

Abstract Abstract
The pathophysiologic alterations of patients with pulmonary arterial hypertension (PAH) are diverse. We aimed to determine novel pathogenic pathways from circulating proteins in patients with PAH. Multianalyte profiling (MAP) was used to measure 90 specifically selected antigens in the plasma of 113 PAH patients and 51 control patients. Erythropoietin (EPO) functional activity was assessed via in vitro pulmonary artery endothelial cell networking and smooth muscle cell proliferation assays. Fifty-eight patients had idiopathic PAH, whereas 55 had other forms of PAH; 5 had heritable PAH, 18 had connective tissue disease (15 with scleroderma and 3 with lupus erythematosis), 13 had portopulmonary hypertension, 6 had PAH associated with drugs or toxins, and 5 had congenital heart disease. The plasma-antigen profile of PAH revealed increased levels of several novel biomarkers, including EPO. Immune quantitative and histochemical studies revealed that EPO not only was significantly elevated in the plasma of PAH patients but also promoted pulmonary artery endothelial cell network formation and smooth muscle cell proliferation. MAP is a hypothesis-generating approach to identifying novel pathophysiologic pathways in PAH. EPO is upregulated in the circulation and lungs of patients with PAH and may affect endothelial and smooth muscle cell proliferation.
Keywords: pulmonary arterial hypertension, plasma proteomics, multianalyte profiling, erythropoietin
Introduction
The normal adult pulmonary circulation is a low-resistance system that accommodates blood flow from the right ventricle at relatively low pressures. In pulmonary arterial hypertension (PAH), increases in afterload result in volume and pressure overload of the right ventricle, leading to right-sided heart failure and premature death. PAH may occur in the absence of identifiable risks, termed idiopathic PAH (IPAH), or with specific mutations or familial patterns (heritable PAH [HPAH]). In addition, other nonidiopathic forms of PAH (NIPAH) may occur in association with portal hypertension (portopulmonary hypertension), HIV infection, drugs or toxins, congenital heart disease, and connective tissue diseases.
At the structural level, PAH is characterized by pulmonary artery vasoconstriction, remodeling, and reduced distensibility. Collectively, these changes may result in selective death and proliferation of subsets of pulmonary artery endothelial cells (ECs), as well as hypertrophy and hyperplasia of vascular smooth muscle cells (SMCs). Remodeling of the adventitial fibroblast layer and the recruitment of circulating inflammatory and EC precursors to the pulmonary vascular wall contribute.1,2 The profile of circulating proteins could provide insight into the pathophysiology of these vascular alterations.
We used quantitative and rapid multianalyte profiling (MAP) to identify, in patients with IPAH or NIPAH and healthy volunteers, specific circulating antigens with potential mechanistic roles in PAH. After finding upregulation of erythropoietin (EPO) in the circulation in PAH, we performed functional studies to document the importance of EPO in the vascular cell proliferation seen in PAH.
Methods
Study population
We recruited 113 patients with PAH from the Pulmonary Vascular Disease Program at the University of Pennsylvania. PAH was diagnosed according to established criteria, including mean pulmonary artery pressure of >25 mmHg, pulmonary vascular resistance of >3 Wood units, left ventricular end-diastolic or pulmonary capillary wedge pressure of <15 mmHg, and absence of other cardiac, pulmonary, or thromboembolic disease. Control plasma was obtained from 51 healthy volunteers. This study was approved by the University of Pennsylvania Institutional Review Board, and all participants provided written informed consent.
Sample collection and analysis
Blood was collected in potassium EDTA (ethylenediaminetetraacetic acid) vacutainers (BD Biosciences, San Jose, CA) from the proximal port of a pulmonary artery catheter or peripheral venipuncture a median of 78 days (IPAH) or 108 days (NIPAH) from catheterization. Samples were maintained upright for 30 minutes, centrifuged, aliquoted, and frozen, and plasma was stored at −80°C. Serum was obtained by adding thrombin (500 IU/mL) and calcium chloride (10% w/v) to plasma and allowing clot formation for 2 hours. Plasma samples were utilized to run a multiplex immunoassay and serum samples to conduct cell-based assays.
Ninety antigens involved in several oncologic, inflammatory, and immune-response pathways were measured in the plasma of PAH patients and controls with a multiplex immunoassay (Human MAP v1.6 platform, RBM, Austin, TX; Table S1).3 Briefly, antigens were calibrated by means of duplicated standard curves, and raw intensity measurements were converted to protein concentrations with proprietary software. Machine performance was verified with quality control samples for each antigen and randomly analyzed to eliminate any sequential bias produced by diagnosis, age, or plasma collection date. Final concentrations were expressed as fold changes.
Formalin-fixed, paraffin-embedded human lung tissue sections from the explanted lungs of 2 patients with IPAH and one with NIPAH who underwent lung transplantation were provided by the Pulmonary Hypertension Breakthrough Initiative consortium. IPAH patient 1 was a 58-year-old white woman, and patient 2 was a 47-year-old white woman. The NIPAH patient was a 28-year-old white woman with connective tissue disease. The control lung tissue was purchased from Biomax US (Rockville, MD; catalog no. BN04011). Sections were stained with a polyclonal antibody against EPO (sc-7956, 1∶100; Santa Cruz Biotechnology, Dallas).
Cell-based assays
Normal rat pulmonary artery ECs were kindly provided by Dr. Troy Stevens of the University of South Alabama. Cells plated onto 24-well plates precoated with 300 μL growth factor–depleted Matrigel (R&D, Minneapolis) were cultured in 10% fetal bovine serum in Dulbecco’s modified Eagle’s medium. IPAH serum from a patient expressing 1,560 pg/mL of EPO (detected by MAP) was added to wells (50% v/v) in the presence or absence of 200 μg of a neutralizing anti-EPO antibody (R&D) or control immunoglobulin G (IgG; DAKO, Glostrup, Denmark). After 24 hours in culture, EC networks were visualized with inverted phase contrast microscopy and quantified (Photoshop CS3). All experiments were performed in triplicate, and 6 images per condition were visualized and quantified.
