress, NFκB
Introduction
Persistent pulmonary hypertension of the newborn (PPHN) is the product of a failed pulmonary vascular transition to extrauterine life and results in elevated pulmonary artery pressures, hypoxemia, and respiratory insufficiency. PPHN is an important cause and consequence of hypoxic respiratory failure in term and late preterm newborns and affects up to 1 in 500 term neonates.1 In addition to being associated with a substantial risk of mortality, PPHN is also associated with significant short-term and long-term morbidities.2-4 Commonly used PPHN therapies include mechanical ventilation, oxygen, and the use of pulmonary vasodilators, such as inhaled nitric oxide (iNO).5,6 Although iNO improves oxygenation and reduces the need for extracorporeal membrane oxygenation, it does not improve survival, and up to 40% of infants do not respond or do not sustain their response to NO.7,8 Although hypoxemia often prompts the administration of high concentrations of oxygen for infants with PPHN, a growing body of literature demonstrates that even brief periods of hyperoxia trigger oxidative stress as well as changes in enzyme activity and vascular reactivity.9-11 Thus, new therapies are needed for newborns with PPHN.
Disruptions of the NO–cyclic guanosine monophosphate (cGMP) signaling pathway play an important role in pathogenesis of PPHN. Activation of NO leads to production of cGMP, a critical second messenger for smooth muscle relaxation. cGMP is in turn inactivated by the cGMP-specific phosphodiesterase, or phosphodiesterase-5 (PDE5), the predominant phosphodiesterase in lung vasculature and an important therapeutic target in PPHN.12 Although high concentrations of oxygen are widely used to promote pulmonary vasodilation in infants with PPHN, new evidence indicates that such therapy may upregulate PDE513,14 and paradoxically increase pulmonary vascular contractility.15 We previously demonstrated that ventilation of healthy neonatal sheep with 100% O2 for 24 hours increases PDE5 protein expression and activity in the resistance pulmonary arteries (PAs).13 Similarly, exposure of cultured fetal PA smooth muscle cells (FPASMCs) to 24 hours of hyperoxia increases PDE5 expression and activity, changes that can be reversed by treatment with the antioxidant N-acetylcysteine.13 Moreover, PPHN may independently alter expression and activity of key enzymes in the NO-cGMP pathway. For example, ventilation with 100% oxygen increases PDE5 activity in PAs of PPHN lambs more than in control lambs,13,14 and basal PDE5 activity is higher in FPASMC isolated from PPHN relative to healthy lambs.16
Hyperoxia produces lung injury in part through increased formation of reactive oxygen species (ROS). One consequence of increased ROS is activation of nuclear factor κ B (NFκB), a redox-sensitive transcription factor that plays an important role in immune and inflammatory responses. NFκB activation has been described in states of increased oxidative stress,17,18 whereas treatment with antioxidants decreases NFκB activity.19-21 NFκB activation has been implicated in clinical diseases that include pulmonary arterial hypertension (PAH) in adults22 and bronchopulmonary dysplasia in preterm infants.23 Recent evidence from a lamb model of PPHN suggests that basal NFκB activity is elevated,24 but the effect of hyperoxia on NFκB activity in PPHN is not known.
Glucocorticoids are used in neonatal intensive care to treat a variety of diseases, including adrenal insufficiency and pressor-resistant hypotension. Although steroids have been used in adults with connective tissue diseases associated with pulmonary arterial hypertension, there are very few data regarding their potential benefits in neonatal pulmonary hypertension. Glucocorticoids have been found to improve oxygenation and attenuate the pulmonary hypertensive response in animal models of meconium aspiration syndrome,25 a common cause of PPHN. In case reports from human neonates, steroids have been reported to decrease hospital stay and duration of oxygen use after meconium aspiration.26-28 More recently, new data have emerged on the use of steroids in animal models of PPHN. Prenatally administered betamethasone attenuated oxidative stress and improved in vitro PA response to vasodilators in a fetal lamb model of pulmonary hypertension.29 Methylprednisolone improved oxygenation and attenuated the pulmonary hypertensive response in a porcine model of meconium aspiration.25 We recently demonstrated improved oxygenation and decreased PDE5 activity in PPHN lambs treated with hydrocortisone.30 Given the wide variety of potential effects of steroids, it is not yet clear what specific mechanisms are responsible for the effects of hydrocortisone on PDE5. We recently reported that NFκB activity is elevated in fetal lambs with pulmonary hypertension.24 Because glucocorticoids have been reported to antagonize NFκB activity,31 and there are possible binding sites for NFκB on the PDE5 promoter,32 we hypothesized that NFκB is a potential target of hydrocortisone in PPHN. The goal of our study was to test the hypothesis that hydrocortisone normalizes PDE5 activity by reducing oxidative stress and NFκB.
Methods
Cell culture
Primary cultures of FPASMCs from intrapulmonary arteries of fetal lambs with PPHN induced by in utero ductal ligation were prepared as previously described.16 All cultures were maintained in Dulbecco modified Eagle medium supplemented with 1 g/L glucose (Mediatech), 10% fetal bovine serum (Hyclone), antibiotics (Mediatech), and antimycotics (Mediatech) at 37°C in a humidified atmosphere with 5% CO2–95% air. All experiments were performed using cells at between passage 2 and 4, because we have previously characterized that these cells maintain their PPHN phenotype through passage 4.16 PPHN FPASMCs were treated in incubators with 21% O2–5% CO2 or 95% O2–5% CO2 with or without 100 nM hydrocortisone (Sigma) or 1 μM helenalin (Calbiochem). On the basis of dose-titration studies (data not shown), we used a 100-nM concentration of hydrocortisone, which is equivalent to levels achieved after physiologic replacement dosing in neonates.33 PPHN FPASMC were harvested for analysis after 24 hours using 1X Mg-lysis buffer (Upstate) supplemented with a protease inhibitor cocktail (Sigma) and a phosphatase inhibitor cocktail (EMD Biosciences). Cell extracts were sonicated, and protein concentration was determined using the Bradford assay.34
PDE5 activity assay
After treatment, PPHN FPASMCs were harvested, and protein was prepared as described above. Protein extracts were purified over a Centri-Spin 10 column to remove any phosphate contamination (Princeton Separations). Protein concentration was determined using the Bradford method. Total protein (5 μg) was assayed for cGMP hydrolytic activity using a commercially available colorimetric cyclic nucleotide phosphodiesterase assay kit (Enzo) with and without sildenafil (Sigma) as described previously.13 Results are shown as the PDE5-specific picomoles of cGMP hydrolyzed per milligram total protein per minute.
