

Abstract Abstract
Pulmonary arterial hypertension (PAH) is a vascular remodeling disease characterized primarily by increased proliferation and resistance to apoptosis in distal pulmonary arteries. Previous literature has demonstrated that the transcription factors NFAT (nuclear factor of activated T cells) and HIF-1α (hypoxia inducible factor 1α) are extensively involved in the pathogenesis of this disease and, more recently, has implicated STAT3 (signal transducer and activator of transcription 3) in their activation. Novel research shows that miR-204, a microRNA recently found to be notably downregulated through induction of PARP-1 (poly [ADP-ribose] polymerase 1) by excessive DNA damage in PAH, inhibits activation of STAT3. Contemporary research also indicates systemic impairment of skeletal muscle microcirculation in PAH and attributes this to a debilitated vascular endothelial growth factor pathway resulting from reduced miR-126 expression in endothelial cells. In this review, we focus on recent research implicating miR-204 and miR-126 in vascular remodeling processes, data that allow a better understanding of PAH molecular pathways and constitute a new hope for future therapy.
Keywords: pulmonary arterial hypertension, microRNA, vascular remodeling, angiogenesis, skeletal muscle
Pulmonary arterial hypertension (PAH) is a severe disorder clinically defined by mean pulmonary arterial pressure of at least 25 mmHg at rest.1 PAH patients display multiple symptoms that are not specific to PAH, including dyspnea, dizziness, and exercise intolerance. Mean age at diagnosis is around 45 years, although onset of symptoms can occur at any age.2 Epidemiologically, it is estimated that between 20 and 50 persons per million suffer from this disease.3 Physiologically, PAH is a vascular remodeling disease of various degrees that affects the adventitia, media, and intima of distal pulmonary arteries, leading to decreased lung perfusion and sustained elevation of pulmonary vascular resistance.4 In response to this resistance, patients develop progressive compensatory right ventricular hypertrophy, which rapidly becomes insufficient and leads to dilatation and failure.5,6 Histologically, PAH is associated with enhanced inflammation, proliferation, and resistance to apoptosis of pulmonary artery smooth muscle cells (PASMCs).7
Despite progress in treatment, medication remains limited and noncurative, and therefore PAH patients typically have poor prognoses (mortality rate of more than 10% after the first year of therapy)8 and quality of life remains severely affected. The pathological mechanisms of PAH establishment need to be better understood and further studied because of their complexities and therapeutic interest. Indeed, knowledge regarding the molecular actors implicated in these impairments increases with each publication, revealing a complex process that remains far from being completely understood.
In the past few years, literature has consistently implicated the role of microRNAs (miRNAs) in PAH. Briefly, miRNAs are single-stranded, evolutionarily conserved, small, noncoding RNAs that are transcribed but not translated.9 The miRNA genes produce primary miRNA transcripts that contain at least one ≈70-nucleotide hairpin loop. These transcripts are transported into the cytoplasm by exportin 5, where they are cleaved by the endonuclease Dicer into an imperfect duplex of 21–23 nucleotides.10 One strand of the duplex is degraded and the other, mature miRNA binds to Dicer and forms a complex with argonaute proteins to form an RNA-induced silencing complex. The miRNAs allow posttranscriptional regulation of gene expression by binding to a target messenger RNA’s 3′-UTR (untranslated region), thereby repressing translation and/or degrading the messenger RNA.11,12
Recently, miRNAs have been widely implicated in both healthy and pathological processes of vascular remodeling, such as wound healing,13 development of pulmonary vessels, and tumor angiogenesis,14 and in the proliferation-apoptosis imbalance15 of cancer. Interestingly, alteration of miRNA expression has been widely found and recognized by the scientific community as a critical actor in PAH establishment. In this review, we focus on the tight link between certain miRNAs and vascular remodeling in PAH.