Human pulmonary artery SMCs (Lonza, Walkersville, MD) were serum starved for 24 hours and then cultured in 2% serum media. PAH serum (10%) was added to cultures in the presence or absence of 10, 50, or 100 IU/mL of recombitant human EPO (rHuEPO; Epoetin alpha, Amgen, Thousand Oaks, CA); 2, 20, or 200 μL of neutralizing anti-EPO antibody (R&D); or 200 μL of control IgG (DAKO). To determine the percentage of cells entering S phase, cells were treated with EdU (5-ethynyl-2′-deoxyuridine), a thymidine analog. EdU-positive cells were detected with a Click-iT EdU assay (Invitrogen, Carlsbad, CA) and visualized with a Nikon Eclipse Ti microscope. All experiments were performed in triplicate, and each result represents a distinct set of experiments.
Statistical analyses
Differential analysis for the multiplex assay was performed with PaGE (Patterns from Gene Expression),4 a permutation t statistic method that controls for multiple testing using false discovery rate.5 Classification analysis was performed using random forests6 in R statistical software. This approach avoids dividing the data into arbitrary training and test sets, thereby maintaining the maximum number of replicates for both training and testing. Thus, the classification error rates reflect the full battery of samples. The classifier was also rebuilt by using a random set of 75 samples, validating on the remaining samples, and validating again after inclusion of 12 additional samples. Furthermore, we constructed a classifier utilizing a subcohort matched for age and sex, which included 20 IPAH patients (mean age 33 years), 8 NIPAH patients (mean age 36 years), and 11 controls (mean age 31 years). We performed a subset analysis of treated and untreated patients, including IPAH (treated: n = 51, untreated: n = 7), NIPAH (treated: n = 50, untreated: n = 5), and total PAH (treated: n = 101, untreated: n = 12). Finally, subjects and proteins were grouped via hierarchical clustering.7 In vitro experiments were analyzed via ANOVA with Duncan’s post hoc analysis.
Results
The study sample included 113 PAH patients and 51 healthy volunteers (controls; Table 1). Fifty-eight patients had IPAH, whereas 55 had NIPAH. Five had HPAH, 18 had connective tissue disease (15 with scleroderma and 3 with lupus erythematosis), 13 had portopulmonary hypertension, 6 had PAH associated with drugs or toxins, and 5 had congenital heart disease. Approximately half of the patients were treated with bosentan, and approximately 30% were receiving prostacyclin analogs.
Table 1.
Demographic and clinical characteristics
| Variable | IPAH (n = 58) | NIPAH(n = 55) | Control (n = 33) |
|---|---|---|---|
| Mean age ± SD, years | 51 ± 15 | 50 ± 11 | 27 ± 6 |
| Female sex | 45 (77) | 40 (72) | 10 (30) |
| Race/ethnicity | |||
| White (not of Hispanic origin) | 47 (81) | 42 (76) | 25 (76) |
| Black (not of Hispanic origin) | 6 (10) | 7 (13) | 4 (12) |
| Hispanic | 4 (7) | 5 (9) | 1 (3) |
| Asian | 1 (2) | 1 (2) | 3 (9) |
| NIPAH | |||
| Heritable PAH | 5 (9) | ||
| Portopulmonary hypertension | 13 (24) | ||
| Drugs and toxins | 6 (11) | ||
| Connective tissue diseases | 18 (33) | ||
| Other | 13 (23) | ||
| WHO functional class | |||
| I | 3 (5) | 0 (0) | |
| II | 19 (33) | 16 (29) | |
| III | 33 (57) | 37 (67) | |
| IV | 3 (5) | 2 (4) | |
| Hemodynamics | |||
| Mean right atrial pressure ± SD, mmHg | 10 ± 7 | 10 ± 6 | |
| Mean pulmonary artery pressure ± SD, mmHg | 47 ± 14 | 45 ± 13 | |
| Mean pulmonary capillary wedge pressure ± SD, mmHg | 11 ± 6 | 10 ± 4 | |
| Mean pulmonary vascular resistance ± SD, dyn s cm−5 | 789 ± 439 | 631 ± 366 | |
| Mean cardiac index ± SD, L/min/m2 | 2.4 ± 0.9 | 2.7 ± 0.9 | |
| Treatment | |||
| Calcium channel blocker | 19 (33) | 14 (25) | |
| Inhaled iloprost | 13 (22) | 7 (13) | |
| Intravenous epoprostenol | 12 (21) | 10 (18) | |
| Ambrisentan | 1 (2) | 0 (0) | |
| Bosentan | 29 (50) | 25 (45) | |
| Sildenafil | 4 (7) | 5 (9) |
Data are no. (%) of patients unless otherwise noted. PAH: pulmonary arterial hypertension; IPAH: idiopathic PAH; NIPAH: nonidiopathic PAH; WHO: World Health Organization.
MAP indicated that 41 antigens were significantly increased (n = 38) or decreased (n = 3) in both PAH groups when compared to the control group (Table 2), after multiple comparisons were accounted for. In addition, levels of 28 antigens distinguished IPAH from NIPAH patients. A heat map, tree-view diagram (Fig. S1) shows specific clusters of antigens within PAH patient or control groups. IPAH, NIPAH, and control groups were separated into subclusters. Random forests classification with the initial 164 samples resulted in a list of antigens ranked by their discriminatory power. The best discriminators of PAH patients from controls are shown in Table 3, and the best discriminators between IPAH and NIPAH patients are shown in Table 4. The prediction algorithm reported an “out-of-bag” error rate of 0 when classifying control versus PAH patients and error rates of 0.17 and 0.25 when classifying controls versus IPAH and NIPAH patients, respectively (Table 5; also see Table S2).