Detection of reactive oxygen species
PPHN FPASMCs were infected with 100 plaque-forming units per cell of a ratiometic green fluorescent protein (roGFP) adenoviral construct as previously described.13 roGFP is a previously characterized ratiometric fluorescent probe sensitive to cellular oxidative stress.35,36 To create this probe, surface-exposed residues in green fluorescent protein (GFP) were replaced with cysteine residues capable of forming disulfide bonds. Assessment of fluorescence ratios therefore provides a real-time measure of cysteine thiol redox status in live cells. roGFP-infected PPHN FPASMCs were exposed to 21% O2–5% CO2 or 95% O2–5% CO2 for 24 hours, with and without 100 nM hydrocortisone, and their oxidative status was subsequently analyzed using multilaser flow cytometry as previously described.13,36
Plasmid DNA transfection and luciferase assays
NFκB promoter activity was determined using the pGL4.32 (luc2P/NF-κB-RE/Hygro) plasmid (Promega). PPHN FPASMC were cotransfected with 4 μg of plasmid DNA and 0.1 μg pRL-CMV Vector (Promega) on a 10-cm2 tissue culture plate at 90% confluence, using lipofectamine (Gibco) according to the manufacturer’s instructions. After 24 hours, cells were split onto 6-well plates and allowed to adhere. Luciferase activity in protein extracts was determined 72 hours after transfection using the Dual-Luciferase Reporter Assay System (Promega) and a Femtomaster FB12 luminometer (Zylux). Activity was normalized to the internal renilla luciferase control to correct for differences in transfection efficiencies. PPHN FPASMCs were treated in incubators with 21% O2–5% CO2 or 95% O2–5% CO2 with or without 100 nM hydrocortisone (Sigma) for 24 hours before luciferase assays.
Western blot analysis
After treatment, PPHN FPASMCs were harvested for total protein (40 μg). Protein concentration was measured using the Bradford assay. PDE5 and NFκB inhibitory protein (IκB) expression were assessed via Western blot, which was performed as previously described.13 Membranes were blocked for 1 hour at room temperature with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween 20 (1X TBST) and incubated overnight at 4°C with primary antibody in 5% milk plus 1X TBST at an appropriate dilution (1:500 for mouse anti-PDE5 [BD Transduction], 1∶500 for goat anti-IκB [Santa Cruz Biotechnology], 1∶1,000 for rabbit anti-Nox1 [Santa Cruz Biotechnology], 1∶1000 for rabbit anti-Nox4 [Santa Cruz Biotechnology], 1∶1,000 for rabbit anti-extracellular superoxide dismutase [ecSOD; Enzo Life Sciences], and 1∶2,000 for mouse β-actin [Sigma]). The membranes were washed and incubated with the appropriate secondary antibody diluted 1∶1,000 in 5% milk plus 1X TBST. Membranes were then washed and exposed via chemiluminescence (Pierce). Bands were analyzed using a Digital Science Image Station (Kodak). Expression was normalized to β-actin. Data are shown as fold relative to 21% untreated FPASMC.
Statistical analysis
All data are expressed as the mean ± SEM. Results were analyzed by one-way analysis of variance with Bonferroni multiple comparison test where appropriate, using Prism software (GraphPad). Statistical significance was set at P < 0.05.
Results
Hydrocortisone normalizes hyperoxia-induced PDE5 activity in PPHN FPASMC
We have recently reported that hydrocortisone treatment of PPHN lambs ventilated with 100% O2 leads to normalization of pulmonary arterial PDE5 expression and activity relative to untreated PPHN lambs.30 In this study, we determined the effects of hydrocortisone treatment on PDE5 in hyperoxia-exposed FPASMC isolated from PPHN lambs. Similar to our recent report,16 24 hours of hyperoxia did not significantly alter PDE5 protein expression in PPHN FPASMC (Fig. 1). We also did not observe any changes in PDE5 expression after hydrocortisone treatment (Fig. 1). In contrast to expression, and in agreement with our previous work,13,16 hyperoxia induced a 2.4 ± 0.4 fold increase in PDE5 activity in PPHN FPASMC (P < 0.05). Hydrocortisone treatment significantly decreased PDE5 activity to levels equivalent to room air–exposed cells (P < 0.05; Fig. 2). Hydrocortisone did not alter PDE5 activity in PPHN FPASMC exposed to normoxia.
Figure 1.
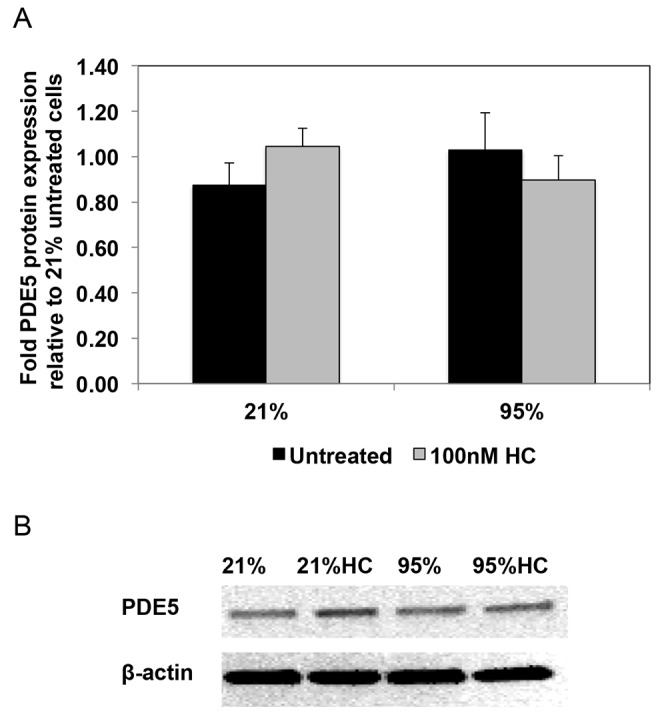
Hydrocortisone (HC) does not affect phosphodiesterase-5 (PDE5) protein expression in fetal pulmonary artery smooth muscle cells (FPASMCs) from lambs with persistent pulmonary hypertension of the newborn (PPHN). A, PPHN FPASMC were exposed to 21% O2–5% CO2 or 95% O2–5% CO2 ± 100 nM HC for 24 hours. Cells were harvested for total protein and subjected to PDE5 Western blot analysis with β-actin normalization. Data are shown as means ± SEM (n = 8 for all groups). B, Representative Western blots are shown for PDE5 and β-actin in PPHN FPASMC.
Figure 2.
