

Abstract Abstract
Right ventricular (RV) afterload consists of both resistive and capacitive (pulsatile) components. Total afterload can be measured directly with pulmonary artery input impedance spectra or estimated, either with lumped-parameter modeling or by pressure-volume analysis. However, the inverse, hyperbolic relationship between resistance and compliance in the lung would suggest that the pulsatile components are a predictable and constant proportion of the resistive load in most situations, meaning that total RV load can be estimated from mean resistive load alone. Exceptions include elevations in left atrial pressures and, to a lesser extent, chronic thromboembolic disease. The pulsatile components may also play a more significant role at normal or near-normal pulmonary artery pressures. Measures of coupling between RV afterload and RV contractility may provide important information not apparent by other clinical and hemodynamic measures. Future research should be aimed at development of noninvasive measures of coupling.
Keywords: pulmonary hypertension, right ventricle, afterload, pulmonary vascular resistance, arterial compliance
Right ventricular (RV) failure is an important cause of morbidity and mortality in pulmonary hypertension (PH).1 Therefore, understanding and assessing the load that the right ventricle must overcome to eject blood is vital. In its purest sense, RV afterload is the RV wall stress that occurs during RV ejection. By LaPlace’s law, wall stress is proportional to the pressure during RV ejection (PEJ) × radius (REJ) of the wall divided by the wall thickness (HEJ). This, of course, is an estimate, since the right ventricle is not a true sphere. Because the ventricular radius during ejection is generally small and wall thickness is relatively constant, the wall stress is generally proportional to the RV pressure during ejection. The RV pressure during ejection is determined by several components: (1) mean resistance, or resistance to blood flow during steady state; (2) the compliance, or blood storage capacity of the vascular system; (3) arterial wave reflections that occur as a result of pulsatile blood flow; and (4) the inertance of blood during ejection. Therefore, clinical measures commonly obtained during right heart catheterization, such as mean pulmonary artery pressure (mPAP) and pulmonary vascular resistance (PVR), may be inadequate descriptions of RV afterload because they do not take into account contributions of pulsatile loading.
Measuring RV afterload
Pulmonary artery input impedance is the most comprehensive description of RV vascular load and takes into account all of the four components mentioned above. Milnor and O’Rourke2 were the first to measure pulmonary artery input impedance in the human, in 1969. Impedance is measured in the frequency domain rather than the time domain, requiring Fourier analysis of simultaneous measures of pressure and flow. A graph of modulus (amplitude of pressure/amplitude of flow) and phase (delay between flow and pressure) is created and plotted against frequency (harmonics), typically multiples of the heart rate. An example of pulmonary impedance spectra is shown in Figure 1. The impedance at the zero harmonic (Z0) is equivalent to resistance at steady state flow, or the ratio of mean pressure to mean flow. As opposed to the systemic circulation where right atrial pressure is negligible compared to systemic mean pressure, the magnitude of left atrial pressure may be significant when compared to mPAP. Most authors have proposed that the left atrial pressure measurement should be subtracted from mPAP, i.e., PVR, before calculation of Z0 and Fourier transformation at other frequencies.2-6 However, other studies have included the effects of left atrial pressure on mean pressure, i.e., total pulmonary resistance, during impedance analysis.7,8 This consideration may be particularly relevant when considering patients with WHO (World Health Organization) group II PH who have elevated left atrial pressures. The impedance at the first harmonic (Z1) accounts for a large portion of total blood flow and is an important fraction of total load.3,9 It is largely influenced by wave reflections and is believed to represent a significant portion of pulsatile load. The sum of impedance at higher frequencies has been termed characteristic impedance (ZC; usually ≥2 Hz; 2–12 Hz in this example). The characteristic impedance is the ratio of blood mass inertia to the proximal vessel compliance. The frequency of the first minimum impedance (fMIN) is a function of pulse wave velocity as well as the distance to the major sites of reflection. Calculation of other useful parameters, such as reflection coefficients and the ratio of forward to backward waves, may be obtained. By integrating the product of pressure and flow obtained in the impedance spectrum, total hydraulic power (power = work performed/unit time) can be estimated.2,9 Oscillatory (pulsatile) power can be calculated by subtracting mean power (mPAP × cardiac output) from total hydraulic power. This oscillatory power generated to account for the pulsatile load is wasted, in the sense that it does not contribute to forward flow. With development of PH, Z0 and Z1 will increase, ZMIN will shift toward higher frequencies, and hydraulic power will increase.10 It should be noted that there is still some controversy about the interpretation of impedance data.
Figure 1.