Table 2.
Multianalyte profiling analysis data for fold change in antigen concentration
| Antigen | IPAH vs. control | NIPAH vs. control | IPAH vs. NIPAH | Swiss-Prot accession no. |
|---|---|---|---|---|
| Adiponectin | 2.4a | 2.4a | 1.0 | Q15848 |
| Beta-2 microglobulin | 3.0a | 3.3a | 0.9 | P23560 |
| Creatine kinase-MB | 1.9a | 1.8a | 1.1 | P06732 |
| ENRAGE | 16.2a | 11.9a | 1.4 | P80511 |
| Eotaxin | 1.9a | 1.8a | 1.0 | P51671 |
| Erythropoietin | 7.9a | 13.6a | 0.6 | P01588 |
| Fibrinogen | 1.5a | 1.4a | 1.1 | P02671 |
| GM-CSF | 3.1a | 2.5a | 1.2 | P04141 |
| IL-2 | 6.2a | 4.9a | 1.3 | P01585 |
| IL-10 | 1.7a | 1.6a | 1.0 | P22301 |
| IL-16 | 5.1a | 4.2a | 1.2 | Q14005 |
| IL-18 | 2.3a | 2.2a | 1.0 | AJ295724.1* |
| IL-1ra | 5.0a | 10.9a | 0.5 | P18510 |
| IL-5 | 1.9a | 1.8a | 1.1 | P05113 |
| IL-8 | 1.7a | 2.0a | 0.9 | P10145 |
| Lipoprotein (a) | 1.6a | 2.3a | 0.7 | P08519 |
| MCP-1 | 2.0a | 1.9a | 1.1 | P13500 |
| MDC | 1.8a | 1.4a | 1.2 | O00626 |
| MIP-1β | 2.1a | 3.2a | 0.7 | P13236 |
| Myeloperoxidase | 8.3a | 12.6a | 0.7 | P05164 |
| Myoglobin | 2.0a | 2.0a | 1.0 | P02144 |
| MMP-9 | 1.4a | 3.1a | 0.5 | P14780 |
| TNF RII | 2.2a | 2.6a | 0.8 | Q92956 |
| TNF-beta | 3.8a | 3.4a | 1.1 | P01374 |
| Von Willebrand factor | 2.4a | 2.3a | 1.1 | P04275 |
| Endothelin-1 | 2.8a | 1.4 | 2.0 | P05305 |
| G-CSF | 1.5a | 1.3 | 1.2 | P09919 |
| IL-3 | 1.3a | 1.0 | 1.3 | P08700 |
| Complement 3 | 1.1 | 1.1a | 1.0 | P01024 |
| Cancer antigen 19–9 | 1.2 | 2.0a | 0.6 | Q9BXJ9 |
| CD40 | 2.0 | 2.2a | 0.9 | Q6P2H9 |
| Fatty acid–binding protein | 4.0 | 4.1a | 1.0 | P05413 |
| Haptoglobin | 1.0 | 1.5a | 0.7 | P00738 |
| IL-1α | 24.5 | 2.2a | 11.1 | P18510 |
| Thyroid-stimulating hormone | 1.3 | 2.6a | 0.5 | P01215 |
| Insulin | 1.3 | 1.7a | 0.8 | P01308 |
| IL-12p40 | 4.2a | 2.7a | 1.6d | P29460 |
| IL-12p70 | 4.5a | 1.4a | 3.2d | P29460 |
| PAPP-A | 3.8a | 1.7a | 2.3d | Q13219 |
| TNF-alpha | 4.4a | 2.6a | 1.7d | P01375 |
| IL-13 | 0.6b | 0.4b | 1.4d | P35225 |
| Alpha-2 macroglobulin | 0.5b | 0.4b | 1.4 | P01023 |
| Factor VII | 0.5b | 0.6b | 0.8 | P08709 |
| Cancer antigen 125 | 3.6a | 13.6a | 0.3c | Q14596 |
| C reactive protein | 7.6a | 14.5a | 0.5c | P02741 |
| ICAM-1 | 1.6a | 2.2a | 0.7c | P05362 |
| IgA | 1.3a | 1.7a | 0.8c | AJ222547.1* |
| IL-6 | 88.2a | 186.1a | 0.5c | P05231 |
| Leptin | 2.4a | 4.0a | 0.6c | P41159 |
| PAI-1 | 1.8a | 2.2a | 0.8c | P05121 |
| TIMP-1 | 1.8a | 2.2a | 0.8c | P01033 |
| VCAM-1 | 1.8a | 2.2a | 0.8c | P19320 |
| Alpha-1 antitrypsin | 1.1 | 1.3a | 0.9c | P07758 |
| IgM | 0.9 | 1.2a | 0.8c | BAA12061* |
| MMP-2 | 1.0 | 1.7a | 0.6c | P08253 |
| Serum amyloid P | 0.9 | 1.2a | 0.8c | P02743 |
| Stem cell factor | 0.8 | 1.2a | 0.7c | P21583 |
| Thyroxine-binding globulin | 1.1 | 1.3a | 0.8c | P05543 |
| Tenascin-C | 1.0 | 1.8a | 0.6c | X78565.1* |
| Tissue factor | 1.1 | 2.6a | 0.4c | P13726 |
| VEGF | 0.8 | 2.0a | 0.4c | P15692 |
| Apolipoprotein A1 | 0.8b | 1.0 | 0.8c | P02647 |
| SGOT | 0.6b | 1.0 | 0.6c | P17174 |
| Apolipoprotein CIII | 0.7b | 0.8 | 1.0 | P02656 |
| IGF-1 | 0.3b | 0.5 | 0.6 | P01343 |
| IL-4 | 0.8 | 0.5b | 1.7d | P05112 |
| Lymphotactin | 1.0 | 0.7b | 1.4d | P47992 |
| IL-15 | 1.3a | 0.9 | 1.5d | P40993 |
IPAH: idiopathic pulmonary hypertension; NIPAH: nonidiopathic pulmonary hypertension. ENRAGE: extracellular newly identified RAGE (receptor for advanced-glycation end products)-binding protein; G-CSF: granulocyte colony-stimulating factor; GM-CSF: granulocyte-macrophage colony-stimulating factor; ICAM: intracellular adhesion molecule; Ig: immunoglobulin; IGF: insulin-like growth factor; IL: interleukin; MCP: monocyte chemotactic protein; MDC: macrophage-derived chemokine; MIP: macrophage inflammatory proetin; MMP: matrix metalloproteinase; PAI: plasminogen activator inhibitor; PAPP: pregnancy-associated plasma protein; SGOT: serum glutamic oxalacetic transaminase; TIMP: tissue inhibitor of metalloproteinases; TNF: tumor necrosis factor; TNF RII: TNF receptor II; VCAM: vascular cell adhesion protein; VEGF: vascular endothelial growth factor. Asterisk indicates NCBI (National Center for Biotechnology Information) number.