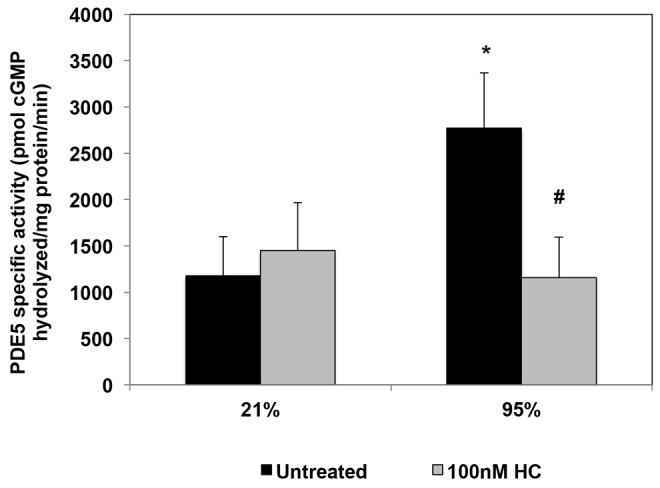
Hydrocortisone (HC) normalizes hyperoxia-induced phosphodiesterase-5 (PDE5) activity in fetal pulmonary artery smooth muscle cells (FPASMCs) from lambs with persistent pulmonary hypertension of the newborn (PPHN). PPHN FPASMC were exposed to 21% O2–5% CO2 or 95% O2–5% CO2 ± 100 nM HC for 24 hours, and total protein was harvested. PDE5-specific activity was measured as the sildenafil-inhibitable fraction of total cyclic guanosine monophosphate (cGMP) hydrolysis and normalized for total milligrams of protein. Data are shown as means ± SEM (n = 6 for 21% untreated and 95% plus 100 nM HC; n = 4 for 21% plus 100 nm HC and 95% untreated; read in duplicates). Asterisk indicates P < 0.05 versus 21% untreated; number sign indicates P < 0.05 versus 95% untreated.
Hydrocortisone attenuates cytosolic oxidative stress in PPHN FPASMC exposed to hyperoxia
We previously reported that the hyperoxia-mediated effects on PDE5 activity are likely mediated via increases in ROS.13 Hydrocortisone treatment of intact ventilated PPHN lambs decreased markers of oxidative stress and decreased PDE5 activity relative to untreated animals.30 In this study, we sought to directly determine the effects of hydrocortisone on oxidative stress in isolated PPHN FPASMC. Because we previously reported higher cytosolic protein thiol oxidation in FPASMC from PPHN lambs compared with control animals,37 we first investigated the effects of hyperoxia and hydrocortisone on cytosolic oxidative stress. After 24-hour exposure of FPASMC to 95% O2, cytosolic oxidative stress, as measured by the redox-sensitive probe roGFP, was increased by 1.7 ± 0.3 fold (P < 0.05; Fig. 3A). Hydrocortisone (100 nM) decreased cytosolic roGFP oxidation in PPHN FPASMC to levels equivalent to room air–exposed cells (P < 0.05; Fig. 3A). Although hyperoxia exposure also significantly increased mitochondrial matrix RoGFP oxidation (1.9 ± 0.2 fold; P < 0.05), hydrocortisone did not blunt oxidant stress in this compartment (Fig. 3B).
Figure 3.
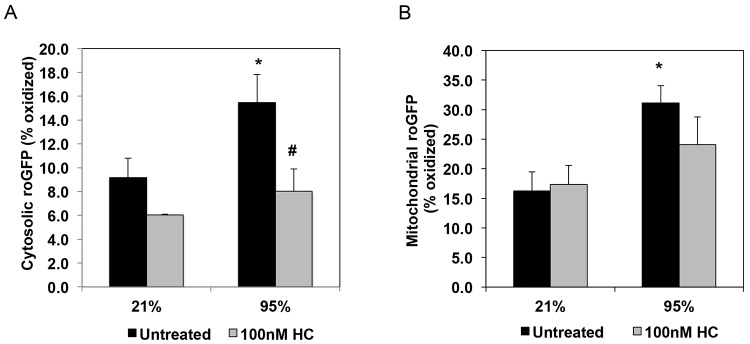
Hydrocortisone (HC) blocks hyperoxia-induced cytosolic reactive oxygen species (ROS) in fetal pulmonary artery smooth muscle cells (FPASMCs) from lambs with persistent pulmonary hypertension of the newborn (PPHN). The redox sensor ratiometic green fluorescent protein (roGFP) was used to assess oxidant levels in the cytosol (A) and mitochondrial matrix (B). A, PPHN FPASMC were exposed to 21% O2–5% CO2 or 95% O2–5% CO2 ± 100 nM HC for 24 hours, and oxidation of the cytosolic roGFP probe was measured (n = 4). B, PPHN FPASMC were exposed to 21% O2–5% CO2 or 95% O2–5% CO2 ± 100 nM HC for 24 hours, and oxidation of the mitochondrial roGFP probe was measured (n = 7). Data are shown as mean ± SEM for percentage maximal oxidation of roGFP. Asterisk indicates P < 0.05 versus 21% untreated; number sign indicates P < 0.05 versus 95% untreated.
Hydrocortisone increases ecSOD protein expression but does not affect Nox4 and Nox1 in hyperoxia-exposed PPHN FPASMC
To elucidate the potential mechanisms by which hydrocortisone attenuates oxidative stress in PPHN FPASMCs, we investigated its effects on the expression of NADPH oxidase subunits, Nox1 and Nox4. Nox1 has been shown to be an important contributor to hyperoxia-induced ROS production in mice,38 whereas Nox4 expression is upregulated in PAs and isolated PASMC from PPHN lambs.24 We observed no significant changes in Nox1 or Nox4 expression with hyperoxia and no effects with hydrocortisone treatment (Fig. 4). Next, we wanted to determine whether hydrocortisone affects the expression of SOD enzymes. The promoters of all SOD enzymes have been reported to contain glucocorticoid response elements.39,40 Although there were no significant changes in either CuZnSOD or MnSOD expression (data not shown), hydrocortisone increased ecSOD protein expression by 71% ± 26% (P < 0.05; Fig. 5) compared with untreated cells.
Figure 4.
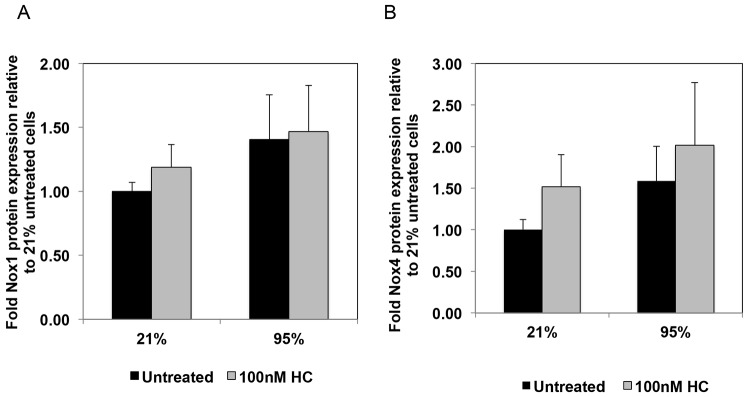
Hydrocortisone (HC) does not affect expression of Nox1 and Nox4 in fetal pulmonary artery smooth muscle cells (FPASMCs) from lambs with persistent pulmonary hypertension of the newborn (PPHN). PPHN FPASMC were exposed to 21% O2–5% CO2 or 95% O2–5% CO2 ± 100 nM HC for 24 hours. Cells were harvested for total protein and subjected to Nox1 (A) and Nox4 (B) Western blot analysis, with β-actin normalization. Data are shown as means ± SEM (Nox1: n = 11 for 21% untreated, 95% untreated, 95% plus 100 nM HC, n = 10 for 21% plus 100 nM HC; Nox4: n = 9 for 21% untreated and 95% untreated, n = 8 for 21% plus 100 nM HC, 95% plus 100 nM HC).