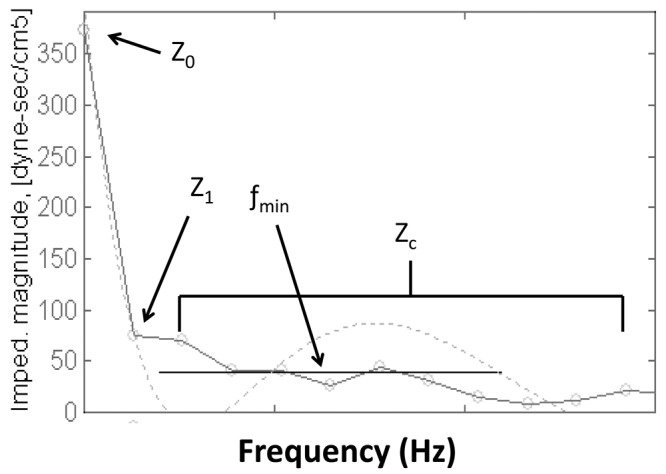
Example of pulmonary artery impedance spectra after Fourier transformation: modulus (amplitude of pressure divided by amplitude of flow) is plotted against frequency. Z0 is equivalent to the ratio mean pressure/mean flow; Z1 (impedance at the first harmonic) represents a significant amount of total blood flow and is largely influenced by wave reflection; ZC (characteristic impedance) is the ratio of blood mass inertia to the proximal vessel compliance; fMIN (frequency at the first impedance minimum) is a function of pulse wave velocity as well as the distance to the major sites of reflection.
Pulmonary impedance is difficult both to measure and to interpret. In order to present impedance data in a more simple and concise format, lumped-parameter models based on electric circuits were developed to mimic impedance and estimate RV afterload. These lumped parameters may also be estimated by means other than impedance spectra that are more easily obtained clinically. One such model is the 3-element Windkessel model developed by Westerhof (Fig. 2),11 which includes resistance, compliance, and characteristic impedance (Fig. 3). In this model, characteristic impedance functions as a proximal, in-series resistor with resistance (resistor) and compliance (capacitor) in parallel. This model and other versions have been shown to be a reasonable way to describe pulmonary input impedance.12 More recently, wave intensity analysis has been used to describe afterload. As opposed to impedance spectra, it is a time domain–based model that represents pressure and velocity waveforms as successive wave fronts, specifically, simultaneous forward and backward waves that can be quantitated.13 In the pulmonary circulation, this type of analysis has been limited to animal studies.14
Figure 2.
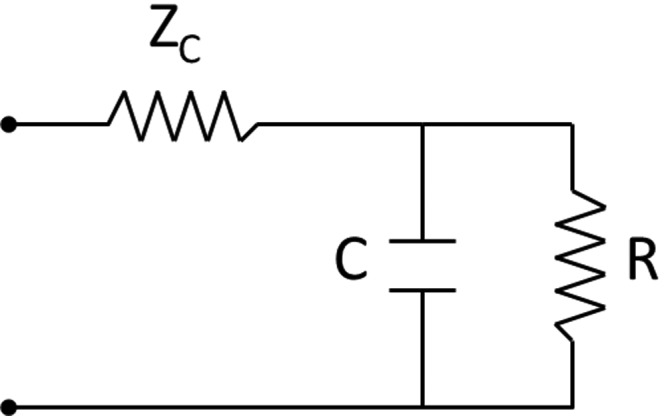
Three-element Windkessel model of lumped parameters used to mimic impedance and estimate ventricular afterload. ZC: characteristic impedance; C: compliance; R: resistance.
Figure 3.

A, Right ventricular (RV) pressure-volume loop. The width of the loop is stroke volume (dashed line). RV afterload can be described as the sum of RV pressures throughout ejection (gray line). B, Effective arterial elastance (EA), a lumped measure of afterload, is the slope of the gray line connecting the points (end-systolic pressures, end-systolic volume) and (0, end-diastolic pressure). EA is calculated as end-systolic pressure/stroke volume. PV: pulmonary valve.
RV afterload can also be described on a pressure-volume loop as the sum of RV systolic pressure occurring throughout ejection. This summation is illustrated in Figure 3A. First described in the left ventricle, effective arterial elastance (EA) is another lumped parameter that should take into account both resistive and pulsatile loading and can be calculated as end-systolic pressure/stroke volume (Fig. 3B).15 This type of analysis has the added benefit of matching afterload to measures of load-independent contractility when preload is reduced (end-systolic elastance, EES), providing information on RV-pulmonary vascular coupling (EES/EA; Fig. 4). Although RV pressure-volume analysis is regularly performed in animals, reports of its use in humans are more limited in the literature.16-18 However, this type of analysis is feasible and valuable. We recently reported its use to compare ventricular contractility and afterload in patients with systemic sclerosis–associated pulmonary arterial hypertension (SScPAH) and idiopathic pulmonary arterial hypertension (IPAH).19 At similar levels of afterload, we found depressed myocardial contractility in the SScPAH group, compared to the IPAH group. Therefore, significant ventricular-vascular uncoupling of the SScPAH group was present, compared to he IPAH group as well as to a group of systemic sclerosis patients without PH. The differences in contractile function were not apparent by other clinical, hemodynamic, or radiologic measures, speaking to the importance of directly measuring ventricular-vascular coupling. Unfortunately, simplified approaches using single-beat estimates to calculate contractile and coupling measures have yielded disappointing results.20 It is clear that the volume intercept (V0) of the end-systolic pressure-volume relationship cannot be assumed to be 0 in pulmonary arterial hypertension.19,20 In keeping with this fact, simplified methods to estimate EES that ignore V0 (mPAP/end-systolic volume [ESV] or systolic pulmonary artery pressure/ESV) are inaccurate.