Molecular contribution of PASMCs to PAH phenotype
Signal transducer and activator of transcription 3 (STAT3) affects multiple downstream processes that encourage PAH pathogenesis through promoting excess cellular proliferation and resistance to apoptosis, and indeed it is found in greater quantity in the PASMCs of PAH patients than in healthy PASMCs (Fig. 1).16 STAT3 is a broad transcription factor, targeted by the nonreceptor tyrosine kinase family member Src,17 which has been shown to play multiple roles in the pathogenesis of cancer18 and several cardiovascular diseases.19 STAT family members are activated through phosphorylation, allowing translocation to the nucleus for transcription regulation activity, induced by cytokines (e.g., interleukin-6),20 growth factors (e.g., platelet-derived growth factor), and agonists (e.g., angiotensin II and endothelin-1).21 An example of the pathogenic potential of STAT3 is survivin, an inhibitor of apoptosis that has elevated levels in both cancer22 and PAH23 and was recently found to be a downstream target of STAT3 through activation of the transcription factor Krüppel-like factor 5.24 STAT3 also suppresses expression of bone morphogenetic protein receptor type II (BMPR2),25,26 a receptor that mediates apoptosis and suppresses Src activation27 and in which a mutation causing dysfunction is a hallmark of heritable PAH.28 Previous studies have found that STAT3 interacts with the promoters of NFAT (nuclear factor of activated T cells) and the provirus integration site for Moloney murine leukemia virus, which in addition to having its own pro-proliferative and antiapoptotic effects, aids in activation of NFATc2;16 thus, in this manner STAT3 induces directly the expression of NFAT and indirectly the activation of NFAT.
Figure 1.

Molecular contribution of pulmonary artery smooth muscle cells (PASMC) to pulmonary arterial hypertension (PAH) phenotype (HIF-1α, NFAT, STAT3): cross links between the major molecular actors implicated in establishment of the PAH-PASMC phenotype. PDGF: platelet-derived growth factor; ET-1: endothelin-1; IL-6: interleukin-6; NFAT: nuclear factor of activated T cells; STAT3: signal transducer and activator of transcription 3; HIF-1α: hypoxia inducible factor 1α; Pim-1: provirus integration site for Moloney murine leukemia virus; KLF5: Krüppel-like factor 5; Bad: Bcl-2-associated death promoter; BMPR2: bone morphogenetic protein receptor type II; Bcl-2: B-cell lymphoma 2; HXK2: hexokinase-2; ROS: reactive oxygen species; PDK: pyruvate dehydrogenase kinase; [K+]i: intracellular [K+]; [Ca2+]i: intracellular [Ca2+]; PPAR: peroxisome proliferator-activated receptor; PDH: pyruvate dehydrogenase. Blue arrows indicate activation and red arrows repression.
Several studies have demonstrated that NFATc2 contributes to the proliferation-apoptosis imbalance that characterizes PAH by affecting both sides of the balance.29-31 NFATc2 has been found to increase B-cell lymphoma 2 expression and colocalization to the mitochondria, leading to mitochondrial hyperpolarization (which prevents release of proapoptotic mediators) and thus enhancing resistance to apoptosis.29,30,32 In addition, through downregulation of K+ channels, especially Kv1.5, NFATc2 (cytoplasmic 2) increases intracellular [K+] ([K+]i) by limiting K+ efflux, thereby inhibiting caspase-mediated apoptosis.29,30,33 On the other side of the balance, Kv1.5 downregulation promotes cell depolarization, causing an influx of Ca2+, which in turn encourages proliferation and vasoconstriction.34 This increase of intracellular Ca2+ ([Ca2+]i) also has a positive feedback effect on NFAT itself by activating calcineurin, which dephosphorylates NFAT, allowing translocation to the nucleus for transcription regulation.35 This K+ channel inhibition is not solely dependent on NFAT but rather is a common theme in many aspects of PAH pathogenesis.