Figure 5.
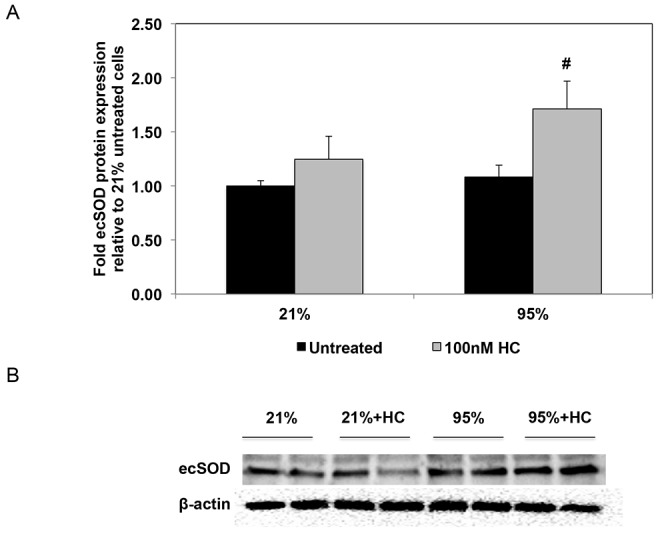
Hydrocortisone (HC) increases expression of extracellular superoxide dismutase (ecSOD) in fetal pulmonary artery smooth muscle cells (FPASMCs) from lambs with persistent pulmonary hypertension of the newborn (PPHN). A, PPHN FPASMC were exposed to 21% O2–5% CO2 or 95% O2–5% CO2 ± 100 nM HC for 24 hours. Cells were harvested for total protein and subjected to ecSOD Western blot analysis, with β-actin normalization. Data are shown as means ± SEM (n = 7 for 21% untreated, 21% plus 100 nM HC, 95% untreated, n = 8 for 95% plus 100 nM HC). Number sign indicates P < 0.05 versus 95% untreated. B, Representative Western blots are shown for ecSOD and β-actin in PPHN FPASMC.
Hydrocortisone decreases NFκB activity induced by exposure of PPHN FPASMC to hyperoxia
The ROS-activated transcription factor NFκB mediates responses to inflammatory and oxidant insults and offers a potential link between oxidant stress and subsequent lung injury. NFκB inhibition has been reported to attenuate hyperoxic lung injury in mice,22 and it decreased pulmonary hypertension and prevented right ventricular hypertrophy in a rat monocrotaline model of pulmonary hypertension.41 These findings led us to hypothesize that NFκB activation may play a role in neonatal pulmonary hypertension.24 Here we demonstrate that hyperoxia significantly increased NFκB-driven luciferase reporter activity in PPHN FPASMC by 2.2 ± 0.1 fold (P < 0.05; Fig. 6). Treatment with hydrocortisone blunted the increase in NFκB reporter activity by 30% ± 7% (P < 0.05; Fig. 6).
Figure 6.
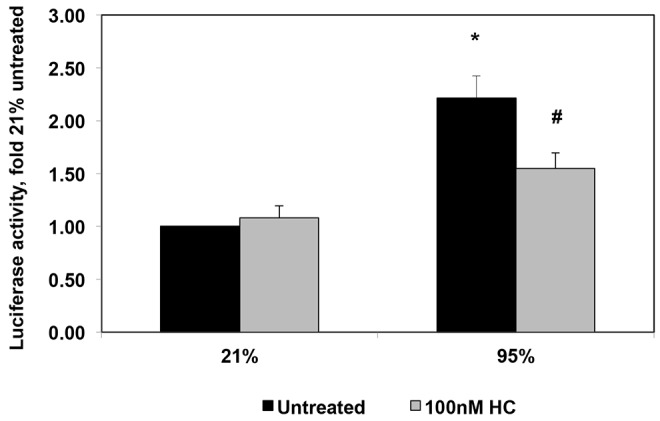
Hydrocortisone (HC) attenuates nuclear factor κ B (NFκB) activity induced by exposure to hyperoxia in fetal pulmonary artery smooth muscle cells (FPASMCs) from lambs with persistent pulmonary hypertension of the newborn (PPHN). PPHN FPASMC were transfected with a plasmid containing consensus NFκB sites upstream of a luciferase reporter. Forty-eight hours after transfection, PPHN FPASMC were exposed to 21% O2–5% CO2 ± 100 nM HC or 95% O2–5% CO2 ± 100 nM HC for 24 hours. Relative light units were determined in a luminometer and normalized to an internal renilla luciferase control plasmid. Data are shown as means ± SEM (n = 4 for all groups). Asterisk indicates P < 0.05 versus 21% untreated; number sign indicates P < 0.05 versus 95% untreated.
Hydrocortisone increases IκB protein expression in PPHN FPASMC
NFκB exists in the cytoplasm in an inactive form associated with regulatory IkB proteins. Following stimulation of the NFkB cascade, the inhibitory IkB is degraded, thereby allowing NFkB translocation into the nucleus where it serves as a transcription factor. Because IκB expression is an important regulator of NFκB activity, we sought to determine the effects of hyperoxia and hydrocortisone treatment on IκB expression. In contrast to previous studies in microvascular endothelial cells, which have shown decreased IκB expression in response to hyperoxia,42 we found that 24 hours of hyperoxia did not alter IκB protein expression in PPHN FPASMC (Fig. 7). In both normoxic and hyperoxic PPHN vascular smooth muscle cells, hydrocortisone treatment significantly increased IκB expression (P < 0.05; Fig. 7).
Figure 7.