Figure 4.

Cartoon illustrating right ventricular pressure-volume loops at varying levels of preload decline. The slope of the gray line connecting the end-systolic pressure points is the end-systolic elastance (EES), a load-independent measure of contractility. The EES can be compared with effective arterial elastance (EA) to assess coupling of right ventricular contractility to pulmonary vasculature load.
The resistance-compliance (R-C) relationship in the lung vasculature
Using the 3-element Windkessel model, Lankhaar and colleagues21 were the first to show that the relationship between resistance and compliance in the lung vasculature is quite different from that in the systemic vasculature. In a study of 29 patients, 10 with IPAH, 9 with chronic thromboembolic disease pulmonary hypertension (CTEPH), and 10 with normal pulmonary pressures, they found that resistance and compliance followed a predictable inverse, hyperbolic relationship. Any change in resistance was met by an inverse change in compliance (Fig. 5). This is notably different from the systemic circulation, where compliance can change independent of resistance, e.g., aging.22,23 In addition, the characteristic impedance appeared to be principally determined by mPAP, since patients with IPAH (and higher mPAP) had higher ZC than those with CTEPH (lower mPAP). Although patients in the latter group presumably had proximal vessel narrowing, overall compliance was lower, which appeared to be the major determinant of ZC. The lumped compliance parameter was estimated three different ways in Lankhaar’s21 study, including stroke volume divided by pulse pressure (SV/PP), which is the simplest clinical measure.24 This lumped parameter should take into account contributions of wave reflections and pulsatile load because of the inclusion of pulse pressure in its denominator. When wave reflections return during RV systole, RV systolic pressure must increase to exceed this added load and eject blood, thereby increasing the pulse pressure. As pulse pressure increases, SV/PP (or lumped compliance) decreases. It is fair to note that this lumped parameter does not account for blood leaving the lung through the microcirculation.25
Figure 5.
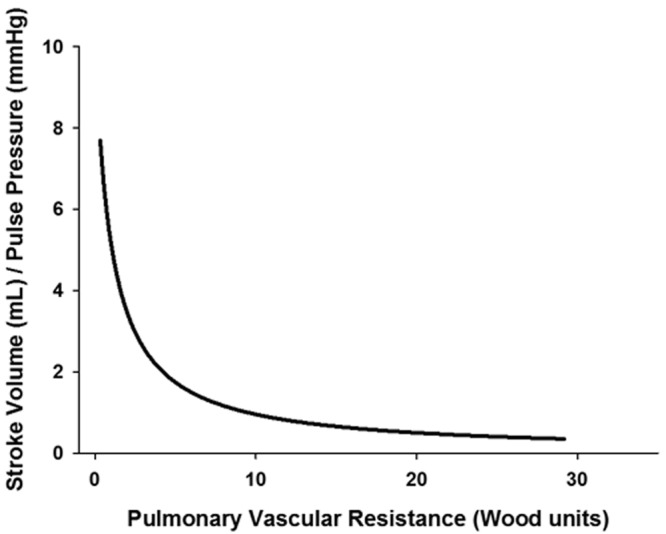
Inverse, hyperbolic relationship between pulmonary vascular resistance and pulmonary vascular compliance.
In a subsequent study by the same group, the R-C relationship did not change in 62 patients (52 with IPAH and 10 with CTEPH) before and after treatment of PH.26 Changes in heart rate appeared to have little meaningful effect on the relationship.27 Our group found a similar inverse, hyperbolic relationship in studying more than 1,000 patients with suspected or known pulmonary arterial hypertension, all of whom by definition had a pulmonary capillary wedge pressure (PCWP) of ≤15 mmHg.28 We showed that this relationship was unaltered by interstitial lung disease and systemic sclerosis and only minimally affected by age.19,28 The inverse, hyperbolic relationship between resistance and compliance implies that the RC time (the product of resistance × compliance) in the lung vasculature is constant. Physiologically, the RC time provides a time constant for the pulmonary arterial diastolic pressure decay. The constant RC time is in stark contrast to the systemic circulation, where resistive and capacitive load can vary independently of each other and the RC time is quite variable.28
The constant RC time in the pulmonary circulation may be explained by one of two scenarios. Sniderman and Fitchett29 suggested that an increase in PVR leads to an increase in pressure within the vasculature, which in turn leads to a decrease in compliance as a result of the nonlinear pressure-diameter relation of the pulmonary arteries.27 The second scenario, suggested by Saouti et al.,25,30 relates to the differences in geographical distributions of compliance in each circulation. In the systemic circulation, the proximal aorta accounts for most of the total arterial compliance, whereas the small, distal vessels are responsible for most of the resistance (Poiseuille’s law states that resistance is inversely proportional to the radius of the vessel to the 4th power).22,23 In the lung, the distal arteries and arterioles that are responsible for resistance also hold most of the capacitance properties of the pulmonary circulation. In fact, the Vonk-Noordegraaf group25,30 estimated that proximal lung vessels may count for only ∼20% of the total vascular compliance. The distal distribution of compliance may be explained by the large number of distal arterioles in the lung, compared to the systemic circulation (roughly 10 times as many), and therefore the increase blood storage capacity at the peripheral lung.25 These differences between the pulmonary and systemic vasculatures are supported by a prior study authored by Yin et al.31 His group showed, via impedance spectra analysis, that nitroprusside lowered systemic vascular resistance and mean arterial pressure without changing the characteristic impedance, yet in the lung, lower PVR and mPAP were accompanied by lower characteristic impedance. Work by Kuo et al.,32 who have developed a model of proximal pulmonary compliance in the sheep, provides further support. A chamber that allows manipulation of compliance was surgically connected to the very proximal pulmonary artery, followed by an in-series variable-resistance clamp, and then reconnected to the distal pulmonary artery and the native lungs. Therefore, distal compliance (lung) of the system remained intact. The authors showed that decreasing proximal compliance to 0 led to only a very small decline in cardiac output. On the other hand, cardiac output could be markedly reduced by increasing resistance.32