Significant increase versus controls.
Significant decrease versus controls.
Significant increase in IPAH versus NIPAH patients.
Significant decrease in IPAH versus NIPAH patients.
Table 3.
Random forests variable-importance values of proteins for classifying pulmonary arterial hypertension patients and control subjects
| Protein | Values |
|---|---|
| Beta-2 microglobulin | 8.548995 |
| IL-16 | 5.426744 |
| C reactive protein | 3.397961 |
| TIMP-1 | 2.617926 |
| MIP-1β | 2.483686 |
| PAI-1 | 2.43742 |
| IL-15 | 2.004102 |
| Lymphotactin | 1.989372 |
| IL-18 | 1.689985 |
| ICAM-1 | 1.591548 |
| VCAM-1 | 1.523685 |
| Cancer antigen 125 | 1.384795 |
| TNF RII | 1.255459 |
| Alpha-2 macroglobulin | 1.22494 |
| Myeloperoxidase | 1.2009 |
| IL-13 | 1.182275 |
| TNF-alpha | 1.145184 |
| Factor VII | 1.101884 |
See Table 2 for abbreviations.
Table 4.
Random forests variable-importance values for classifying idiopathic PAH patients, nonidiopathic PAH patients, and control subjects
| Protein | Values |
|---|---|
| Beta-2 microglobulin | 7.795031 |
| SGOT | 5.026216 |
| IL-16 | 4.108143 |
| C reactive protein | 3.583839 |
| PAPP-A | 3.422608 |
| Cancer antigen 125 | 3.386835 |
| VEGF | 2.739381 |
| TIMP-1 | 2.670368 |
| ICAM-1 | 2.407055 |
| PAI-1 | 2.330771 |
| IL-15 | 2.281829 |
| Tissue factor | 2.246911 |
| Fatty acid–binding protein | 2.011522 |
| MIP-1β | 1.992987 |
| Lymphotactin | 1.749701 |
| Stem cell factor | 1.682598 |
| TNF-alpha | 1.663006 |
| MMP-2 | 1.657007 |
| Factor VII | 1.635494 |
| Tenascin-C | 1.562257 |
| TNF RII | 1.509915 |
| Myeloperoxidase | 1.502262 |
| IL-18 | 1.454997 |
| VCAM-1 | 1.436826 |
| IL-4 | 1.41256 |
| Adiponectin | 1.267817 |
| GM-CSF | 1.182589 |
| IL-12p70 | 1.170181 |
| CD40 | 1.107959 |
| Alpha-2 macroglobulin | 1.053929 |
| TNF-beta | 1.05057 |
PAH: pulmonary arterial hypertension. See Table 2 for other abbreviations.
Table 5.
“Out-of-bag” random forests error rates for classifications
| Actual | Predicted control | Predicted PAH | Predicted IPAH | Predicted NIPAH | Error |
|---|---|---|---|---|---|
| Control | 51 | 0 | 0 | 0 | 0 |
| PAH | 0 | 113 | 0 | ||
| IPAH | 1 | 48 | 9 | 0.17 | |
| NIPAH | 0 | 8 | 47 | 0.25 |
PAH: pulmonary arterial hypertension; IPAH: idiopathic PAH; NIPAH: nonidiopathic PAH.
Sensitivity analyses using the other validation strategies described in “Methods” differentiated PAH from controls with 100% accuracy. To account for the differences in age and sex between the PAH patients and controls and the potential effect of treatment, classifiers were rebuilt with age-matched samples for women only and for treated and untreated patients. Results were unchanged, and treated and untreated patients did not show distinct profiles (data not shown).
EPO protein levels are increased in plasma and lung tissue in PAH. Levels of EPO were increased by 7.9- and 13.6-fold in IPAH and NIPAH patients, respectively, compared to controls (Table 2). EPO immunostaining was greater in IPAH and NIPAH lung tissue sections, compared to that in controls (Fig. 1). Furthermore, EPO was significantly increased in the vascular SMC layer of remodeled large, medium, and small pulmonary arteries in PAH lung tissue (Fig. 1F–1H). Accompanying the increased EPO expression, PAH lesions exhibited a marked increase in inflammatory cells resembling hemosiderin-laden macrophages and demonstrating strong immunoreactivity for EPO.
Figure 1.

Expression and localization of erythropoietin (EPO) is increased in pulmonary arterial hypertension lung tissue. EPO is highly expressed in smooth muscle cells and endothelial cells of arteries showing muscular hypertrophy and plexiform lesions. EPO is also localized in alveolar epithelial cells. Inflammatory cells show positive staining for EPO in pulmonary arterial hypertension (PAH). IPAH: idiopathic PAH; NIPAH: nonidiopathic PAH. Scale bar: 80 μm.