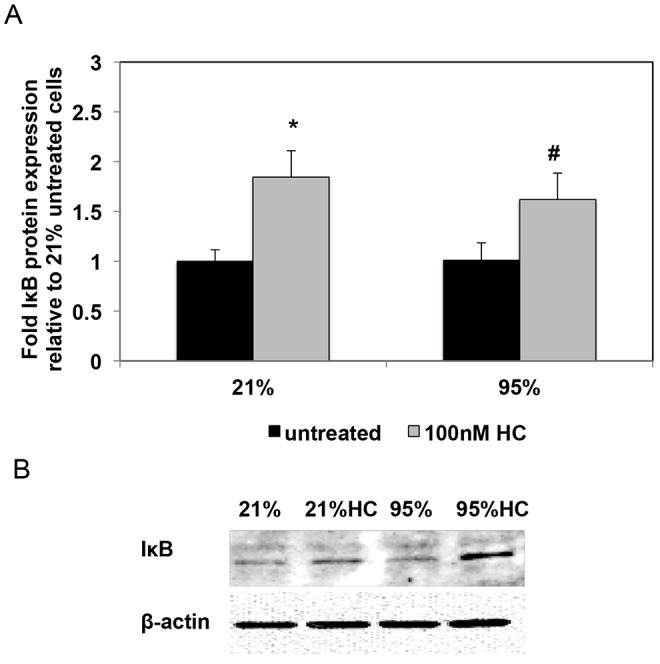
Hydrocortisone (HC) increases protein levels of the nuclear factor κ B (NFκB) inhibitory protein (IκB). A, Fetal pulmonary artery smooth muscle cells (FPASMCs) from lambs with persistent pulmonary hypertension of the newborn (PPHN) were exposed to 21% O2–5% CO2 or 95% O2–5% CO2 ± 100 nM HC for 24 hours. Cells were harvested for total protein and subjected to IκB Western blot analysis, with β-actin normalization. Data are shown as means ± SEM (n = 7 for all groups). Asterisk indicates P < 0.05 versus 21% untreated; number sign indicates P < 0.05 versus 95% untreated. B, Representative Western blots are shown for IκB and β-actin in PPHN FPASMC.
NFκB inhibition decreases hyperoxia-induced PDE5 activity but does not affect PDE5 protein expression in PPHN FPASMC
Based on our observations that hyperoxia upregulates both PDE5 activity and NFκB reporter activity in PPHN FPASMC, we next sought to determine the effects of NFκB inhibition on PDE5 expression and activity. Treatment with an inhibitor of NFκB, helenalin, did not affect PDE5 protein expression (Fig. 8A); however, it decreased hyperoxia-induced PDE5 activity to a similar extent as treatment with hydrocortisone (decrease by 69% ± 11% with helenalin and by 63% ± 17% with hydrocortisone; P < 0.05; Fig. 8B). Treatment of FPASMC with the combination of helenalin and hydrocortisone decreased PDE5 activity by 54% ± 15%, similar to levels seen with either agent alone (Fig. 8B).
Figure 8.

Helenalin (Hel), an nuclear factor κ B (NFκB) inhibitor, blocks hyperoxia-induced phosphodiesterase-5 (PDE5) activity in fetal pulmonary artery smooth muscle cells (FPASMCs) from lambs with persistent pulmonary hypertension of the newborn (PPHN). PPHN FPASMC were exposed to 21% O2–5% CO2 or 95% O2–5% CO2 ± 100 nM hydrocortisone (HC) ± 1 μM helenalin for 24 hours. A, Protein extracts were subjected to PDE5 Western blot analysis, with β-actin normalization. Data are shown as means ± SEM (n = 4 for all groups). B, PDE5-specific activity was measured as the sildenafil-inhibitable fraction of total cGMP hydrolysis and normalized for milligram of protein. Data are shown as means ± SEM (n = 4–8; read in duplicates). Asterisk indicates P < 0.05 versus 21% untreated; number sign indicates P < 0.05 versus 95% untreated.
Discussion
Although corticosteroids have been widely used in the neonatal intensive care unit for the treatment of pulmonary diseases such as bronchopulmonary dysplasia and have been suggested as possible therapy for neonates with meconium aspiration syndrome, little is known about their effects in PPHN. We recently reported that hydrocortisone improves oxygenation, decreases oxidative stress, and normalizes PDE5 activity in neonatal lambs with PPHN secondary to antenatal ductal ligation.30 In this study, we explored the molecular mechanisms for these effects by studying the effects of hydrocortisone on FPASMC isolated from PPHN lambs. We found that treatment with hydrocortisone decreased hyperoxia-induced PDE5 activity, decreased cytosolic oxidative stress, increased ecSOD expression, decreased NFκB reporter activity, and increased IκB expression. Our findings also suggest that the hydrocortisone-induced reduction in NFκB activation is at least partly responsible for the decrease in PDE5 activity.
We have reported a number of important differences between PASMC from PPHN lambs and PASMC from healthy control lambs, including higher basal PDE5 activity, lower cGMP response to NO, higher basal levels of oxidant stress in the mitochondrial matrix and cytosol, and higher NFκB activity.16,24,37 Given these differences, we used FPASMC isolated from PPHN lambs as a tool to investigate the aberrant vascular signaling pathways characteristic of PPHN and to determine the effect of hyperoxia on these pathways. Consequently, we investigated the effects of hydrocortisone in PPHN FPASMC to gain mechanistic insights into its beneficial effects on vascular function in intact PPHN lambs. First, we determined the effects of treatment with physiologic concentrations of hydrocortisone on PDE5 expression and activity. Unlike our reported findings in cells from normal late-gestation fetal lambs,13,16 hyperoxia did not increase PDE5 protein expression in FPASMC from PPHN lambs (Fig. 1). However, in contrast to expression, PDE5 activity was dramatically upregulated by hyperoxia, and hydrocortisone completely blocked this increase (Fig. 2). This is similar to our recent findings that hydrocortisone normalizes PDE5 activity in resistance PAs of PPHN lambs ventilated with 100% O2 for 24 hours.30
We previously reported that PDE5 activity is upregulated by increased ROS and that this effect is reversed by treatment with antioxidants in intact neonatal PPHN lambs and in smooth muscle cells isolated from them.13,14,16 Therefore, we next investigated the effects of hyperoxia with and without hydrocortisone on oxidative stress in two subcellular compartments, using a previously characterized redox-sensitive probe, roGFP. As might be expected, exposure of PPHN FPASMC to hyperoxia for 24 hours resulted in increased oxidative stress, both in the cytosol and in the mitochondrial matrix (Fig. 3). We recently reported that hydrocortisone decreases vascular oxidative stress in ventilated lambs with PPHN,30 and in this study, we now report that hydrocortisone treatment selectively decreases cytosolic oxidative stress in pulmonary vascular smooth muscle without affecting mitochondrial matrix ROS (Fig. 3). We hypothesized that this effect of hydrocortisone was due to either decreased production of ROS or induction of antioxidant enzymes. Although we observed a trend toward hyperoxia-mediated increases in expression of NADPH subunits, Nox1 and Nox4, we did not find any significant effects of hydrocortisone treatment on their expression (Fig. 4). Hydrocortisone treatment did, however, increase expression of one of the superoxide dismutase enzymes, ecSOD (Fig. 5). Interestingly, we previously demonstrated that PPHN is associated with diminished ecSOD activity.37 We hypothesize that increased expression of ecSOD in steroid-treated FPASMC might reduce oxidant stress in response to hyperoxia, in agreement with studies in rodents that have shown that overexpression of ecSOD attenuates hyperoxic lung injury.43
One potential consequence of increased oxidative stress is activation of NFκB, a key transcription factor implicated in the pathogenesis of conditions associated with increased ROS,19,44 including several pulmonary diseases such as primary pulmonary hypertension45 and the development of chronic lung disease of prematurity.18,46,47 NFκB inhibition has been shown to prevent monocrotaline-induced pulmonary hypertension in rodents,41 and we recently reported that its activity is elevated in fetal lambs with chronic intrauterine pulmonary hypertension.24 In this study, we found that hyperoxia exposure significantly increased NFκB activity in PPHN FPASMC and that this effect was attenuated by treatment with hydrocortisone (Fig. 6). Interestingly, a recent report demonstrated NFkB inhibition by dexamethasone in tracheobronchial lavage fluid of premature infants with respiratory distress.47
To understand the mechanism responsible for hydrocortisone’s effects on NFkB, we studied protein expression of IκB, an important inhibitor of NFκB. In contrast to earlier studies that have demonstrated IκB degradation in hyperoxia in endothelial cells,42,48 24 hours of hyperoxia did not induce significant differences in IκB expression in PPHN FPASMC (Fig. 7). In agreement with decreased NFκB reporter activity, we observed that hydrocortisone increased IκB protein expression in both normoxic and hyperoxic PPHN FPASMC (Fig. 7). This is similar to a previous report that another steroid, methylprednisolone, increases IκB expression in pulmonary endothelial cells.48 Of note, although hydrocortisone increased IκB expression in both normoxia and hyperoxia-exposed cells, decreased NFκB activity was observed only in hyperoxia-exposed cells. The level of NFκB activity in normoxia-treated cells was similar for both hydrocortisone-treated and untreated cells, regardless of the induction of IκB expression by hydrocortisone. This suggests that the basal NFκB activity present in normoxic cells is essential for cell functioning and cannot be further inhibited by increased IκB expression.