Another consequence of the pulmonary R-C relationship is the proportionality of pulmonary pressures. As long as the left atrial pressure is constant and low, there is a linear relationship between mPAP, systolic PAP, and diastolic PAP.25,27,33 Pulse pressure is therefore also proportional to mPAP. From this proportionality, it should follow that the pulsatile (or oscillatory) load should be a predictable portion of the resistive load. Saouti et al.34 have estimated that the RV oscillatory power is 23% of the total hydraulic RV power in patients with and without PH. In keeping with this finding, total RV power can be estimated from mean power, which is easily calculated from standard right heart catheterization measures (total power = 1.3 × mean power; mean power = mPAP × cardiac output).34 The pulmonary R-C relationship and its implications for predictable pulsatile loading may suggest that more cumbersome impedance measurements are not necessary in pulmonary arterial hypertension. There are, however, several scenarios where the R-C relation may not hold true.
Exceptions to the rule
Elevated left atrial pressure
The effects of rising or falling left atrial pressure on human RV afterload were studied in a series of experiments by Kussmaul and Laskey4,6,35,36 using pulmonary artery input impedance. To study the effect of a rising PCWP, the left anterior descending artery was temporarily occluded in 9 patients undergoing coronary angioplasty.6 All but one of these patients had normal resting pulmonary pressures (mPAP < 25 mmHg). The rise in PCWP was out of proportion to the rise in mPAP (13 vs. 9 mmHg), leading to a decreased transpulmonary gradient. Although cardiac output fell slightly, PVR (Z0) still decreased by approximately 35%. This PVR decline was attributed to the passive pressure recruiting more “resistance” vessels, which is supported by animal studies of acute left atrial pressure elevations.8,37 Despite lower PVR, Z1 actually increased by ∼50%, suggesting increases in pulsatile afterload. Notably, heart rate remained unchanged. The source of the increased pulsatile load was less clear. The fMIN remained unaltered, suggesting that there was no change in pulse wave velocity or distance to the reflection sites.6 Similarly, the characteristic impedance ZC remained unchanged. Assuming that no change occurred during angioplasty to alter blood viscosity (which would change the inertia), the authors suggested that either the proximal pulmonary vessels both stiffened and dilated or that neither occurred. In a second study, the same group measured impedance before and after mitral valvuloplasty in 16 patients with severe mitral stenosis.35 In this cohort, PVR and ZC were unchanged after valvuloplasty, but the associated fall in PCWP coincided with a fall in significant decrease in Z1.
Studies of acute, mechanically induced pulmonary venous hypertension in dogs have yielded similar results: a falling PVR despite an overall rise in mPAP and no statistical difference in ZC (Z1 was not reported).8 A model of chronic venous PH, on the other hand, led to an increased PVR and an expected rise in ZC, compared with controls.8 As mentioned above, a study of pulmonary arterial hypertension patients by Lankhaar et al.21 has shown that ZC closely follows mPAP because of its effect on overall lung vasculature compliance. Therefore, in pulmonary venous hypertension, it appears that the effects of an elevated PCWP on mPAP may balance out, or “counter,” the lower PVR; otherwise, characteristic impedance should actually fall. It should be noted that the above studies investigated those patients with normal or mildly elevated resting pulmonary pressures. It has been suggested that only those patients with less severe PH retain the ability to recruit pulmonary vascular beds during acute increases in left atrial pressure.35,38 This ability may be lost as the chronicity and severity of PH increase, and therefore, elevations in left atrial pressure may not lower the transpulmonary gradient or PVR. In this case, we would expect to see elevations in both Z1 and ZC with increasing left atrial pressure.
Using the unique pulmonary R-C relationship, our group recently showed that elevations in left atrial pressures lead to lower compliance than one would predict from the PVR alone. This causes a shift in the R-C curve to the left (Fig. 6) and a lower RC time, suggesting increased pulsatile afterload.28 This shift in the R-C curve and lower RC time held true in patients with both acute and chronic PCWP elevations. The RC time declined in proportion to the degree of PCWP elevation, and in situations of markedly elevated PCWP, the lowering of the RC time was quite dramatic.28 In a group of patients with advanced heart failure, Dupont and colleagues39 illustrated that heart failure treatment (which lowered PCWP) increased compliance more than one would predict from the associated fall in PVR, thereby increasing the RC time constant and reducing pulsatile load. They also showed that compliance (which essentially bundled the effects of PVR and PCWP) was a better predictor than resistance of RV dysfunction and long-term prognosis.
