

Abstract Abstract
Endothelin-1 is a potent mediator of sepsis-induced pulmonary hypertension (PH). The pulmonary vascular effects of selective blockade of endothelin receptor subtype A (ETAR) during endotoxemia remain unknown. We hypothesized that selective ETAR antagonism attenuates endotoxin-induced PH and improves pulmonary artery (PA) vasoreactivity. Adult male Sprague-Dawley rats (250–450 g) received lipopolysaccharide (LPS; Salmonella typhimurium; 20 mg/kg intraperitoneally) or vehicle 6 hours before hemodynamic assessment and tissue harvest. The selective ETAR antagonist sitaxsentan (10 or 20 mg/kg) or vehicle was injected intravenously 3 hours after receipt of LPS. Right ventricular systolic pressure, mean arterial pressure (MAP), cardiac output (CO), oxygenation (P/F ratio), and serum bicarbonate were measured. Bronchoalveolar lavage (BAL) cell differential and lung wet-to-dry ratios were obtained. Endothelium-dependent and endothelium-independent vasorelaxations were determined in isolated PA rings. PA interleukin (IL)-1β, IL-6, tumor necrosis factor α (TNF-α), and inducible nitric oxide synthase (iNOS) messenger RNA (mRNA) were measured. LPS caused PH, decreased MAP, CO, and serum bicarbonate, and increased PA IL-1β, IL-6, TNF-α, and iNOS mRNA. Sitaxsentan attenuated sepsis-induced PH and increased MAP. The P/F ratio, CO, serum bicarbonate, and BAL neutrophilia were not affected by sitaxsentan. In isolated PA rings, while not affecting phenylephrine-induced vasocontraction or endothelium-dependent relaxation, sitaxsentan dose-dependently attenuated LPS-induced alterations in endothelium-independent relaxation. PA cytokine mRNA levels were not significantly attenuated by ETAR blockade. We conclude that ETAR blockade attenuates endotoxin-induced alterations in systemic and PA pressures without negatively affecting oxygenation. This protective effect appears to be mediated not by attenuation of sepsis-induced cardiac dysfunction, acidosis, or alveolar inflammation but rather by improved endothelium-independent vasorelaxation.
Keywords: sitaxsentan, sepsis, vasoreactivity, cytokines, endothelium
Introduction
Severe sepsis is a common and serious clinical problem.1 Despite recent advances in the understanding of the pathophysiology of this syndrome, therapeutic options are limited, and mortality remains high.2-4 The direct toxic effect of severe infection on the systemic vasculature results in vasodilation, hypotension, and hypoperfusion and is a major contributor to sepsis-induced mortality. However, in the pulmonary vascular compartment, sepsis increases vasomotor tone and causes significant pulmonary hypertension (PH).5 These vascular effects are due to the disparate effects of endotoxin, which causes vasodilation in the systemic vascular compartment and vasoconstriction in the pulmonary vascular bed.6 Increases in pulmonary artery (PA) pressure and right ventricular (RV) afterload may then culminate in clinically significant PH and/or RV dysfunction. Importantly, a critical role of PH and RV dysfunction in sepsis and sepsis-induced acute respiratory distress syndrome (ARDS) has recently been recognized.7-10 Given the worse outcomes reported for PH and RV dysfunction in acute illness,10,11 there is a clinical need to alleviate PA pressures in sepsis while avoiding worsening of systemic hypotension.
A main causative agent in sepsis-induced PH is endothelin-1 (ET-1).12-14 ET-1 is upregulated in sepsis, with levels inversely correlated with clinical outcomes.13,15-17 Importantly, there is a strong correlation between serum ET-1 levels and increased PA pressure.12 ET-1 is also a potent proinflammatory mediator that directly stimulates production of other proinflammatory cytokines, such as interleukin (IL)-1β, IL-6, tumor necrosis factor α (TNF-α), interferon-γ, inducible nitric oxide synthase (iNOS), and IL-4.18 Of note, several of these cytokines (e.g., IL-1β, iNOS) have also been implicated in endotoxin-induced PA dysfunction,6 and in this context, inhibition of ET-1 signaling has been demonstrated to improve outcomes in experimental sepsis.19
ET-1 acts via two receptors that are highly expressed in the pulmonary vasculature: endothelin receptor A (ETAR) and endothelin receptor B (ETBR). Both ETAR and ETBR are expressed on PA smooth muscle cells (PASMCs), where they mediate PA vasoconstriction.20 However, ETBR is also located on PA endothelial cells (PAECs), where it mediates vasodilation.20 The latter effect is due to increased production of vasodilator agents, such as nitric oxide (NO) and prostacyclins, as well as to facilitation of ET-1 clearance.20 Importantly, the proinflammatory effects of ET-1 purportedly are mediated primarily by ETAR, with subsequent activation of protein kinase C signaling.12,21,22 While it has long been thought that only ETBR is present on PAECS, recent data demonstrated that ETAR is present on PAECs as well.23 This finding is of particular importance, because PAECs have been identified as critical mediators in the pathogenesis of sepsis and ARDS,24 thus suggesting a contribution of endothelial ETAR to the pathogenesis of these syndromes.
In light of the pulmonary vasodilator properties of ETBR and the vasoconstrictive and proinflammatory properties of ETA, selective inhibition of ETAR during endotoxemia is conceptually appealing. Indeed, two previous studies demonstrated beneficial effects of selective ETAR inhibitors during endotoxemia.19,25 However, mechanistic studies investigating the effects of ETAR inhibition on pulmonary vascular tone and proinflammatory markers in the pulmonary vasculature have not yet been performed.
We set out to investigate the effects of the selective ETAR antagonist sitaxsentan on hemodynamic and physiologic parameters in a rat model of endotoxemia. Sitaxsentan was previously approved for treatment of pulmonary arterial hypertension (PAH) in Europe, Canada, and Australia. It was withdrawn from the market in 2010 secondary to liver safety concerns, but because of its high affinity for ETAR,26 it remains a valuable tool for studying the effectiveness of selective ETAR blockade in preclinical investigations. Since endotoxemia is associated with impaired PA vasorelaxation and increased PA proinflammatory cytokine levels,6 we specifically evaluated the effects of sitaxsentan on endothelium-dependent and endothelium-independent PA vasoreactivity as well as on PA proinflammatory cytokine and iNOS expression. We hypothesized that during experimental endotoxemia, selective ETAR blockade will improve cardiopulmonary hemodynamics and PA vasoreactivity without exhibiting detrimental effects on systemic hemodynamics or oxygenation.