Finally, we investigated the effects of NFκB inhibition on PDE5 activity after hyperoxia exposure. Although the link between NFκB and PDE5 has not been well studied, analysis of the PDE5 promoter demonstrates putative NFκB binding sites.32 In addition, other investigators have shown that treatment of human PASMCs with an inhibitor of IκB degradation reduces PDE5 transcript levels.49 In our studies, we observed no differences in PDE5 protein with either hydrocortisone or with NFκB inhibitor (Fig. 8A). PDE5 activity, however, was attenuated by treatment with either hydrocortisone or NFκB inhibitor alone as well as treatment with both agents (Fig. 8B). This decrease resulted in activity levels similar to basal activity in cells cultured under normoxic conditions (Fig. 8B).
The data presented here, taken together with our findings in ventilated lambs with PPHN, strongly suggest a possible role for glucocorticoids in the treatment of PPHN. We speculate that, by increasing ecSOD expression, steroids reduce ROS-mediated induction of NFκB activity and PDE5 activity (Fig. 9). The speculation that increases in ecSOD expression attenuate NFκB are in agreement with other studies that have demonstrated that ecSOD overexpression blocks NFκB activity in mouse epithelial cells subjected to oxidative stress.50 However, questions remain about the interplay between steroids, oxidant stress, PDE5, and NFκB. Although there are possible NFκB binding sites on the PDE5 promoter, we did not find evidence for NFκB upregulation of PDE5 expression under conditions of increased oxidative stress. We also found no significant changes in PDE5 expression after hydrocortisone or with NFκB inhibition, which suggests that the effects of these agents on PDE5 activity may be via posttranslational modification. It is also unclear whether anti-inflammatory properties of hydrocortisone play a role in decreasing PDE5 activity. Although the role of inflammation in PPHN is not well understood, inflammation is increasingly being recognized as an important factor in adult idiopathic pulmonary arterial hypertension.51 A recent report also suggests that PDE inhibitors may have direct anti-inflammatory effects in hyperoxic lung injury.52
Figure 9.
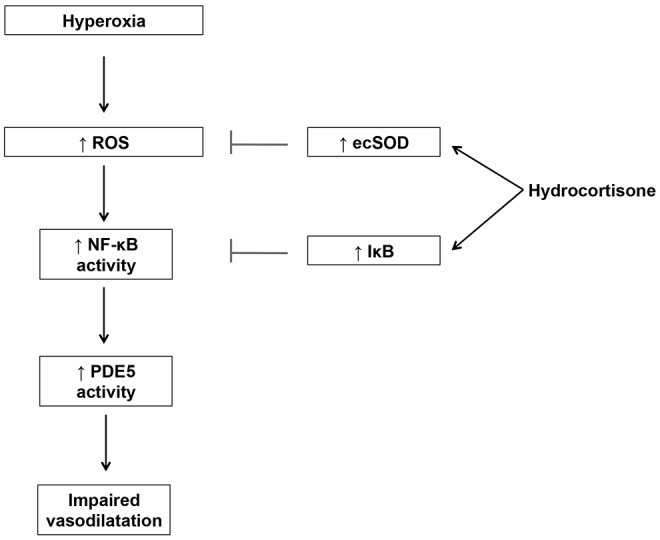
Model for the effects of hydrocortisone on phosphodiesterase-5 (PDE5) in fetal pulmonary artery smooth muscle cells (FPASMCs) from lambs with persistent pulmonary hypertension of the newborn (PPHN). Exposure to hyperoxia leads to increased reactive oxygen species (ROS) production, resulting in increased nuclear factor κ B (NFκB) activity with subsequent induction of PDE5 activity. Hydrocortisone disrupts this pathway by increasing extracellular superoxide dismutase (ecSOD) expression, leading to attenuation of ROS signaling and normalization of PDE5 activity. IκB: NFκB inhibitory protein.
There are several limitations to our study. All of our experiments were conducted in FPASMC isolated from lambs with pulmonary vascular remodeling due to chronic intrauterine pulmonary hypertension. As we have previously demonstrated,16 PPHN FPASMC have a different phenotype from cells isolated from healthy fetal lambs. Therefore, they provide a valuable tool to investigate the dysregulated signaling pathways that produce PPHN and to determine the cellular effects of therapeutic agents, such as hydrocortisone. We also acknowledge that, although our studies provide new insights into potential pathways by which hydrocortisone reduces PDE5 activity, the exact molecular mechanisms by which hyperoxia activates PDE5 activity remain unclear. Additional studies are needed to determine whether the changes we have observed with hydrocortisone are genomic in nature, because steroids have been shown to have both genomic and nongenomic effects.