Figure 6.
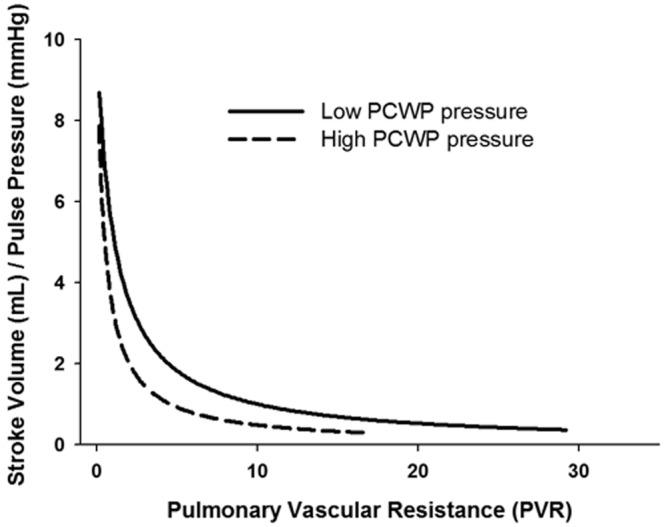
Shift of the pulmonary resistance-compliance curve to the left with elevations in pulmonary capillary wedge pressure (PCWP).
Chronic thromboembolic disease pulmonary hypertension (CTEPH)
The afterload imposed on the right ventricle in chronic thromboembolic disease—in particular, when proximal obstruction is present—remains somewhat controversial. Several studies of pulmonary input impedance in animals have suggested elevations in low- (Z1) and high-frequency (ZC) impedance in models of CTEPH (external pulmonary artery constriction or ensnarement).40,41 Castelain42 noted earlier return of wave reflections by analysis of high-fidelity pulmonary artery waveforms. In contrast to these findings, data from the Vonk-Noordegraaf group,30 obtained from 23 CTEPH patients, suggested that the pulmonary RC time remained constant within each individual lung (whether the lung was subject to high or low flow) as well as when both lungs were considered together. In that study, the pulse pressure method (not SV/PP) was used to estimate compliance; it typically yields lower values than other methods of compliance estimation, including the SV/PP method. In addition, total pulmonary resistance (mPAP/cardiac output) was used in calculation of the RC time constants.30 More recently, Mackenzie Ross and colleagues43 published a large retrospective analysis comparing RC times in IPAH (n = 78), proximal CTEPH (n = 91) before and after pulmonary endarterectomy (PEA), and distal CTEPH (n = 53). Proximal CTEPH was defined as patients who achieved normal mPAP of ≤25 mmHg after PEA. RC times were lowest in the proximal-CTEPH group (0.49 ± 0.11 s), followed by the distal-CTEPH group (0.55 ± 0.12 s), and highest in the IPAH group (0.63 ± 0.14 s). These differences remained significant after adjustment for age, PCWP, and mPAP. The same methods for calculating compliance (SV/PP) and PVR (mPAP−PCWP/cardiac output) were used in all patients. These data seem plausible, as some decline in RC time with proximal obstruction would be expected, since approximately 20% of the total compliance does reside in the proximal vessels.25 Overall, the decline in RC time was modest. Perhaps most interesting is that the RC time actually decreased to a greater extent in those patients who underwent successful PEA (0.38 ± 11 s). Despite lowering of their mPAP to ≤25 mmHg, compliance did not rise to match the decline in resistance. The reason for the decline in RC time is unclear, but the authors speculate that structural changes after surgical PEA (removal of intima and media and/or subsequent healing) may alter the compliance properties.43
Normal pulmonary pressures
Studies investigating the RC time in the normal pressure range have typically consisted of a small number of patients, and most have used total pulmonary resistance rather than PVR to measure the RC time.21,26 More in-depth examination of the RC time in our study of more than 1,000 patients with known or suspected PAH reveals a lower RC time in patients with normal mPAP (<25 mmHg) and normal PVR (<3 Wood units), compared to those with higher mPAP and higher PVR (Table 1; Fig. 7). The median RC time values at low mPAP and low PVR are similar to those reported by MacKenzie Ross et al.43 after PEA. The reason for the lower calculated RC time at low pulmonary pressures is unknown, but there are several possibilities.
Table 1.
RC time in patients with and without pulmonary hypertension
| Group | n | RC time, median (IQR) | P valuea |
|---|---|---|---|
| mPAP < 25 mmHg | 415 | 0.36 (0.29–0.47) | <0.001 |
| mPAP ≥ 25 mmHg | 593 | 0.53 (0.45–0.63) | <0.001 |
| PVR < 3 Wood units | 442 | 0.36 (0.29–0.47) | <0.001 |
| PVR ≥ 3 Wood units | 566 | 0.54 (0.46–0.64) | <0.001 |
RC time: product of resistance × compliance; IQR: interquartile range; mPAP: mean pulmonary artery pressure; PVR: pulmonary vascular resistance.