Methods
Animal care
Experiments were performed in male Sprague-Dawley rats (250–450 g; Charles River Labs, Wilmington, MA). All rats received humane care according to the protocols outlined in the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals (NIH no. 85-23; revised 1985). All animal protocols were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee. Animals were allowed ad lib. access to food and water throughout the experiment.
Chemicals and reagents
All reagents were obtained from Sigma (St. Louis, MO) unless specified otherwise. Krebs-Henseleit (KH) solution was created in deionized distilled H2O containing (in mmol/L): NaCl 127, KCl 4.7, NaHCO3 17, MgSO4 1.17, KH2PO4 1.18, CaCl2 2.5, and d-glucose 5.5. The final pH of all solutions was 7.35–7.45. Sitaxsentan (provided by Pfizer; New York) was dissolved in phosphate-buffered saline (PBS), with HCl added to obtain a pH of 6.7 per the manufacturer’s instructions.
Experimental procedure
Animals were injected with endotoxin (Salmonella typhimurium lipopolysaccharide [LPS], 20 mg/kg intraperitoneally) or vehicle (PBS, 1.0 mL intraperitoneally).6,27 As reported previously, within 3 hours of LPS administration rats developed a septic phenotype with occurrence of lethargy and diarrhea.6,27 Sitaxsentan (10 mg/kg; a dose previously shown to block ETAR28) or vehicle was administered via tail vein injection 3 hours after LPS or LPS vehicle administration. For ex vivo vasoreactivity studies, we also examined a higher sitaxsentan dose (20 mg/kg). Six hours after LPS or vehicle administration, animals either underwent hemodynamic evaluation or en bloc removal of heart and lungs, with subsequent PA isolation for vasoreactivity studies or real-time polymerase chain reaction (PCR).6,27 PAs from animals undergoing hemodynamic assessment were not used for vasoreactivity studies or PCR in order to avoid potential confounding effects of the surgical intervention on these end points. Final n was 4–5/group.
Hemodynamic evaluation and bronchoalveolar lavage
Animals were anesthetized by inhaled isoflurane 1%–2% in oxygen via nose cone and then orotracheally intubated with a 14-gauge catheter, as previously described.29 Animals were placed on a servo-controlled heated tray to maintain temperature at 37°C. The left carotid artery and right external jugular vein were cannulated with PE-50 (polyethylene) tubing and a 2-Fr Millar catheter (Millar Instruments, Houston, TX), respectively, via cutdowns. Lungs were ventilated with 100% oxygen (tidal volume: 6 mL/kg; rate: 60–80/min) and isoflurane to maintain general anesthesia. End-expiratory pressure was kept at 3 cm H2O. Mean arterial pressure (MAP) and airway pressures were measured continuously. Blood gases were sampled periodically (i-STAT analyzer; Abbott Point of Care, Princeton, NJ) via the arterial catheter. The ratio of the arterial oxygen tension (PaO2) to the fraction of inspired oxygen (FiO2) was calculated (expressed as P/F ratio). Ventilation was adjusted to maintain PaCO2 at 35–40 Torr. Normal saline boluses (10 mL/kg) were given as needed to replace blood loss from sampling. A thoracotomy was made in the left second intercostal space, through which the ascending aorta was isolated. A flow probe was placed around the aortic arch for continuous cardiac output (CO) monitoring (2.5PSL probe and TS420 monitor; Transonic, Ithaca, NY). After flow probe placement, animals were returned to the supine position, and the Millar catheter was advanced into the right ventricle under pressure waveform guidance. RV systolic pressure (RVSP) was measured continuously. After all in vivo end points were obtained, animals were killed via exsanguination. A medial sternotomy was performed, and the rib cage was spread open. The right upper lobe of each animal was tied off and removed for lung wet-to-dry ratio determination. Whole-lung bronchoalveolar lavage (BAL) was performed by instilling aliquots of 1 mL NaCl 0.9% via the 14-gauge catheter, followed by immediate aspiration. Filling and emptying of the lung was confirmed by visual inspection. Three consecutive aliquots were used per animal.
Wet-to-dry lung weight ratio
The right upper lobe of each animal was tied off, blotted dry of surface fluid, and then weighed. Tissue was placed in a convection oven at 95°C for 24 hours and then weighed again.
Isolated PA ring preparation and vasoreactivity study protocol
Animals were anesthetized with pentobarbital (150 mg/kg intraperitoneally). Heart and lungs were removed en bloc via medial sternotomy and immediately placed in ice-cold modified KH solution bubbled with 100% O2. Under a dissecting microscope, the PAs were isolated and removed of all extraneous tissue. PAs were sectioned into 2–3-mm rings and either snap-frozen for real-time PCR or suspended between steel hooks and force transducers (ADInstruments, Colorado Springs, CO) for measurement of isometric force displacement, as previously reported.6,27,30 Briefly, PA rings were immersed in KH solution at 37°C and bubbled with 95% O2/5% CO2. Force displacement was recorded with an AD Instruments Powerlab data recorder connected to a Dell computer (Round Rock, TX) running Labchart software. PA rings were allowed to equilibrate at 0.75 g of force for 60 min, with the bath being changed every 15 minutes. After equilibration, PA rings were tested for integrity by evaluating their contractile response to 80 mmol/L of KCl. Rings that reached at least 1.2 g of force were kept for experimentation; otherwise, they were discarded. KCl was removed, and a new KH bath was instituted. Viability was then tested by contracting rings with 1 μM of phenylephrine (PE). Rings that did not exhibit a force increase of at least 0.2 g after the PE test were discarded. Endothelial integrity was tested with 1 μM of acetylcholine (ACh). PA rings that did not achieve a decrease of at least 50% from PE contraction were discarded. ACh was then washed out through multiple changes of the KH bath, and rings were allowed to equilibrate back to baseline. Vasoconstrictor response was tested by exposing rings to increasing concentrations of PE (10−8M to 10−5M). At maximal PE-induced contraction (10−5M), endothelium-dependent vasorelaxation was evaluated via ACh dose response (10−8M to 10−5M). Rings were washed out with a new KH bath and allowed to equilibrate back to baseline. PE precontraction was then repeated, and endothelium-independent vasorelaxation was evaluated via a sodium nitroprusside (SNP) dose curve (10−8M to 10−5M). Force displacement was measured for each PE, ACh, and SNP concentration once force generation reached a plateau (after approximately 5 minutes per dose). Vasoconstrictor responses after stimulation with PE are expressed as percent change from baseline tension of 750 mg. Vasorelaxor responses after ACh and SNP are expressed as percent change from maximum PE precontraction. The time from mounting the PA segments to performing vasorelaxation studies was approximately 120 minutes.