We next determined whether EPO promoted EC networking, a feature of PAH pathobiology,1 within a reconstituted basement membrane assay. The use of 10% IPAH serum, as well as coincubation with control IgG, resulted in the formation of a robust network composed of interconnected pulmonary artery ECs and displaying prominent and multiple cellular extensions (Fig. 2A, top and middle). When these same cultures were coincubated with an EPO-neutralizing antibody, EC networking was significantly attenuated (Fig. 2A, bottom, and Fig. 2B). Thus, EPO in PAH patient serum appears to have bioactivity on pulmonary ECs in vitro.
Figure 2.

Erythropoietin (EPO) from pulmonary arterial hypertension (PAH) serum promotes pulmonary artery endothelial cell (EC) networking in vitro. A, Pulmonary artery EC in 10% idiopathic PAH (IPAH) serum (top), with control immunoglobulin G (IgG; middle), or with 200 uL EPO-neutralizing antibody (antiEPO; bottom). B, Cell coverage area; a single asterisk indicates P < 0.05; a double asterisk indicates P > 0.05.
To assess the effect of circulating EPO in PAH on SMC proliferation, human pulmonary artery SMCs were exposed to either rHuEPO or PAH patient serum containing high levels of EPO. Both rHuEPO and IPAH serum promoted SMC proliferation in a dose-dependent manner (Fig. 3A, 3B). Coincubation of serum-treated cultures with 200 μL of EPO-neutralizing antibody attenuated SMC proliferation in a dose-dependent manner (Fig. 3C), whereas control IgG had no effect (Fig. 3B). Anti-EPO 2 and 20 μL did not reverse the 10% IPAH group to “control” levels.
Figure 3.
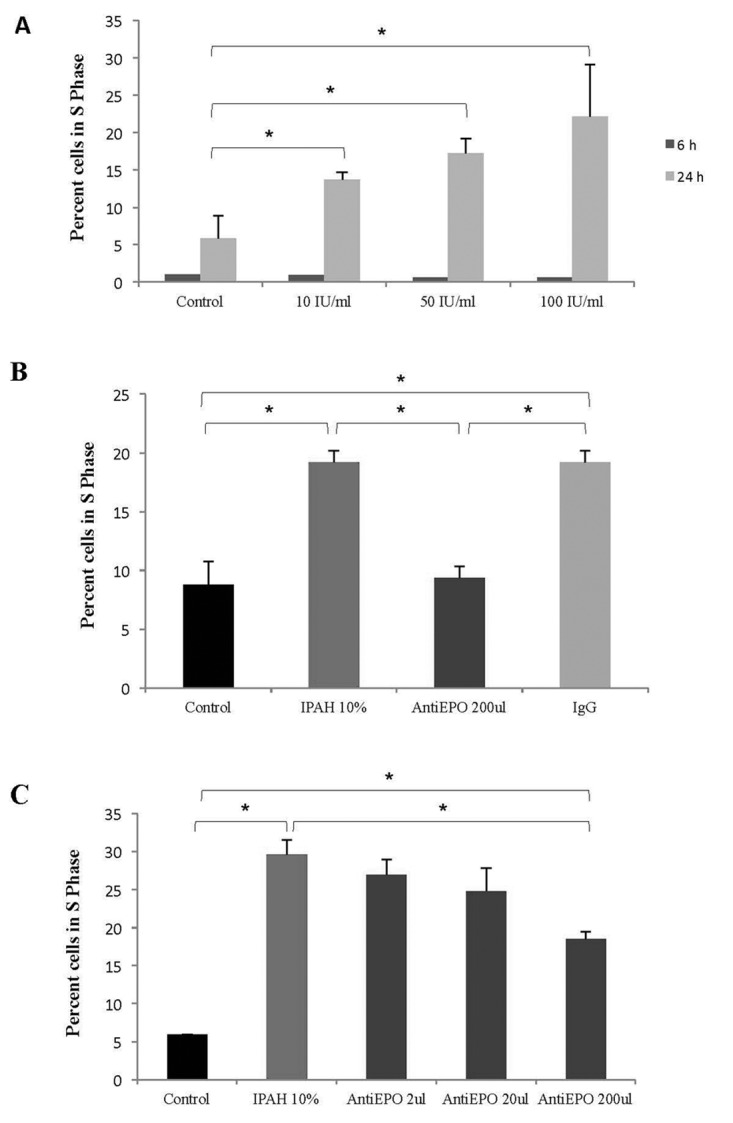
Erythropoietin (EPO) from pulmonary arterial hypertension (PAH) serum promotes pulmonary artery smooth muscle cell (SMC) proliferation in vitro. A, Increased concentrations of recombitant human EPO (10, 50, or 100 IU/mL) promote SMC proliferation in a dose-dependent manner. B, C, Treatment of SMCs with 10% idiopathic PAH (IPAH) serum increases their proliferation, which was inhibited by EPO-neutralizing antibody in a dose-dependent manner. Immunoglobulin G (IgG) control antibody did not affect SMC proliferation (B). Asterisk indicates P < 0.05.
Discussion
We have used a high-throughput hypothesis-generating technique to identify novel antigens differentially expressed in the blood of almost 150 patients with PAH, a much larger sample than in other published proteomic studies in PAH.8-13 We showed that EPO is significantly elevated in PAH plasma and expressed in pulmonary vasculature and that it promotes vascular networking and cell proliferation in pulmonary artery ECs and SMCs, respectively. Since PAH is inherently a complex phenotype, we believe that a single biochemical or protein signature approach is unlikely to capture the disease complexity for diagnostic or clinical purposes. However, this study demonstrates the ability to translate data from hypothesis-generating studies into confirmatory functional experiments using tissue from patients with PAH.