Comparison by Mann-Whitney rank-sum test.
Figure 7.

RC time (product of resistance × compliance) plotted against pulmonary vasculature resistance (PVR; A) and mean pulmonary artery pressures (mPAP; B). Groups are separated into normal and elevated PVR (A) and normal and elevated mPAP (B), with linear regression lines drawn for each group.
The so-called waterfall effect in the lung occurs when either the alveolar pressure or the elastic/muscular recoil of the vessel wall itself exceeds the left atrial pressure. The pressure has been termed the “critical” pressure. When this occurs, left atrial pressure is no longer the downstream pressure affecting resistance,31,37 and therefore using the PCWP to calculate PVR would lead to an overestimation of true resistance.44 However, this phenomenon would actually lead to an erroneously high RC time rather than a lower one.
It is also possible that the effects of measurement error, particularly for the PCWP, lead to more pronounced effects at low pulmonary artery pressures. Consider two patients, both with a cardiac output of 5 L/min and a true left atrial pressure of 5 mmHg. Patient 1 has an mPAP of 15 mmHg, and patient 2 has an mPAP of 40 mmHg. The PCWP is erroneously measured at 10 mmHg because of incomplete wedging of the pulmonary artery catheter. Patient 1 will have a measured PVR of 1 Wood unit instead of 2 Wood units, whereas patient 2 will have a measured PVR of 6 versus 7 Wood units. Because of the steepness of the R-C curve at lower resistances, the effect on the RC time will be much greater for patient 1.
Another potential contributor is that pulmonary vasculature may reach a “maximal” compliance, such that further decreases in resistance (at already low resistances) will have no effect on overall compliance. Thus, as resistance declines, compliance is constant, lowering the RC time. We see the same effect at very high resistances, where compliance never actually falls to 0. This requires a constant in the hyperbolic decay equation.19
Finally, at lower mean pressures, compliance and pulsatile afterload may constitute a more important fraction on the total afterload. This has been suggested by some studies, including Milnor’s2 original impedance measures in humans.45 In Milnor’s2 3 subjects without PH, the pulsatile component accounted for approximately one-third of the total power, whereas it accounted for 24% in the patients with PH. Others have suggested that pulsatile load may account for up to 50% at normal pressures.29 Despite the possible increased contribution of pulsatile load, the total RV load likely remains low because of low pulmonary pressures and resistance.
Potential unknown determinants
Despite the overall consistent R-C relationship, there remains significant scatter around the hyperbolic curve fits and RC times, even in WHO group I PH.19 It is possible that a lot of this scatter is due to measurement error, but it also remains possible that other factors that have yet to be discovered affect compliance independent of resistance.
Summary
In summary, proper assessment of RV afterload is important when treating a patient with PH. Because of the unique relationship between resistance and compliance in the lung vasculature, the total RV afterload can be predictably estimated from mean resistance in most cases. In cases of WHO group II PH, CTEPH, and possibly low or borderline pulmonary pressures, the pulsatile component of afterload appears higher than that predicted by mean resistance. In WHO group II PH patients, the RC time can be lowered substantially, suggesting a robust increase in RV afterload. In CTEPH, this effect appears more modest. In situations of normal or near-normal pulmonary pressure, it is likely that the overall load remains low. How best to quantify the effects of PCWP on pulsatile and total load requires further study, and other methods to effectively measure afterload in this population may be useful. Finally, the use of invasive pressure-volume assessment to understand coupling between RV contractility and the load imposed by the vasculature is feasible. A noninvasive strategy to measure coupling would be clinically useful and should be the direction of future research.
Source of Support: RJT is supported by funding from the National Heart, Lung, and Blood Institute (grants 1R01HL114910-01 and L30 HL110304).
Conflict of Interest: None declared.