PA real-time PCR
Relative quantitative real-time PCR was used to assess expression of the iNOS, TNF-α, IL-1β, and IL-6 genes in the PA. Tissues were homogenized, and total RNA was extracted using RNEasy Plus Mini Kit (SA Biosciences, Valencia, CA) according to the manufacturer’s instructions. RNA was then reverse transcribed into complementary DNA (cDNA) using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA). Messenger RNA (mRNA) expression was monitored with the Applied Biosystems 7500 Real-Time PCR System. Briefly, 10 ng of template was used per real-time PCR reaction, and amplification was detected by incorporation of SYBR green dye. IL-1β, IL-6, and iNOS (SA Bioscience) gene expression was measured, with β-actin (SA Bioscience) as internal control. The following validated primer sets were used to measure TNF-α gene expression and the GAPDH (glyceraldehyde 3-phosphate dehydrogenase) internal control: TNF-α: 5′-AAATGGGCTCCCTCTCATCAGTTC-3′ and 5′-TCTGCTTGGTTGGTTTGCTACGAC-3′; GAPDH: 5′-TGCCAAGTATGATGACATCAAGAAG-3′ and 5′-AGCCCAGGATGCCCTTTAGT-3′.31 Analysis was limited to PA rings from animals that did not undergo surgery and whose PAs were not used for vasoreactivity assessment.
Statistical analysis
Values are reported as means ± SEM, except for results from relative quantitative real-time PCR, where values are expressed as fold change with high-end errors and low-end errors, according to the ΔΔCt method.32 Comparisons were made by one-way analysis of variance (ANOVA) with post hoc Bonferroni correction or by unpaired Student’s t test (Prism 6; Graphpad Software, San Diego, CA), as appropriate. Normality testing was performed by Kolgorov-Smirnov testing. Nonparametric data were analyzed with the Kruskal-Wallis test. PA ring data were analyzed with two-way ANOVA for repeated measures, with post hoc Tukey correction for multiple comparisons. Statistical significance was accepted for P < 0.05.
Results
Selective ETAR blockade attenuates endotoxin-induced PH and systemic hypotension
We first investigated whether selective ETAR antagonist treatment attenuates endotoxin-induced alterations in the pulmonary and systemic vasculature. As expected, endotoxin administration caused PH, while systemic blood pressure decreased (Fig. 1A, 1B). Since changes in RVSP or PA pressure must always be evaluated in the context of systemic blood pressure,33 we calculated the ratio between RVSP and MAP and observed a significant increase with endotoxin (Fig. 1C). As expected, endotoxin administration also resulted in a significant decrease in CO (Fig. 1D). Endotoxin-mediated effects on pulmonary and systemic hemodynamics were both attenuated by sitaxsentan. In particular, sitaxsentan administration was associated with a significant decrease in endotoxin-induced PH and a significant increase in MAP (Fig. 1A, 1B). This resulted in a marked improvement in the RVSP/MAP ratio (Fig. 1C). However, CO was not affected by sitaxsentan, suggesting that sitaxsentan’s protective effects on RVSP and MAP are not mediated through beneficial effects on cardiac performance.
Figure 1.

Sitaxsentan treatment attenuates endotoxin-induced alterations in pulmonary and systemic hemodynamics. Adult male Sprague-Dawley rats were injected with lipopolysaccharide (LPS; 20 mg/kg intraperitoneally) or LPS vehicle (phosphate-buffered saline [PBS], 1 mL intraperitoneally). Three hours later, animals received either sitaxsentan (10 mg/kg via tail vein) or sitaxsentan vehicle (PBS, 1 mL via tail vein). Right ventricular systolic pressure (RVSP; A), mean arterial pressure (MAP; B), and cardiac output (CO; D) were measured 3 hours after administration of sitaxsentan or its vehicle. Note the LPS-induced decreases in MAP and increases in RVSP (A) and RVSP/MAP (C). Sitaxsentan administration was associated with a significant increase in MAP and a decrease in RVSP and RVSP/MAP. Values are means + SEM. N = 4–5/group. A single asterisk indicates P < 0.05 and 3 asterisks P < 0.001 versus LPS vehicle groups; a single number sign (#) indicates P < 0.05 and 3 number signs P < 0.001 versus sitaxsentan vehicle. P = 0.09 for LPS+sitax versus LPS veh. veh+veh: LPS vehicle + sitxsentan vehicle; veh+sitax: LPS vehicle + sitxsentan; LPS+veh: LPS + sitaxsentan vehicle; LPS+sitax: LPS + sitxsentan.
Protective effects of ETAR blockade on pulmonary hemodynamics are not associated with negative effects on pulmonary gas exchange
Pulmonary vasodilator therapy in the setting of lung disease or sepsis may worsen oxygenation by counteracting physiological hypoxic pulmonary vasoconstriction.34,35 We therefore measured the P/F ratio in all animals. Notably, when compared with vehicle treatment in endotoxemic rats, sitaxsentan treatment did not worsen oxygenation (Fig. 2), suggesting that there was no increase in ventilation-perfusion mismatch with selective ETAR blockade. However, it should be noted that endotoxemia did not result in a significant decrease in the P/F ratio, likely because of the short time frame between LPS administration and the death of the animals (6 hours).
Figure 2.