It has been demonstrated that active STAT3 is required for hypoxia inducible factor 1α (HIF-1α) expression.36 HIF-1α is activated by a low–reactive oxygen species (ROS) environment and has been shown, alone and in conjunction with STAT3, to induce high levels of vascular endothelial growth factor (VEGF) expression, thereby promoting proliferation.37 HIF-1α activation has also been shown to result in downregulation of Kv1.538 and thus shares downstream effects with NFAT, including increased [Ca+]i promoting vasoconstriction and proliferation and [K+]i impeding apoptosis.38 HIF-1α activation directs cell metabolism toward a glycolysis mechanism through increased expression of glycolysis initiators such as hexokinase-2 (HXK2)39,40 and pyruvate dehydrogenase kinase (PDK).41 Recently, HXK2 translocation, promoted by HIF-1α, was shown to cause mitochondrial membrane hyperpolarization and therefore resistance to apoptosis.42 PDK is an inhibitor of pyruvate dehydrogenase and thus inhibits the influx of pyruvate into the mitochondria. This shifts the cell toward glycolysis by decreasing the action of Krebs’s cycle, which causes mitochondrial hyperpolarization and reduced production of ROSs, both of which obstruct release of proapoptotic mediators from the mitochondrial transition pore, an efflux channel.43 In fact, inhibition of PDK by dichloroacetate has been shown to reverse the PAH phenotype in an accepted animal model.44 Decreased ROS levels also inhibit Kv1.5 channel function,45 leading to proliferation and resistance to apoptosis through mechanisms described above, and in addition, they further activate HIF-1α, producing a positive feedback mechanism on HIF-1α.38
miR-204 played a key role in the PAH-PASMC phenotype
As described above, activation of Src-STAT3 is a critical player in aberrant NFAT, BMPR2, and HIF-1α expression leading to the PAH-PASMC pro-proliferative and antiapoptotic phenotype. The next step was to determine how STAT3 was regulated. Interestingly, an increasing number of studies and data have strongly implicated miRNA in proliferation-apoptosis process regulation. To determine whether miRNAs are aberrantly expressed in human PAH-PASMC, TaqMan low-density arrays were performed. Seven miRNAs were aberrantly expressed; 6 of these were upregulated (miR-450a, 145, 302b, 27b, 367, and 138), some strongly implicated in PAH etiology,46,47 whereas only one, miR-204, was downregulated in PAH patients, as compared to controls.
Courboulin et al.48 showed that miR-204 expression is mainly confined to PASMCs in the lung. Furthermore, miR-204 expression was decreased in PAH human lung and correlated with PAH severity. Intriguingly, miR-204 has been found to be downregulated in several types of tumors, and it has been proposed that downregulation could contribute to tumor growth.14 Interestingly, miR-204 has already been shown to be decreased in plexiform vasculopathy of severe PAH in humans,49 predicted to be a disease-modifying miRNA in PAH,50 and established as a critical actor in a hypoxia-induced pulmonary hypertension mouse model.51 Abated miR-204 levels were also associated with decreased apoptosis, enhanced cell proliferation,52 and membrane depolarization,53,54 the most common alterations observed in PAH-PASMC. In addition, it has been shown that this pro-proliferative phenotype is associated with activation of the Src-STAT3 and NFAT pathway (Fig. 2),16 suggesting a putative link between miR-204 downregulation, NFAT activation, and cell proliferation. Moreover, Courboulin et al.48 showed that STAT3 activation (p-STAT3) was increased upon miR-204 downregulation, despite the finding that neither NFAT nor Src-STAT3 were predicted direct targets of miR-204. However, Joshi et al.55 documented that miR-204 regulates SHP2 by directly targeting its 3′-UTR, which, by activating Src, increases STAT3 activation. Furthermore, this signaling model has been confirmed by an in vitro approach: by increasing the miR-204 amount in PAH-PASMC, Courboulin et al.48 were able to reverse the pro-proliferative and antiapoptotic phenotype of diseased cells.
Figure 2.