References
- 1.Campo A, Mathai SC, Le Pavec J, Zaiman AL, Hummers LK, Boyce D, Housten T, et al. Outcomes of hospitalisation for right heart failure in pulmonary arterial hypertension. Eur Respir J 2011;38:359–367. [DOI] [PubMed]
- 2.Milnor WR, Conti CR, Lewis KB, O’Rourke MF. Pulmonary arterial pulse wave velocity and impedance in man. Circ Res 1969;25:637–649. [DOI] [PubMed]
- 3.Murgo JP, Westerhof N. Input impedance of the pulmonary arterial system in normal man: effects of respiration and comparison to systemic impedance. Circ Res 1984;54:666–673. [DOI] [PubMed]
- 4.Kussmaul WG, Wieland JM, Laskey WK. Pressure-flow relations in the pulmonary artery during myocardial ischaemia: implications for right ventricular function in coronary disease. Cardiovasc Res 1988;22:627–638. [DOI] [PubMed]
- 5.Haneda T, Nakajima T, Shirato K, Onodera S, Takishima T. Effects of oxygen breathing on pulmonary vascular input impedance in patients with pulmonary hypertension. Chest 1983;83:520–527. [DOI] [PubMed]
- 6.Kussmaul WG, Wieland J, Altschuler J, Laskey WK. Pulmonary impedance and right ventricular-vascular coupling during coronary angioplasty. J Appl Physiol 1993;74:161–169. [DOI] [PubMed]
- 7.Mills CJ, Gabe IT, Gault JH, Mason DT, Ross J Jr., Braunwald E, Shillingford JP. Pressure-flow relationships and vascular impedance in man. Cardiovasc Res 1970;4:405–417. [DOI] [PubMed]
- 8.Hopkins RA, Hammon JW, McHale PA, Smith PK, Anderson RW. An analysis of the pulsatile hemodynamic responses of the pulmonary circulation to acute and chronic pulmonary venous hypertension in the awake dog. Circ Res 1980;47:902–910. [DOI] [PubMed]
- 9.Milnor W, Bergel D, Bargainer JD. Hydraulic power associated with pulmonary blood flow and its relation to heart rate. Circ Res 1966;3:467–480. [DOI] [PubMed]
- 10.O’Rourke MF. Vascular impedance in studies of arterial and cardiac function. Physiol Rev 1982;62:570–623. [DOI] [PubMed]
- 11.Westerhof N, Elzinga G, Sipkema P. An artificial arterial system for pumping hearts. J Appl Physiol 1971;31:776–781. [DOI] [PubMed]
- 12.Grant BJ, Paradowski LJ. Characterization of pulmonary arterial input impedance with lumped parameter models. Am J Physiol Heart Circ Physiol 1987;252:H585–H593. [DOI] [PubMed]
- 13.Parker KH, Jones CJH. Forward and backward running waves in the arteries: analysis using the method of characteristics. J Biomech Eng 1990;112:322–326. [DOI] [PubMed]
- 14.Hollander EH, Wang JJ, Dobson GM, Parker KH, Tyberg JV. Negative wave reflections in pulmonary arteries. Am J Physiol Heart Circ Physiol 2001;281:H895–H902. [DOI] [PubMed]
- 15.Kelly RP, Ting CT, Yang TM, Liu CP, Maughan WL, Chang MS, Kass DA. Effective arterial elastance as index of arterial vascular load in humans. Circulation 1992;86:513–521. [DOI] [PubMed]
- 16.Bishop A, White P, Groves P, Chaturvedi R, Brookes C, Redington A, Oldershaw P. Right ventricular dysfunction during coronary artery occlusion: pressure-volume analysis using conductance catheters during coronary angioplasty. Heart 1997;78:480–487. [DOI] [PMC free article] [PubMed]
- 17.Bishop A, White P, Oldershaw P, Chaturvedi R, Brookes C, Redington A. Clinical application of the conductance catheter technique in the adult human right ventricle. Int J Cardiol 1997;58:211–221. [DOI] [PubMed]
- 18.Kuehne T, Yilmaz S, Steendijk P, Moore P, Groenink M, Saeed M, Weber O, et al. Magnetic resonance imaging analysis of right ventricular pressure-volume loops: in vivo validation and clinical application in patients with pulmonary hypertension. Circulation 2004;110:2010–2016. [DOI] [PubMed]
- 19.Tedford RJ, Mudd JO, Girgis RE, Mathai SC, Zaiman AL, Housten-Harris T, Boyce D, et al. Right ventricular dysfunction in systemic sclerosis–associated pulmonary arterial hypertension. Circ Heart Failure 2013;6:953–963. [DOI] [PMC free article] [PubMed]
- 20.Trip P, Kind T, van de Veerdonk MC, Marcus JT, de Man FS, Westerhof N, Vonk-Noordegraaf A. Accurate assessment of load-independent right ventricular systolic function in patients with pulmonary hypertension. J Heart Lung Transplant 2013;32:50–55. [DOI] [PubMed]
- 21.Lankhaar J, Westerhof N, Faes TJC, Marques KMJ, Marcus, JT, Postmus PE, Vonk-Noordegraaf A. Quantification of right ventricular afterload in patients with and without pulmonary hypertension. Am J Physiol Heart Circ Physiol 2006;291:H1731–H1737. [DOI] [PubMed]
- 22.Sipkema P, Latham R, Westerhof N, Rubal B, Slife D. Isolated aorta setup for hemodynamic studies. Ann Biomed Eng 1990;18:491–503. [DOI] [PubMed]
- 23.Stergiopulos N, Segers P, Westerhof N. Use of pulse pressure method for estimating total arterial compliance in vivo. Am J Physiol Heart Circ Physiol 1999;276:H424–H428. [DOI] [PubMed]
- 24.Stergiopulos N, Meister JJ, Westerhof N. Evaluation of methods for estimation of total arterial compliance. Am J Physiol Heart Circ Physiol 1995;268:H1540–H1548. [DOI] [PubMed]