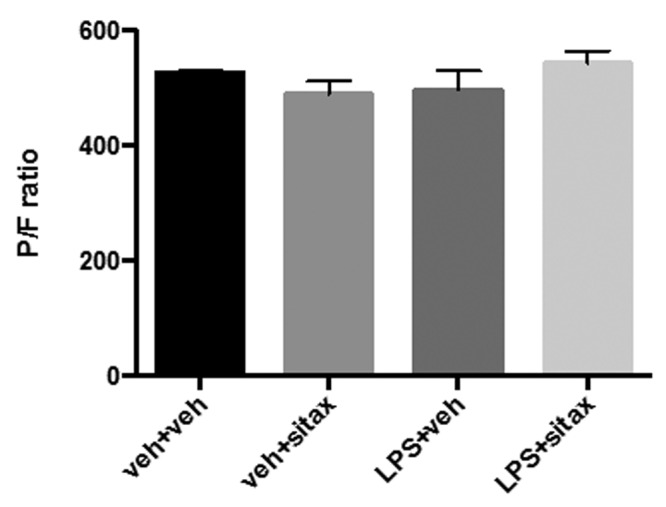
Sitaxsentan treatment during endotoxemia is not associated with worsening oxygenation. Rats were treated with lipopolysaccharide (LPS), LPS vehicle, sitaxsentan, or sitaxsentan vehicle, as outlined in Figure 1. Partial pressure of arterial oxygen (PaO2) was measured via arterial blood gas analysis. The fraction of inspired oxygen (FiO2) was set at 100%. Oxygenation is expressed as the PaO2/FiO2 (P/F) ratio. Note that sitaxsentan treatment during endotoxemia did not negatively affect oxygenation, suggesting the lack of additional ventilation-perfusion mismatch. Values are means + SEM. N = 4–5/group. veh+veh: LPS vehicle + sitxsentan vehicle; veh+sitax: LPS vehicle + sitxsentan; LPS+veh: LPS + sitaxsentan vehicle; LPS+sitax: LPS + sitxsentan.
Protective effects of ETAR blockade on pulmonary hemodynamics are not mediated by attenuation of alveolar inflammation
We did not observe a significant increase in lung wet-to-dry ratio after LPS administration (Fig. 3A), suggesting that the animals exhibited pulmonary vascular dysfunction even in the absence of overt signs of lung injury. We did, however, note an increase in BAL neutrophilia after LPS administration, suggesting early, and possibly evolving, lung injury (Fig. 3B). Interestingly, BAL neutrophilia was not attenuated by sitaxsentan treatment (Fig. 3B), suggesting that early alveolar inflammation is not a significant target of sitaxsentan during endotoxemia.
Figure 3.
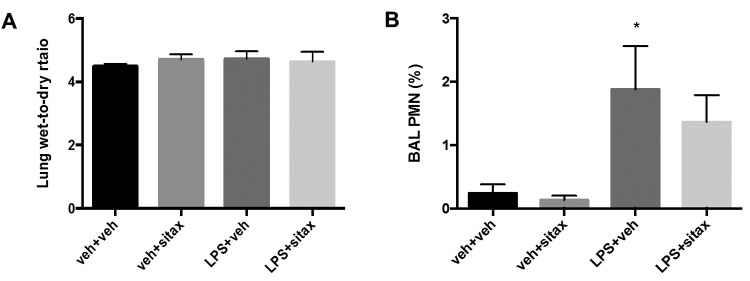
Protective sitaxsentan effects during endotoxemia are not mediated by attenuation of alveolar inflammation. Rats were treated with lipopolysaccharide (LPS), LPS vehicle, sitaxsentan, or sitaxsentan vehicle, as outlined in Figure 1. Lung wet-to-dry ratio (A) was affected by neither LPS nor sitaxsentan. Note, however, that LPS administration was associated with a small but significant increase in bronchoalveolar lavage (BAL) neutrophil count (B), suggesting evolving lung injury. Neutrophilic alveolitis was not attenuated by sitaxsentan. Values are means + SEM. N = 4–5/group. Asterisk indicates P < 0.05 versus LPS vehicle. veh+veh: LPS vehicle + sitxsentan vehicle; veh+sitax: LPS vehicle + sitxsentan; LPS+veh: LPS + sitaxsentan vehicle; LPS+sitax: LPS + sitxsentan; PMN: polymorphonuclear cells.
Favorable effects of ETAR blockade on pulmonary and systemic hemodynamics are not mediated through beneficial effects on metabolic acidosis
Sepsis is frequently associated with metabolic acidosis.3 Since acidosis may result in vasomotor dysfunction with vasodilation in the systemic vascular bed and vasoconstriction in the pulmonary circulation,3,36 we evaluated whether the protective effects of sitaxsentan on hemodynamic parameters are associated with improvements in serum bicarbonate levels. As expected, endotoxin administration resulted in decreased bicarbonate levels (Fig. 4). However, this was not affected by sitaxsentan (Fig. 4), indicating that sitaxsentan’s effects on pulmonary and systemic parameters are not mediated by improvements in acid-base status or end-organ function.
Figure 4.
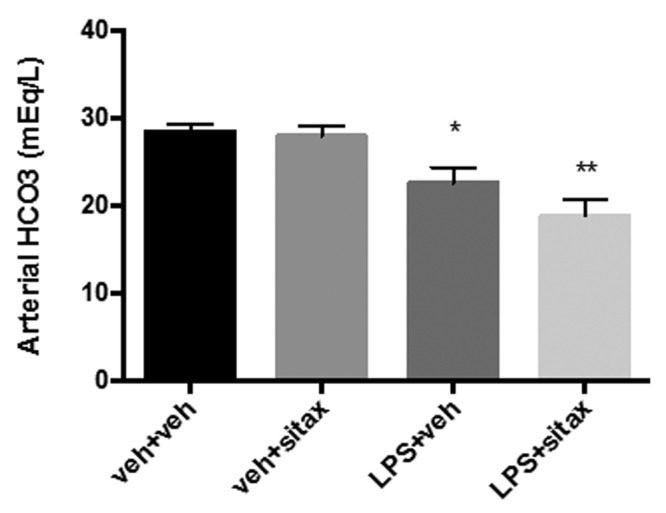
Protective sitaxsentan effects during endotoxemia are not mediated by increasing serum bicarbonate levels. Rats were treated with lipopolysaccharide (LPS), LPS vehicle, sitaxsentan, or sitaxsentan vehicle, as outlined in Figure 1, and serum bicarbonate levels were measured via arterial blood gas analysis. Note the LPS-induced decrease in bicarbonate levels, indicating development of metabolic acidosis. Bicarbonate levels were not affected by sitaxsentan. Values are means + SEM. N = 4–5/group. A single asterisk indicates P < 0.05 and a double asterisk P < 0.01 versus LPS vehicle groups. veh+veh: LPS vehicle + sitxsentan vehicle; veh+sitax: LPS vehicle + sitxsentan; LPS+veh: LPS + sitaxsentan vehicle; LPS+sitax: LPS + sitxsentan.