In conclusion, we have demonstrated that hydrocortisone normalizes PDE5 activity, decreases cytosolic oxidative stress, increases ecSOD expression, and increases IκB expression and decreases NFκB activity in FPASMC cultured from PPHN lambs. We propose a mechanism by which hydrocortisone attenuates ROS production by induction of ecSOD, which results in decreased NFκB activity and normalization of PDE5 activity (Fig. 9). These results confirm our initial in vivo findings that hydrocortisone has a direct effect on NO-cGMP signaling in vascular smooth muscle30 and represent the first description of NFκB-mediated regulation of PDE5 activity in the setting of neonatal pulmonary hypertension. Our findings provide mechanistic insights for the improvement in oxygenation observed when PPHN lambs are treated with hydrocortisone. Furthermore, the finding that hydrocortisone leads to decreased PDE5 activity supports its potential use as a therapeutic agent in PPHN, a disease associated with increased oxidative insults and aberrations of the NO signaling pathway, including PDE5. Clinical trials to evaluate the role of hydrocortisone in PPHN are warranted.
Acknowledgments
We thank Dr. Paul T. Schumacker for providing the roGFP virus.
Source of Support: This work was supported by a Child Health Research Career Development Award of Lurie Children’s Hospital (to M.P.) and the National Institutes of Health (R03 HD060138-01 to S.L., K08 HL086715 to K.N.F., HL109478 to K.N.F., and HL54705 to R.H.S.). This work was also supported by the Northwestern University Flow Cytometry Facility and a Cancer Center Support Grant (National Cancer Institute CA060553).
Conflict of Interest: None declared.
References
- 1.Walsh-Sukys MC, Tyson JE, Wright LL, et al. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: practice variation and outcomes. Pediatrics 2000;105:14–20. [DOI] [PubMed]
- 2.Konduri GG, Kim UO. Advances in the diagnosis and management of persistent pulmonary hypertension of the newborn. Pediatr Clin North Am 2009;56:579–600. [DOI] [PMC free article] [PubMed]
- 3.Steinhorn RH. Neonatal pulmonary hypertension. Pediatr Crit Care Med 2010;11:S79–S84. [DOI] [PMC free article] [PubMed]
- 4.Lakshminrusimha S. The pulmonary circulation in neonatal respiratory failure. Clin Perinatol 2012;39:655–683. [DOI] [PMC free article] [PubMed]
- 5.Steinhorn RH. Therapeutic approaches using nitric oxide in infants and children. Free Rad Biol Med 2011;51:1027–1034. [DOI] [PMC free article] [PubMed]
- 6.Steinhorn RH. Pharmacotherapy for pulmonary hypertension. Pediatr Clin North Am 2012;59:1129–1146. [DOI] [PMC free article] [PubMed]
- 7.Clark RH, Kueser TJ, Walker MW, et al. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical Inhaled Nitric Oxide Research Group. New Engl J Med 2000;342:469–474. [DOI] [PubMed]
- 8.Neonatal Inhaled Nitric Oxide Study Group. Inhaled nitric oxide in full-term and nearly full-term infants with hypoxic respiratory failure. New Engl J Med 1997;336:597–604. [DOI] [PubMed]
- 9.Lakshminrusimha S, Russell JA, Steinhorn RH, et al. Pulmonary arterial contractility in neonatal lambs increases with 100% oxygen resuscitation. Pediatr Res 2006;59:137–141. [DOI] [PMC free article] [PubMed]
- 10.Vento M, Saugstad OD. Oxygen supplementation in the delivery room: updated information. J Pediatr 2011;158:e5–e7. [DOI] [PubMed]
- 11.Lakshminrusimha S, Steinhorn RH, Wedgwood S, et al. Pulmonary hemodynamics and vascular reactivity in asphyxiated term lambs resuscitated with 21 and 100% oxygen. J Appl Physiol 2011;111:1441–1447. [DOI] [PMC free article] [PubMed]
- 12.Farrow KN, Steinhorn RH. Phosphodiesterases: emerging therapeutic targets for neonatal pulmonary hypertension. Handb Exp Pharmacol 2011:251–277. [DOI] [PMC free article] [PubMed]
- 13.Farrow KN, Groh BS, Schumacker PT, et al. Hyperoxia increases phosphodiesterase 5 expression and activity in ovine fetal pulmonary artery smooth muscle cells. Circ Res 2008;102:226–233. [DOI] [PMC free article] [PubMed]
- 14.Farrow KN, Lakshminrusimha S, Czech L, et al. SOD and inhaled nitric oxide normalize phosphodiesterase 5 expression and activity in neonatal lambs with persistent pulmonary hypertension. Am J Physiol Lung Cell Mole Physiol 2010;299:L109–L116. [DOI] [PMC free article] [PubMed]
- 15.Lakshminrusimha S, Swartz DD, Gugino SF, et al. Oxygen concentration and pulmonary hemodynamics in newborn lambs with pulmonary hypertension. Pediatr Res 2009;66:539–544. [DOI] [PMC free article] [PubMed]
- 16.Farrow KN, Wedgwood S, Lee KJ, et al. Mitochondrial oxidant stress increases PDE5 activity in persistent pulmonary hypertension of the newborn. Respir Physiol Neurobiol 2010;174:272–281. [DOI] [PMC free article] [PubMed]
- 17.Michiels C, Minet E, Mottet D, Raes M. Regulation of gene expression by oxygen: NF-kappaB and HIF-1, two extremes. Free Rad Biol Med 2002;33:1231–1242. [DOI] [PubMed]
- 18.Wright CJ, Zhuang T, La P, Yang G, Dennery PA. Hyperoxia-induced NF-kappaB activation occurs via a maturationally sensitive atypical pathway. Am J Physiol Lung Cell Mol Physiol 2009;296:L296–L306. [DOI] [PMC free article] [PubMed]
- 19.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J 1991;10:2247–2258. [DOI] [PMC free article] [PubMed]
- 20.Amore A, Formica M, Giacchino F, et al. N-acetylcysteine in hemodialysis diabetic patients resets the activation of NF-kB in lymphomonocytes to normal values. J Nephrol 2013;26:778–786. [DOI] [PubMed]
- 21.Blackwell TS, Blackwell TR, Holden EP, Christman BW, Christman JW. In vivo antioxidant treatment suppresses nuclear factor-kappa B activation and neutrophilic lung inflammation. J Immunol 1996;157:1630–1637. [PubMed]
- 22.Liu ZQ, Liu B, Yu L, Wang XQ, Wang J, Liu HM. Simvastatin has beneficial effect on pulmonary artery hypertension by inhibiting NF-kappaB expression. Mol Cell Biochem 2011;354:77–82. [DOI] [PubMed]