- 25.Saouti N, Westerhof N, Postmus PE, Vonk-Noordegraaf A. The arterial load in pulmonary hypertension. Eur Respir Rev 2010;19:197–203. [DOI] [PMC free article] [PubMed]
- 26.Lankhaar J, Westerhof N, Faes TJC, Gan CTJ, Marques KM, Boonstra A, van den Berg FG, Postmus PE, Vonk-Noordegraaf A. Pulmonary vascular resistance and compliance stay inversely related during treatment of pulmonary hypertension. Eur Heart J 2008;29:1688–1695. [DOI] [PubMed]
- 27.Kind T, Faes TC, Vonk-Noordegraaf A, Westerhof N. Proportional relations between systolic, diastolic and mean pulmonary artery pressure are explained by vascular properties. Cardiovasc Eng Technol 2011;2:15–23. [DOI] [PMC free article] [PubMed]
- 28.Tedford RJ, Hassoun PM, Mathai SC, Girgis RE, Russell SD, Thiemann DR, Cingolani OH, et al. Pulmonary capillary wedge pressure augments right ventricular pulsatile loading. Circulation 2012;125:289–297. [DOI] [PMC free article] [PubMed]
- 29.Sniderman AD, Fitchett DH. Vasodilators and pulmonary arterial hypertension: the paradox of therapeutic success and clinical failure. Int J Cardiol 1988;20:173–181. [DOI] [PubMed]
- 30.Saouti N, Westerhof N, Helderman F, Marcus JT, Stergiopuos N, Westerhof, BE, Boonstra A, Postmus PE, Vonk-Noordegraaf A. RC time constant of single lung equals that of both lungs together: a study in chronic thromboembolic pulmonary hypertension. Am J Physiol Heart Circ Physiol 2009;297:H2154–H2160. [DOI] [PubMed]
- 31.Yin FC, Guzman PA, Brin KP, Maughan WL, Brinker JA, Traill TA, Weiss JL, Weisfeldt ML. Effect of nitroprusside on hydraulic vascular loads on the right and left ventricle of patients with heart failure. Circulation 1983;67:1330–1339. [DOI] [PubMed]
- 32.Kuo AS, Sato H, Reoma JL, Cook KE. The relationship between pulmonary system impedance and right ventricular function in normal sheep. Cardiovasc Eng 2009;9:153–160. [DOI] [PubMed]
- 33.Chemla D, Castelain V, Provencher S, Humbert M, Simonneau G, Hervé P. Evaluation of various empirical formulas for estimating mean pulmonary artery pressure by using systolic pulmonary artery pressure in adults. Chest 2009;135:760–768. [DOI] [PubMed]
- 34.Saouti N, Westerhof N, Helderman F, Marcus JT, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Right ventricular oscillatory power is a constant fraction of total power irrespective of pulmonary artery pressure. Am J Respir Crit Care Med 2010;182:1315–1320. [DOI] [PubMed]
- 35.Kussmaul WG, Altschuler JA, Herrmann HC, Laskey WK. Effects of pacing tachycardia and balloon valvuloplasty on pulmonary artery impedance and hydraulic power in mitral stenosis. Circulation 1992;86:1770–1779. [DOI] [PubMed]
- 36.Kussmaul WG, Altschuler JA, Matthai WH, Laskey WK. Right ventricular-vascular interaction in congestive heart failure: importance of low-frequency impedance. Circulation 1993;88:1010–1015. [DOI] [PubMed]
- 37.McDonald IG, Butler J. Distribution of vascular resistance in the isolated perfused dog lung. J Appl Physiol 1967;23:463–474. [DOI] [PubMed]
- 38.Janicki JS, Weber KT, Likoff MJ, Fishman AP. The pressure-flow response of the pulmonary circulation in patients with heart failure and pulmonary vascular disease. Circulation 1985;72:1270–1278. [DOI] [PubMed]
- 39.Dupont M, Mullens W, Skouri HN, Abrahams Z, Wu Y, Taylor DO, Starling RC, Tang WHW. Prognostic role of pulmonary arterial capacitance in advanced heart failure. Circ Heart Failure 2012;5:778–785. [DOI] [PMC free article] [PubMed]
- 40.Calvin JE, Baer RW, Glantz SA. Pulmonary artery constriction produces a greater right ventricular dynamic afterload than lung microvascular injury in the open chest dog. Circ Res 1985;56:40–56. [DOI] [PubMed]
- 41.Pagnamenta A, Vanderpool R, Brimioulle S, Naeije R. Proximal pulmonary arterial obstruction decreases the time constant of the pulmonary circulation and increases right ventricular afterload. J Appl Physiol 2013;114:1586–1592. [DOI] [PubMed]
- 42.Castelain V, Hervé P, Lecarpentier Y, Duroux P, Simonneau G, Chemla D. Pulmonary artery pulse pressure and wave reflection in chronic pulmonary thromboembolism and primary pulmonary hypertension. J Am Coll Cardiol 2001;37:1085–1092. [DOI] [PubMed]
- 43.MacKenzie Ross RV, Toshner MR, Soon E, Naeije R, Pepke-Zaba J. Decreased time constant of the pulmonary circulation in chronic thromboembolic pulmonary hypertension. Am J Physiol Heart Circ Physiol 2013;305:H259–H264. [DOI] [PMC free article] [PubMed]
- 44.McGregor M, Sniderman A. On pulmonary vascular resistance: the need for more precise definition. Am J Cardiol 1985;55:217–221. [DOI] [PubMed]
- 45.Laskey WK, Ferrari VA, Palevsky HI, Kussmaul WG. Ejection characteristics in primary pulmonary hypertension. Am J Cardiol 1993;71:1111–1114. [DOI] [PubMed]