Selective ETAR blockade improves endothelium-independent vasorelaxation in isolated PA rings from endotoxemic rats
Having established that the favorable effects of selective ETAR blockade on pulmonary hemodynamics are not caused by improved cardiac function or by beneficial effects on alveolar inflammation or acid-base parameters, we evaluated whether sitaxsentan’s protective effects are associated with direct favorable changes in PA vasoreactivity. We therefore isolated PA rings from endotoxemic and LPS-treated rats with or without sitaxsentan and measured PE-induced vasocontraction as well as endothelium-dependent and endothelium-independent vasorelaxation. Interestingly, we observed a biphasic effect of LPS on PE-induced vasocontraction, with LPS decreasing PA contraction at lower PE concentrations but increasing it at the highest PE concentration. As expected, we observed a 20%–30% decrease in both endothelium-dependent and endothelium-independent vasorelaxation in PA rings from LPS-treated animals, indicating that PA vasorelaxation is significantly impaired during endotoxemia (Fig. 5B, 5C). Interestingly, while the LPS-induced alterations in vasocontraction and endothelium-dependent vasorelaxation were not affected by sitaxsentan administration (Fig. 5B, 5C), sitaxsentan significantly attenuated the LPS-induced decrease in endothelium-independent vasorelaxation (Fig. 5C). There was a dose-dependent effect, whereby 10 mg/kg of sitaxsentan only tended to reverse whereas a 20-mg/kg dose significantly reversed LPS-induced decreases in endothelium-independent vasorelaxation, as indicated by a shift of the SNP dose-response curve toward normal range (Fig. 5C). This suggests that sitaxsentan restores PA vasoreactivity during endotoxemia in a manner that is independent of endothelium-derived NO release.
Figure 5.

Sitaxsentan (sitax) administration attenuates endotoxin-induced decreases in endothelium-independent vasorelaxation in isolated pulmonary artery (PA) rings. Rats were treated with lipopolysaccharide (LPS), LPS vehicle (LPS veh), sitaxsentan 10 mg intravenously (sitax 10), sitaxsentan 20 mg/kg intravenously (sitax 20), or sitaxsentan vehicle (sitax veh; equivalent to a 20 mg/kg dose). Sitaxsentan or vehicle was administered 3 hours after administration of LPS or LPS vehicle. Main PAs were harvested 3 hours after administration of sitaxsentan or its vehicle, cut into 2–3-mm segments, and placed in physiologic organ bath for measurement of phenylephrine (PE)-induced vasocontraction, as well as endothelium-dependent and endothelium-independent vasorelaxation. A, PE-induced vasoconstriction was evaluated by adding increasing concentrations (10−8M to 10−5M) to the organ bath and is expressed as percent change from baseline tension. Vasorelaxation was then measured in maximally PE-precontracted rings (10−5M) and is expressed as percent change from PE precontraction. B, Endothelium-dependent vasorelaxation was evaluated by adding increasing concentrations of acetylcholine (ACh; 10−8M to 10−5M) to the organ bath. C, Endothelium-independent vasorelaxation was assessed by adding increasing concentrations of sodium nitroprusside (SNP; 10−8M to 10−5M). Note that sitaxsentan dose-dependently improved endothelium-independent vasorelaxation in PA rings from LPS-treated animals. Values are means ± SEM. N = 4 animals/group. A double asterisk indicates P < 0.01, 3 asterisks P < 0.001, and 4 asterisks P < 0.0001 for LPS+sitax veh versus LPS veh+sitax veh; a single number sign (#) indicates P < 0.05 and a double number sign P < 0.01 for LPS+sitax 20 versus LPS+veh.
Selective ETAR blockade does not significantly attenuate LPS-induced increases in PA proinflammatory cytokine mRNA levels
Local cytokine release alters vascular reactivity.27 Given the known anti-inflammatory effects of selective ETAR antagonism on airway smooth muscle cells in asthma,18 we evaluated whether sitaxsentan attenuates levels of proinflammatory cytokines in the PA by measuring IL-1β, TNF-α, and IL-6 mRNA levels as well as iNOS mRNA. As expected, LPS elevated mRNA expression of these proinflammatory mediators in the PA (Fig. 6). While sitaxsentan treatment (10 mg/kg) tended to be associated with decreased IL-1β and TNF-α expression in the PA (Fig. 6A, 6B), this did not reach statistical significance. Sitaxsentan did not affect PA IL-6 and iNOS mRNA levels (Fig. 6C, 6D). Taken together, these data suggest that the beneficial, endothelium-independent effects of ETAR antagonism on pulmonary hemodynamics and PA vasoreactivity during endotoxemia are not due to a direct effect on PA proinflammatory cytokine levels.
Figure 6.

Effect of sitaxsentan treatment on endotoxin-induced increases in pulmonary artery (PA) proinflammatory cytokine transcripts. Rats were treated with lipopolysaccharide (LPS), LPS vehicle, sitaxsentan, or sitaxsentan vehicle, as outlined in Figure 1. Main PAs were harvested 3 hours after administration of sitaxsentan or its vehicle. Interleukin (IL)-1β (A), tumor necrosis factor α (TNF-α; B), IL-6 (C), and inducible nitric oxide synthase (iNOS; D) messenger RNA (mRNA) was measured in PA homogenates via real-time polymerase chain reaction. Gene expression is expressed as fold change versus LPS vehicle + sitaxsentan vehicle–treated (veh+veh) group. Note the LPS-induced increase in transcript of all cytokines and the trend for sitaxsentan to decrease IL-1β and TNF-α mRNA expression. Error bars represent high- and low-end errors. N = 4/group. A single asterisk indicates P < 0.05, a double asterisk P < 0.01, and 3 asterisks P < 0.001 versus the LPS vehicle groups. veh+veh: LPS vehicle + sitxsentan vehicle; veh+sitax: LPS vehicle + sitxsentan; LPS+veh: LPS + sitaxsentan vehicle; LPS+sitax: LPS + sitxsentan.