Interleukins (IL-)2, 5, 6, 8, 10, 16, and 18 and myeloperoxidase were upregulated in PAH, and many of these cytokines are associated with outcomes in PAH.14 IL-6 showed the largest change, with an 88-fold increase in IPAH and a 186-fold increase in NIPAH. IL-6 increases MAPKs (mitogen-activated protein kinases), c-myc, survivin, and bcl-2, with both proproliferative and antiapoptotic effects, and transgenic mice overexpressing IL-6 develop pulmonary hypertension.15 These findings are consistent with studies showing increased circulating levels of IL-614,16 and increases in chemokine and chemokine receptor gene expression in PAH.17
Matrix metalloproteinase (MMP)-2, MMP-9, and tenascin-C (TN-C) were elevated in NIPAH but not in IPAH patient plasma. TN-C is activated and/or induced in multiple forms of clinical and experimental PAH18-22 and may contribute to PAH via its ability to cross modulate the activity of epithelial growth factor (EGF) receptors, as well as via its control of cell migration and programmed cell death. Furthermore, ENRAGE (extracellular newly identified RAGE-binding protein, S100A12) showed a 16.2-fold increase in IPAH patients and an 11.9-fold increase in NIPAH patients. Interestingly, the receptor for advanced-glycation end products (RAGE) and its ligands (specifically S100A12) have been implicated in neointimal formation and vascular SMC proliferation following vascular injury.23 Female mice overexpressing S100A4/Mts1 had an increased risk of severe pulmonary vascular pathology.24 In addition, advanced-glycation end products induce significant SMC migration and increase vascular permeability by promoting an inflammatory response in ECs.25
We focused on the functional importance of EPO, since this protein showed one of the highest increases in IPAH (7.9-fold) and NIPAH ( ˜13.6-fold) patients when compared to controls. EPO increases ET-1 release from ECs, and EPO receptors play an important role in neoangiogenesis through upregulation of the vascular EGF–EGF receptor system, as well as via recruitment of hematopoietic progenitor cells to ischemic tissue.26-28 Interestingly, recent data29 demonstrate the role of EPO as a hypoxia-inducible factor 1α (HIF-1α)–inducible myeloid-activating factor in PAH. Little is known about the effects of EPO on human pulmonary artery SMCs. Exposing cultures to high concentrations of EPO from PAH patient serum increased the networking activity of pulmonary artery ECs and increased SMC proliferation.
While there is a lack of consistent data on EPO production in healthy adult lungs, EPO receptor expression has been detected in the lung mesothelium, chondrocytes, alveolar cells, vascular ECs, smooth muscle fibers, macrophages, and neutrophils.30 The source of increased EPO in PAH is not clear, although we found inflammatory cells located in the alveolar wall to be highly immunoreactive to EPO. Our findings are intriguing, given that other factors involved in stem cell mobilization and/or recruitment (including granulocyte-macrophage colony-stimulating factor and MMP-9) were also increased in IPAH and/or NIPAH patients (Table 2).
Our study has some limitations. Antigen levels may have been affected by PAH therapy, but subset analyses did not suggest differences in results by treatment. In addition, this study did not examine prognosis, which would require a much larger number of patients, to account for the multiple comparisons, and a significant amount of follow-up time. Even though NIPAH patients seemed to show more extreme changes in protein expression than IPAH patients, there were not enough patients in each NIPAH subgroup to detect differences. Similarly, men and ethnic minorities constituted subgroups too small for useful subset analyses. We did not find associations between protein signatures and severity of PAH; however, this may be attributable to the plasma protein and hemodynamic assessments being performed at separate times in many subjects. Finally, the detection of antigens was conducted only in the adult population; our findings may not be generalizable to a pediatric population.
In conclusion, we used the plasma antigen profile in PAH patients to suggest pathogenic targets for functional studies. Upregulation of EPO in plasma, in addition to in vitro lung effects, suggests that EPO may be an important factor in the development or progression of PAH. Future studies of the antigens characterized in our study and the role of EPO in PAH are warranted.
Acknowledgments
We would like to acknowledge the patients and volunteers who participated in this study. We would also like to thank Dr. Scott Diamond, Sandra Kaplan, Chris Archer-Chicko, Dr. Jorge Mercado, Dr. Mercedes Lioni, Dr. Mathieu Tamby, Patricia del Rosario, and Andrew Hsi. Tissue samples were provided under the Pulmonary Hypertension Breakthrough Initiative (PHBI). Other investigators may have received specimens from the same subjects. We are especially grateful to Dr. Marlene Rabinovitch (Stanford University) and Dr. George Noon (Baylor) in the PHBI.
Appendix. Supplemental material
Figure S1.

Heat map expression profile and clustering diagram of antigen levels in idiopathic pulmonary arterial hypertension (IPAH), non-idiopathic pulmonary arterial hypertension (NIPAH), and controls. See Table S1 for antigen names.
Table S1.