- 23.Bourbia A, Cruz MA, Rozycki HJ. NF-kappaB in tracheal lavage fluid from intubated premature infants: association with inflammation, oxygen, and outcome. Arch Dis Child Fetal Neonatal Ed 2006;91:F36–F39. [DOI] [PMC free article] [PubMed]
- 24.Wedgwood S, Lakshminrusimha S, Czech L, Schumacker PT, Steinhorn RH. Increased p22phox/Nox4 expression is involved in remodeling through hydrogen peroxide signaling in experimental persistent pulmonary hypertension of the newborn. Antioxid Redox Signal 2013;18:1765–1776. [DOI] [PMC free article] [PubMed]
- 25.Soukka H, Halkola L, Aho H, Rautanen M, Kero P, Kaapa P. Methylprednisolone attenuates the pulmonary hypertensive response in porcine meconium aspiration. Pediatr Res 1997;42:145–150. [DOI] [PubMed]
- 26.Basu S, Kumar A, Bhatia BD, Satya K, Singh TB. Role of steroids on the clinical course and outcome of meconium aspiration syndrome-a randomized controlled trial. J Trop Pediatr 2007;53:331–337. [DOI] [PubMed]
- 27.Tripathi S, Saili A. The effect of steroids on the clinical course and outcome of neonates with meconium aspiration syndrome. J Trop Pediatr 2007;53:8–12. [DOI] [PubMed]
- 28.Ward M, Sinn J. Steroid therapy for meconium aspiration syndrome in newborn infants. Cochrane Database Syst Rev 2003:CD003485. [DOI] [PMC free article] [PubMed]
- 29.Chandrasekar I, Eis A, Konduri GG. Betamethasone attenuates oxidant stress in endothelial cells from fetal lambs with persistent pulmonary hypertension. Pediatr Res 2008;63:67–72. [DOI] [PubMed]
- 30.Perez M, Lakshminrusimha S, Wedgwood S, et al. Hydrocortisone normalizes oxygenation and cGMP regulation in lambs with persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol 2012;302:L595–L603. [DOI] [PMC free article] [PubMed]
- 31.Almawi WY, Melemedjian OK. Negative regulation of nuclear factor-kappaB activation and function by glucocorticoids. J Mol Endocrinol 2002;28:69–78. [DOI] [PubMed]
- 32.Pyne NJ, Murray F, Tate R, MacLean MR. Molecular determinants in pulmonary hypertension: the role of PDE5. In: Beavo JA, Francis SH, Houslay MD, eds. Cyclic Nucleotide Phosphodiesterases in Health and Disease. Boca Raton, FL: CRC/Taylor & Francis, 2007:510–511.
- 33.Watterberg KL, Scott SM. Evidence of early adrenal insufficiency in babies who develop bronchopulmonary dysplasia. Pediatrics 1995;95:120–125. [PubMed]
- 34.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254. [DOI] [PubMed]
- 35.Hanson GT, Aggeler R, Oglesbee D, et al. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem 2004;279:13044–13053. [DOI] [PubMed]
- 36.Waypa GB, Marks JD, Guzy R, et al. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res 2010;106:526–535. [DOI] [PMC free article] [PubMed]
- 37.Wedgwood S, Lakshminrusimha S, Fukai T, Russell JA, Schumacker PT, Steinhorn RH. Hydrogen peroxide regulates extracellular superoxide dismutase activity and expression in neonatal pulmonary hypertension. Antioxid Redox Signal 2011;15:1497–1506. [DOI] [PMC free article] [PubMed]
- 38.Carnesecchi S, Deffert C, Pagano A, et al. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am J Respir Crit Care Med 2009;180:972–981. [DOI] [PMC free article] [PubMed]
- 39.Yoshioka T, Kawamura T, Meyrick BO, et al. Induction of manganese superoxide dismutase by glucocorticoids in glomerular cells. Kidney Int 1994;45:211–219. [DOI] [PubMed]
- 40.Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med 2003;167:1600–1619. [DOI] [PubMed]
- 41.Sawada H, Mitani Y, Maruyama J, et al. A nuclear factor-kappaB inhibitor pyrrolidine dithiocarbamate ameliorates pulmonary hypertension in rats. Chest 2007;132:1265–1274. [DOI] [PubMed]
- 42.Wright CJ, Agboke F, Chen F, La P, Yang G, Dennery PA. NO inhibits hyperoxia-induced NF-kappaB activation in neonatal pulmonary microvascular endothelial cells. Pediatr Res 2010;68:484–489. [DOI] [PMC free article] [PubMed]
- 43.Folz RJ, Abushamaa AM, Suliman HB. Extracellular superoxide dismutase in the airways of transgenic mice reduces inflammation and attenuates lung toxicity following hyperoxia. J Clin Invest 1999;103:1055–1066. [DOI] [PMC free article] [PubMed]
- 44.Schreck R, Albermann K, Baeuerle PA. Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic Res Commun 1992;17:221–237. [DOI] [PubMed]
- 45.Iwasaki T, Takahashi T, Shimizu H, et al. Increased pulmonary heme oxygenase-1 and delta-aminolevulinate synthase expression in monocrotaline-induced pulmonary hypertension. Curr Neurovasc Res 2005;2:133–139. [DOI] [PubMed]
- 46.Aghai ZH, Kode A, Saslow JG, et al. Azithromycin suppresses activation of nuclear factor-kappa B and synthesis of pro-inflammatory cytokines in tracheal aspirate cells from premature infants. Pediatr Res 2007;62:483–488. [DOI] [PubMed]
- 47.Aghai ZH, Kumar S, Farhath S, et al. Dexamethasone suppresses expression of nuclear factor-kappaB in the cells of tracheobronchial lavage fluid in premature neonates with respiratory distress. Pediatr Res 2006;59:811–815. [DOI] [PubMed]
- 48.Suzuki Y, Nishio K, Takeshita K, et al. Effect of steroid on hyperoxia-induced ICAM-1 expression in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol 2000;278:L245–L252. [DOI] [PubMed]
- 49.Murray F, MacLean MR, Pyne NJ. Increased expression of the cGMP-inhibited cAMP-specific (PDE3) and cGMP binding cGMP-specific (PDE5) phosphodiesterases in models of pulmonary hypertension. Br J Pharmacol 2002;137:1187–1194. [DOI] [PMC free article] [PubMed]
- 50.Ahmed MN, Codipilly C, Hogg N, Auten RL. The protective effect of overexpression of extracellular superoxide dismutase on nitric oxide bioavailability in the lung after exposure to hyperoxia stress. Exp Lung Res 2011;37:10–17. [DOI] [PubMed]
- 51.Savai R, Pullamsetti SS, Kolbe J, et al. Immune/inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:897–908. [DOI] [PubMed]
- 52.de Visser YP, Walther FJ, Laghmani El H, Boersma H, van der Laarse A, Wagenaar GT. Sildenafil attenuates pulmonary inflammation and fibrin deposition, mortality and right ventricular hypertrophy in neonatal hyperoxic lung injury. Respir Res 2009;10:30. [DOI] [PMC free article] [PubMed]