Discussion
This study demonstrates that selective ETAR blockade during experimental endotoxemia is associated with a significant improvement in pulmonary and systemic hemodynamics. These favorable effects are accompanied by the novel finding that selective ETAR blockade is associated with improved endothelium-independent PA vasorelaxation. Viewed in light of additional data demonstrating that ETAR blockade does not significantly affect CO or acid-base status, this suggests that the protective effects of this strategy are not due to direct cardiac effects or effects on end-organ function. The lack of sitaxsentan effects on BAL neutrophil counts also suggests a lack of effect on alveolar inflammation. The sitaxsentan-induced improvement in endothelium-independent vasorelaxation suggests that selective ETAR blockade may directly target the pulmonary vasculature. While PA proinflammatory cytokine transcript tended to be decreased with sitaxsentan, our study was underpowered to allow for any conclusions as to whether attenuation of cytokine production is a mechanism that contributes, at least in part, to the beneficial effects of sitaxsentan on PA vasorelaxation. Other potential mechanisms would be a direct effect on PASMCs or effects on cytokines/chemokines other than the ones measured here. Interestingly, premature mortality (i.e., before the scheduled death at 6 hours) was 50% lower in the sitaxsentan-treated LPS group (1 of 13 animals [7.7%]) than in the untreated LPS group (3 of 18 animals [16.7%]). While this did not reach statistical significance, these survival data are consistent with the life-prolonging effects effect of the ETAR antagonist ETR-P1/fl, reported in a neonatal sepsis model.19 Future studies in our model with longer time courses and dose responses will specifically investigate ETAR blockade effects on survival.
Pulmonary vasodilator therapy during endotoxemia may cause peripheral vasodilation or worsen pulmonary ventilation-perfusion mismatch.7,8 However, we did not detect systemic hypotension or hypoxemia, suggesting that selective ETAR blockade during endotoxemia is both feasible and safe. On the contrary, sitaxsentan administration similarly resulted in a significant increase in MAP (a highly desired effect in endotoxemia/sepsis). Previous studies demonstrated a decrease in serum proinflammatory cytokines with ETAR blockade,19 and it is conceivable that direct effects on systemic vasomotor tone may have contributed to the beneficial effects on MAP. Alternatively, the improvement in MAP may be a direct consequence of improved pulmonary vascular blood flow.
Even though we noted an increase in BAL neutrophils and in mRNA of proinflammatory cytokines, there was no overt evidence of lung injury occurring in our model. This was likely a result of the relatively short time period between LPS administration and the death of the animals (6 hours), which captured early inflammatory changes that may not yet have translated into changes in oxygenation (P/F ratio) or lung wet-to-dry ratio. Definite conclusions about sitaxsentan’s potential for worsening ventilation-perfusion mismatch and hypoxemia during established lung injury are therefore not possible. Future studies will focus on longer time courses with more severe lung injury. We do not suggest, however, that the septic phenotype observed in our animals was not severe enough to cause lung injury, as all animals exhibited lethargy and diarrhea, a change in vital signs (Fig. 1), a decrease in serum bicarbonate (Fig. 4), and even premature mortality. Rather, an increase in PA proinflammatory cytokine transcript before the onset of frank lung injury is consistent with the pathophysiologic changes leading to indirect ARDS in the setting of sepsis.37
Our novel findings are consistent with previously reported systemic hemodynamic improvements with nonselective ET receptor antagonist treatment during sepsis19,38,39 and with other studies that reported improvement in lung injury–associated PH with ET receptor antagonist treatment in a model of ARDS induced by nebulized Escherichia coli.40 The beneficial effect of ETAR antagonism in our studies is further corroborated by data from other laboratories showing that ET-1, ETAR, and/or ETBR levels are rapidly upregulated after LPS administration.41,42 In contrast to other studies,12,18,19 our study failed to demonstrate significant anti-inflammatory effects of ETAR antagonism. However, in a different endotoxemia study, while attenuating lung injury and decreasing protein kinase C activation, ET-1 receptor blockade increased plasma TNF-α and IL-8 levels.43 Importantly, prior studies did not investigate whether ET receptor antagonists exert anti-inflammatory effects specifically in the PA (the vascular compartment directly responsible for development of endotoxin-induced PH).7,9,24 We show that treatment with a selective ETAR antagonist does not result in a significant decrease in pathogenetically relevant proinflammatory cytokine mRNA expression in the PA. Compartment- and receptor-specific effects of ET-1 antagonists may explain the disparity in the currently published literature.
Sitaxsentan’s vasorelaxor effects were not mediated in an endothelium-dependent (and therefore NO-mediated) fashion. This finding is of particular importance, as increased iNOS activity, with resulting increases in NO levels, is a critical contributor to the systemic hypotension that is frequently observed during sepsis.3 The lack of endothelium-mediated/NO-mediated vasorelaxation may therefore explain why sitaxsentan administration during endotoxemia is not associated with worsening systemic hypotension. Since sepsis is characterized by marked endothelial dysfunction,24 the finding of endothelium-independent protection is of critical importance; this implies that selective ETAR antagonism may represent a valuable therapeutic option even in the absence of a functioning endothelium.
Our results differ from those of another study, which showed worsening MAP with the use of a selective ETAR antagonist.42 However, several significant differences exist between the studies with regard to endotoxin administration, time course, and type of antagonist used. Unique pharmacological properties of different ETAR antagonists may contribute to these differences. For example, significant pharmacological differences exist between sitaxsentan, used here, and ambrisentan, an ETAR antagonist used in previous studies.19,20 One aspect is that sitaxsentan is a much more selective antagonist for ETAR than is ambrisentan (6,500∶1 in vitro selectivity for sitaxsentan vs. 4,000∶1 for ambrisentan).26 Furthermore, differences exist in half-life (15 hours for ambrisentan vs. 10 hours for sitaxsentan), time to steady state (6 days for sitaxsentan; 3–4 days for ambrisentan), cytochrome P450 interactions (inhibition of CYP450 2C9 by sitaxsentan but not by ambrisentan), and effects on plasma ET-1 levels (decreased with sitaxsentan but increased with ambrisentan).26 Finally, since endotoxemia results in differential expression of ET-1 and its receptor subtypes, depending on the organ system involved,41,42 it is also possible that differences in distribution, quantity, and/or ratio of ETAR to ETBR determine the overall effect of ET-antagonism on MAP.