Antigens used in multianalyte profiling (MAP)
| Adiponectin | Growth hormone | MCP-1 |
| Alpha-1 antitrypsin | GM-CSF | MDC |
| Alpha-2 macroglobulin | Glutathione S-transferase | MIP-1α |
| Alpha-fetoprotein | Haptoglobin | MIP-1β |
| Apolipoprotein A1 | ICAM-1 | MMP-2 |
| Apolipoprotein CIII | IgA | MMP-3 |
| Apolipoprotein H | IgE | MMP-9 |
| Beta-2 microglobulin | IGF-1 | Myeloperoxidase |
| Brain-derived neurotrophic factor | IgM | Myoglobin |
| Complement 3 | IL-10 | PAI-1 |
| Cancer antigen 125 | IL-12p40 | Prostatic acid phosphatase |
| Cancer antigen 19–9 | IL-12p70 | PAPP-A |
| Calcitonin | IL-13 | Prostate-specific antigen, free |
| CD40 | IL-15 | RANTES |
| CD40 ligand | IL-16 | Serum amyloid P |
| Carcinoembryonic antigen | IL-18 | Stem cell factor |
| Creatine kinase-MB | IL-1α | SGOT |
| C reactive protein | IL-1β | SHBG |
| EGF | IL-1ra | Thyroxine-binding globulin |
| ENA-78 | IL-2 | Tenascin-C |
| Endothelin-1 | IL-3 | Tissue factor |
| ENRAGE | IL-4 | TIMP-1 |
| Eotaxin | IL-5 | TNF RII |
| Erythropoietin | IL-6 | TNF-alpha |
| Fatty acid–binding protein | IL-7 | TNF-beta |
| Factor VII | IL-8 | Thrombopoietin |
| Ferritin | Insulin | Thyroid-stimulating hormone |
| FGF basic | Leptin | VCAM-1 |
| Fibrinogen | Lipoprotein (a) | VEGF |
| G-CSF | Lymphotactin | Von Willebrand factor |
Human MAP v1.6 platform (RMB [Rules Based Medicine], a Clinical Laboratory Improvement Amendments–certified laboratory in Austin, TX). EGF: epidermal growth factor; ENA: epithelial-derived neutrophil-activating protein; ENRAGE: extracellular newly identified RAGE (receptor for advanced-glycation end products)-binding protein; FGF: fibroblast growth factor; G-CSF: granulocyte colony-stimulating factor; GM-CSF: granulocyte-macrophage colony-stimulating factor; ICAM: intracellular adhesion molecule; Ig: immunoglobulin; IGF: insulin-like growth factor; IL: interleukin; MCP: monocyte chemotactic protein; MDC: macrophage-derived chemokine; MIP: macrophage inflammatory proetin; MMP: matrix metalloproteinase; PAI: plasminogen activator inhibitor; PAPP: pregnancy-associated plasma protein; RANTES: regulated upon activation, normally T-cell expressed, and secreted; SGOT: serum glutamic oxalacetic transaminase; SHBG: sex hormone–binding globulin; TIMP: tissue inhibitor of metalloproteinases; TNF: tumor necrosis factor; TNF RII: TNF receptor II; VCAM: vascular cell adhesion protein; VEGF: vascular endothelial growth factor.
Table S2.
“Out-of-bag” random forests error rates for classifications with age-matched samples from 20 women with IPAH, 11 women with NIPAH, and 10 female controls
| Actual | Predicted control | Predicted PAH | Predicted IPAH | Predicted NIPAH | Error |
|---|---|---|---|---|---|
| Control | 9 | 1 | 1 | 0 | 0.1 |
| PAH | 0 | 31 | 0 | ||
| IPAH | 0 | 19 | 1 | 0.17 | |
| NIPAH | 0 | 5 | 6 | 0.25 |
PAH: pulmonary arterial hypertension; IPAH: idiopathic PAH; NIPAH: nonidiopathic PAH.
Source of Support: Support for this work came from the Pulmonary Hypertension Breakthrough Initiative (funding for the PHBI is provided by the Cardiovascular Medical Research Education Fund); National Institutes of Health grants HL079196, HL086719, and HL103844; a Development Partners’ Junior Faculty Development Award from GlaxoSmithKline; and a McCabe Fellowship Award.
Conflict of Interest: SMK reports grants from Actelion, United Therapeutics, Gilead, Lung Rx, Pfizer, Ikaria, the Pulmonary Hypertension Association, Merck, and GeNO. HIP reports grants from Actelion, United Therapeutics, Gilead, Lung Rx, Pfizer, Ikaria, the Pulmonary Hypertension Association, Merck, and GeNO; he has served on study Data and Safety Monitoring Boards for Aires and Pfizer and as a consultant for Bayer and Actelion. DBT reports fees from the Pulmonary Hypertension Association and Cornerstone and grants from Actelion; in addition, he has a pending patent, filed but not completed/pursued by the University of Pennsylvania, related to blood-based approaches to diagnosing PAH pending. RTZ reports receiving grants, fees, and nonfinancial support from United Therapeutics and grants and nonfinancial support from Actelion; he has served on a Scientific Board for United Therapeutics. The other authors report no conflicts of interest relevant to this work.
References
- 1.Davie NJ, Gerasimovskaya EV, Hofmeister SE, Richman AP, Jones PL, Reeves JT, Stenmark KR. Pulmonary artery adventitial fibroblasts cooperate with vasa vasorum endothelial cells to regulate vasa vasorum neovascularization: a process mediated by hypoxia and endothelin-1. Am J Pathol 2006;168:1793–1807. [DOI] [PMC free article] [PubMed]
- 2.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Stenmark KR. Circulating mononuclear cells with a dual, macrophage-fibroblast phenotype contribute robustly to hypoxia-induced pulmonary adventitial remodeling. Chest 2005;128(6 Suppl.):583S–584S. [DOI] [PubMed]
- 3.Bertenshaw GP, Yip P, Seshaiah P, Zhao J, Chen TH, Wiggins WS, Mapes JP, Mansfield BC. Multianalyte profiling of serum antigens and autoimmune and infectious disease molecules to identify biomarkers dysregulated in epithelial ovarian cancer. Cancer Epidemiol Biomark Prev 2008;17:2872–2881. [DOI] [PubMed]
- 4.Grant GR, Liu J, Stoeckert CJ Jr. A practical false discovery rate approach to identifying patterns of differential expression in microarray data. Bioinformatics 2005;21:2684–2690. [DOI] [PubMed]
- 5.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 2001;125:279–284. [DOI] [PubMed]
- 6.Breiman L. Random forest. Mach Learn 2001;45(1):5–32.