Our study has several limitations. First, because of the scarce availability of PA tissue from each animal, we had to limit our biochemical analysis to mRNA analysis. However, since the primary mechanism for increasing proinflammatory cytokine levels is by transcriptional regulation,44 one would expect protein levels to follow the pattern of mRNA expression. Second, we used a higher sitaxsentan dose in some of the animals whose PAs were harvested for ex vivo vasorelaxation studies. We did this because we observed a clear, but not statistically significant, trend with the lower dose. However, it is well described that conductance vessels such as the ones used in ex vivo relaxation studies frequently require higher drug concentrations to elucidate effects, as compared to the concentrations needed to act on smaller, resistance-type vessels in vivo.45 Despite this minor limitation, the PA ring model is considered an excellent tool for mechanistic vasoreactivity studies, since it (1) eliminates potentially confounding effects of extravascular influences on vasomotor tone (e.g., circulating mediators, neural activity) and (2) allows for evaluation of influences that are localized to the vessel wall (e.g., endothelium-derived relaxing and/or contracting factors, as investigated in our study).46 Third, we were unable to decipher the exact mechanisms of sitaxsentan-induced improvements in endothelium-independent PA vasoreactivity. The downward trend observed for TNF-α and IL-1β transcript, even during the 3-hour time frame, suggests potential anti-inflammatory effects of sitaxsentan and is consistent with anti-inflammatory effects of ET receptor blockade on airway smooth muscle,18 but additional investigations with more animals, a longer time frame, and evaluation of different sitaxsentan doses and administration time points are needed.
Conclusions. Treatment with the selective ETAR antagonist sitaxsentan in LPS-induced endotoxemia is associated with improvement in pulmonary and systemic hemodynamics. The associated increase in endothelium-independent PA relaxation suggests that these effects are mediated, at least in part, by a direct effect on the pulmonary vasculature.
Source of Support: This work was funded by an investigator-initiated research grant from Pfizer (WS301931; TL), a Veterans Affairs Merit Review Award (IP), and a Calvin H. English Chair of Respiratory Disease (IP).
Conflict of Interest: TL is currently receiving a 2011 ASPIRE (Advancing Science through Pfizer-Investigator Research Exchange) Young Investigator Research Award (WS1953334) from Pfizer and funding from the 2012 Gilead Sciences Research Scholars Program in Pulmonary Arterial Hypertension. No conflicts of interest are declared for any other author.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001;29(7):1303–1310. [DOI] [PubMed]
- 2.Russell JA. Management of sepsis. N Engl J Med 2006;355(16):1699–1713. [DOI] [PubMed]
- 3.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003;348(2):138–150. [DOI] [PubMed]
- 4.Skrupky LP, Kerby PW, Hotchkiss RS. Advances in the management of sepsis and the understanding of key immunologic defects. Anesthesiology 2011;115(6):1349–1362. [DOI] [PMC free article] [PubMed]
- 5.Lambermont B, Kolh P, Detry O, Gerard P, Marcelle R, D’Orio V. Analysis of endotoxin effects on the intact pulmonary circulation. Cardiovasc Res 1999;41(1):275–281. [DOI] [PubMed]
- 6.Tsai BM, Wang M, Pitcher JM, Kher A, Meldrum DR. Disparate IL-1β and iNOS gene expression in the aorta and pulmonary artery after endotoxemia. Surg Infect 2006;7(1):21–27. [DOI] [PubMed]
- 7.Chan CM, Klinger JR. The right ventricle in sepsis. Clin Chest Med 2008;29(4):661–676. [DOI] [PubMed]
- 8.Lahm T, McCaslin CA, Wozniak TC, Ghumman W, Fadl YY, Obeidat OS, Schwab K, Meldrum DR. Medical and surgical treatment of acute right ventricular failure. J Am Coll Cardiol 2010;56(18):1435–1446. [DOI] [PubMed]
- 9.Price LC, McAuley DF, Marino PS, Finney SJ, Griffiths MJ, Wort SJ. Pathophysiology of pulmonary hypertension in acute lung injury. Am J Physiol Lung Cell Mol Physiol 2012;302(9):L803–L815. [DOI] [PMC free article] [PubMed]
- 10.Bull TM, Clark B, McFann K, Moss M. Pulmonary vascular dysfunction is associated with poor outcomes in patients with acute lung injury. Am J Respir Crit Care Med 2010;182(9):1123–1128. [DOI] [PMC free article] [PubMed]
- 11.Kaw R, Pasupuleti V, Deshpande A, Hamieh T, Walker E, Minai OA. Pulmonary hypertension: an important predictor of outcomes in patients undergoing non-cardiac surgery. Respir Med 2011;105(4):619–624. [DOI] [PubMed]
- 12.Wanecek M, Weitzberg E, Rudehill A, Oldner A. The endothelin system in septic and endotoxin shock. Eur J Pharmacol 2000;407(1–2):1–15. [DOI] [PubMed]
- 13.Sanai L, Haynes WG, MacKenzie A, Grant IS, Webb DJ. Endothelin production in sepsis and the adult respiratory distress syndrome. Intensive Care Med 1996;22(1):52–56. [DOI] [PubMed]
- 14.Yanagisawa M, Kurihara H, Kimura S, Goto K, Masaki T. A novel peptide vasoconstrictor, endothelin, is produced by vascular endothelium and modulates smooth muscle Ca2+ channels. J Hypertens Suppl 1988;6(4):S188–S191. [DOI] [PubMed]
- 15.Sugiura M, Inagami T, Kon V. Endotoxin stimulates endothelin-release in vivo and in vitro as determined by radioimmunoassay. Biochem Biophys Res Commun 1989;161(3):1220–1227. [DOI] [PubMed]
- 16.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988;332(6163):411–415. [DOI] [PubMed]
- 17.Brauner JS, Rohde LE, Clausell N. Circulating endothelin-1 and tumor necrosis factor-α: early predictors of mortality in patients with septic shock. Intensive Care Med 2000;26(3):305–313. [DOI] [PubMed]
- 18.Finsnes F, Lyberg T, Christensen G, Skjonsberg OH. Effect of endothelin antagonism on the production of cytokines in eosinophilic airway inflammation. Am J Physiol Lung Cell Mol Physiol 2001;280(4):L659–L665. [DOI] [PubMed]