- 7.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 1998;95:14863–14868. [DOI] [PMC free article] [PubMed]
- 8.Abdul-Salam VB, Paul GA, Ali JO, Gibbs SR, Rahman D, Taylor GW, Wilkins MR, Edwards RJ. Identification of plasma protein biomarkers associated with idiopathic pulmonary arterial hypertension. Proteomics 2006;6:2286–2294. [DOI] [PubMed]
- 9.Abdul-Salam VB, Wharton J, Cupitt J, Berryman M, Edwards RJ, Wilkins MR. Proteomic analysis of lung tissues from patients with pulmonary arterial hypertension. Circulation 2010;122:2058–2067. [DOI] [PubMed]
- 10.Meyrick BO, Friedman DB, Billheimer DD, Cogan JD, Prince MA, Phillips JA III, Loyd JE. Proteomics of transformed lymphocytes from a family with familial pulmonary arterial hypertension. Am J Respir Crit Care Med 2008;177:99–107. [DOI] [PMC free article] [PubMed]
- 11.Yeager ME, Colvin KL, Everett AD, Stenmark KR, Ivy DD. Plasma proteomics of differential outcome to long-term therapy in children with idiopathic pulmonary arterial hypertension. Proteomics Clin Appl 2012;6:257–267. [DOI] [PMC free article] [PubMed]
- 12.Yu M, Wang XX, Zhang FR, Shang YP, Du YX, Chen HJ, Chen JZ. Proteomic analysis of the serum in patients with idiopathic pulmonary arterial hypertension. J Zhejiang Univ Sci B 2007;8:221–227. [DOI] [PMC free article] [PubMed]
- 13.Zhang J, Zhang Y, Li N, Liu Z, Xiong C, Ni X, Pu Y, Hui R, He J, Pu J. Potential diagnostic biomarkers in serum of idiopathic pulmonary arterial hypertension. Respir Med 2009;103:1801–1806. [DOI] [PubMed]
- 14.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010;122:920–927. [DOI] [PubMed]
- 15.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res 2009;104:236–244. [DOI] [PMC free article] [PubMed]
- 16.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 1995;151:1628–1631. [DOI] [PubMed]
- 17.Bull TM, Coldren CD, Moore M, Sotto-Santiago SM, Pham DV, Nana-Sinkam SP, Voelkel NF, Geraci MW. Gene microarray analysis of peripheral blood cells in pulmonary arterial hypertension. Am J Respir Crit Care Med 2004;170:911–919. [DOI] [PubMed]
- 18.Jones PL, Cowan KN, Rabinovitch M. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am J Pathol 1997;150:1349–1360. [PMC free article] [PubMed]
- 19.Ihida-Stansbury K, McKean DM, Lane KB, Loyd JE, Wheeler LA, Morrell NW, Jones PL. Tenascin-C is induced by mutated BMP type II receptors in familial forms of pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2006;291:L694–L702. [DOI] [PubMed]
- 20.Jones PL, Rabinovitch M. Tenascin-C is induced with progressive pulmonary vascular disease in rats and is functionally related to increased smooth muscle cell proliferation. Circ Res 1996;79:1131–1142. [DOI] [PubMed]
- 21.Cowan KN, Jones PL, Rabinovitch M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest 2000;105:21–34. [DOI] [PMC free article] [PubMed]
- 22.Ivy DD, McMurtry IF, Colvin K, Imamura M, Oka M, Lee DS, Gebb S, Jones PL. Development of occlusive neointimal lesions in distal pulmonary arteries of endothelin B receptor-deficient rats: a new model of severe pulmonary arterial hypertension. Circulation 2005;111:2988–2996. [DOI] [PMC free article] [PubMed]
- 23.Zhou Z, Wang K, Penn MS, Marso SP, Lauer MA, Forudi F, Zhou X, et al. Receptor for AGE (RAGE) mediates neointimal formation in response to arterial injury. Circulation 2003;107:2238–2243. [DOI] [PubMed]
- 24.Dempsie Y, Nilsen M, White K, Mair KM, Loughlin L, Ambartsumian N, Rabinovitch M, Maclean MR. Development of pulmonary arterial hypertension in mice over-expressing S100A4/Mts1 is specific to females. Respir Res 2011;12:159. [DOI] [PMC free article] [PubMed]
- 25.Higashi T, Sano H, Saishoji T, Ikeda K, Jinnouchi Y, Kanzaki T, Morisaki N, Rauvala H, Shichiri M, Horiuchi S. The receptor for advanced glycation end products mediates the chemotaxis of rabbit smooth muscle cells. Diabetes 1997;46:463–472. [DOI] [PubMed]
- 26.Buemi M, Senatore M, Gallo GC, Crasci E, Campo S, Sturiale A, Coppolino G, Bolignano D, Frisina N. Pulmonary hypertension and erythropoietin. Kidney Blood Press Res 2007;30:248–252. [DOI] [PubMed]
- 27.Carlini RG, Dusso AS, Obialo CI, Alvarez UM, Rothstein M. Recombinant human erythropoietin (rHuEPO) increases endothelin-1 release by endothelial cells. Kidney Int 1993;43:1010–1014. [DOI] [PubMed]
- 28.Nakano M, Satoh K, Fukumoto Y, Ito Y, Kagaya Y, Ishii N, Sugamura K, Shimokawa H. Important role of erythropoietin receptor to promote VEGF expression and angiogenesis in peripheral ischemia in mice. Circ Res 2007;100:662–669. [DOI] [PubMed]
- 29.Farha S, Asosingh K, Xu W, Sharp J, George D, Comhair S, Park M, et al. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood 117:3485–3493. [DOI] [PMC free article] [PubMed]
- 30.Yasuda Y, Hara S, Hirohata T, Koike E, Yamasaki H, Okumoto K, Hoshiai H. Erythropoietin-responsive sites in normal and malignant human lung tissues. Anat Sci Int 85:204–213. [DOI] [PubMed]