- 19.Goto T, Hussein MH, Kato S, Daoud GA-H, Kato T, Kakita H, Mizuno H, et al. Endothelin receptor antagonist attenuates inflammatory response and prolongs the survival time in a neonatal sepsis model. Intensive Care Med 2010;36(12):2132–2139. [DOI] [PubMed]
- 20.Trow TK, Taichman DB. Endothelin receptor blockade in the management of pulmonary arterial hypertension: selective and dual antagonism. Respir Med 2009;103(7):951–962. [DOI] [PubMed]
- 21.Filep JG, Fournier A, Foldes-Filep E. Acute pro-inflammatory actions of endothelin-1 in the guinea-pig lung: involvement of ETA and ETB receptors. Br J Pharmacol 1995;115(2):227–236. [DOI] [PMC free article] [PubMed]
- 22.Tsai BM, Wang M, Turrentine MW, Mahomed Y, Brown JW, Meldrum DR. Hypoxic pulmonary vasoconstriction in cardiothoracic surgery: basic mechanisms to potential therapies. Ann Thorac Surg 2004;78(1):360–368. [DOI] [PubMed]
- 23.Hall SM, Davie N, Klein N, Haworth SG. Endothelin receptor expression in idiopathic pulmonary arterial hypertension: effect of bosentan and epoprostenol treatment. Eur Respir J 2011;38:851–860. [DOI] [PubMed]
- 24.Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 2012;18(8):1217–1223. [DOI] [PMC free article] [PubMed]
- 25.Wanecek M, Oldner A, Rudehill A, Sollevi A, Alving K, Weitzberg E. EndothelinA-receptor antagonism attenuates pulmonary hypertension in porcine endotoxin shock. Eur Respir J 1999;13(1):145–151. [DOI] [PubMed]
- 26.Opitz CF, Ewert R, Kirch W, Pittrow D. Inhibition of endothelin receptors in the treatment of pulmonary arterial hypertension: does selectivity matter? Eur Heart J 2008;29:1936–1948. [DOI] [PMC free article] [PubMed]
- 27.Tsai BM, Wang M, Pitcher JM, Kher A, Brown JW, Meldrum DR. Endothelium-dependent pulmonary artery vasorelaxation is dysfunctional in males but not females after acute lung injury. Surgery 2005;138(1):78–84. [DOI] [PubMed]
- 28.Tilton RG, Munsch CL, Sherwood SJ, Chen S-J, Chen Y-F, Wu C, Block N, Dixon RAF, Brock TA. Attenuation of pulmonary vascular hypertension and cardiac hypertrophy with sitaxsentan sodium, an orally active ETA receptor antagonist. Pulm Pharmacol Therap 2000;13(2):87–97. [DOI] [PubMed]
- 29.Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, Justice MJ, et al. 17β-estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med 2012;185(9):965–980. [DOI] [PMC free article] [PubMed]
- 30.Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Tan J, Meldrum DR. Selective estrogen receptor-α and estrogen receptor-β agonists rapidly decrease pulmonary artery vasoconstriction by a nitric oxide-dependent mechanism. Am J Physiol Regul Integr Comp Physiol 2008;295(5):R1486–R1493. [DOI] [PMC free article] [PubMed]
- 31.Peinnequin A, Mouret C, Birot O, Alonso A, Mathieu J, Clarençon D, Agay D, Chancerelle Y, Multon E. Rat pro-inflammatory cytokine and cytokine related mRNA quantification by real-time polymerase chain reaction using SYBR green. BMC Immunol 2004;5:3. [DOI] [PMC free article] [PubMed]
-
32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the /article/back/ref-list/ref/mixed-citation/inline-formula
 method. Methods 2001;25(4):402–408. [DOI] [PubMed]
method. Methods 2001;25(4):402–408. [DOI] [PubMed] - 33.Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation 2009;120(11):992–1007. [DOI] [PubMed]
- 34.Radermacher P, Santak B, Wust HJ, Tarnow J, Falke KJ. Prostacyclin and right ventricular function in patients with pulmonary hypertension associated with ARDS. Intensive Care Med 1990;16(4):227–232. [DOI] [PubMed]
- 35.Blanco I, Gimeno E, Munoz PA, Pizarro S, Gistau C, Rodriguez-Roisin R, Roca J, Barberà JA. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am J Respir Crit Care Med 2010;181(3):270–278. [DOI] [PubMed]
- 36.Balanos GM, Talbot NP, Dorrington KL, Robbins PA. Human pulmonary vascular response to 4 h of hypercapnia and hypocapnia measured using Doppler echocardiography. J Appl Physiol 2003;94(4):1543–1551. [DOI] [PubMed]
- 37.Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet 2007;369(9572):1553–1564. [DOI] [PubMed]
- 38.Konrad D, Haney M, Johansson G, Wanecek M, Weitzberg E, Oldner A. Cardiac effects of endothelin receptor antagonism in endotoxemic pigs. Am J Physiol Heart Circ Physiol 2007;293(2):H988–H996. [DOI] [PubMed]
- 39.Kuklin VN, Kirov MY, Evgenov OV, Sovershaev MA, Sjöberg J, Kirova SS, Bjertnaes LJ. Novel endothelin receptor antagonist attenuates endotoxin-induced lung injury in sheep. Crit Care Med 2004;32(3):766–773. [DOI] [PubMed]
- 40.Persson BP, Rossi P, Weitzberg E, Oldner A. Inhaled tezosentan reduces pulmonary hypertension in endotoxin-induced lung injury. Shock 2009;32(4):427–434. [DOI] [PubMed]
- 41.Jesmin S, Yamaguchi N, Zaedi S, Nusrat Sultana S, Iwashima Y, Sawamura A, Gando S. Time-dependent expression of endothelin-1 in lungs and the effects of TNF-α blocking peptide on acute lung injury in an endotoxemic rat model. Biomed Res 2011;32(1):9–17. [DOI] [PubMed]
- 42.Ishimaru S, Shichiri M, Mineshita S, Hirata Y. Role of endothelin-1/endothelin receptor system in endotoxic shock rats. Hypertens Res 2001;24(2):119–126. [DOI] [PubMed]
- 43.Kuklin V, Kirov M, Sovershaev M, Skogen V, Ytrehus K, Bjertnaes L. Tezosentan-induced attenuation of lung injury in endotoxemic sheep is associated with reduced activation of protein kinase C. Crit Care 2005;9(3):R211–R217. [DOI] [PMC free article] [PubMed]
- 44.Huang W, Tang Y, Li L. HMGB1, a potent proinflammatory cytokine in sepsis. Cytokine 2010;51(2):119–126. [DOI] [PubMed]
- 45.Thomas P, Dixon MS, Winterton SJ, Sheridan DJ. Acute haemodynamic effects of cromakalim in patients with angina pectoris. Br J Clin Pharmacol 1990;29(3):325–331. [DOI] [PMC free article] [PubMed]
- 46.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev 2012;92(1):367–520. [DOI] [PMC free article] [PubMed]