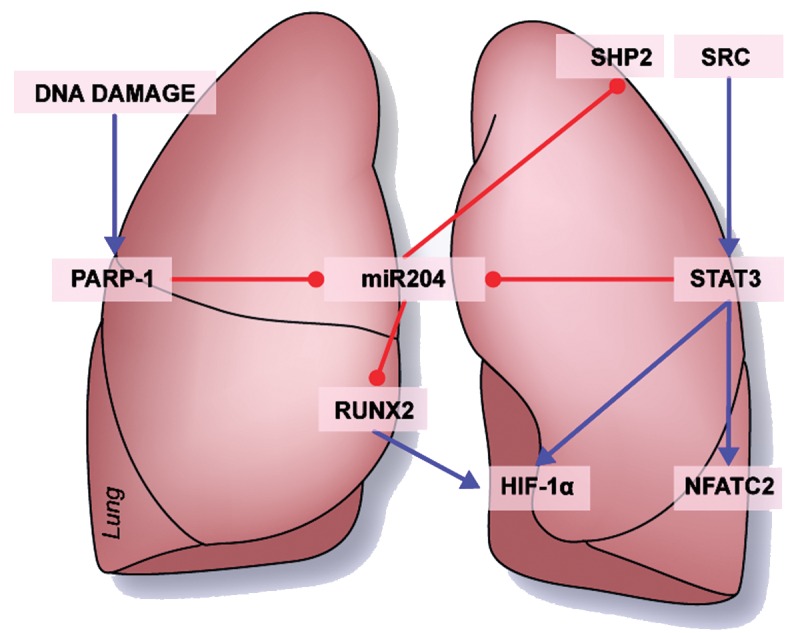
Key role played by miR-204 in pulmonary arterial hypertension pulmonary artery smooth muscle cell phenotype: proposed model of DNA damage leading to miR-204 reduction implicated in STAT3, NFAT, and HIF-1α upregulation. PARP-1: poly (ADP-ribose) polymerase 1; miR: microRNA; STAT3: signal transducer and activator of transcription 3; RUNX2: Runt-related transcription factor 2; HIF-1α: hypoxia inducible factor 1α; NFATC2: nuclear factor of activated T cells, cytoplasmic 2. Blue arrows indicate activation and red arrows repression.
As described above, PASMCs of PAH patients are also characterized by abnormal activation of HIF-1α.38 Interestingly, our laboratory confirmed previous findings in the literature regarding the implication of Runt-related transcription factor 2 (RUNX2) in normoxic HIF-1α activation.56-58 Indeed, we demonstrated that in vitro upregulation of RUNX2 results in HIF-1α activation while its downregulation results in HIF-1α inhibition in human PASMCs.59 Interestingly, it was shown that RUNX2 is one of the most conserved predicted targets of miR-20460 and is regulated by this miRNA in systemic vascular disease.61 As expected, we documented that in vitro downregulation of miR-204 in healthy PASMCs results in RUNX2 and HIF-1α activation, while miR-204 upregulation in PAH-PASMC results in decreased RUNX2 and HIF-1α activation.56,59 These effects are associated with decreases in both PASMC proliferation and apoptosis resistance in PAH.
Interestingly, we showed that miR-204 was decreased in the PASMCs of in vivo monocrotaline (MCT)-induced and Sugen hypoxia-induced PAH rat models.30,62 Furthermore, we observed that in vivo rescue of miR-204, using mimic-204 nebulization, reversed MCT- and Sugen hypoxia-induced PAH63 in the same model. Remarkably, we confirmed our previous in vitro observations. First, animal treatment significantly decreased proliferation and resistance to apoptosis and reduced vascular remodeling in PASMCs of distal pulmonary arteries. Second, rats treated with synthetic miR-204 displayed significant decreases in SHP2, P-STAT3, and NFATc248 activation and additionally displayed inhibition of RUNX2/HIF-1α.56,59 As usual, animal models that did not fully reproduce the pathologic modifications of pulmonary vessels found in the various forms of human PAH limited these findings. The sameness of the observations in the Sugen and MCT models strongly confirmed and validated our results.
DNA damage and miR-204
It is widely established that inflammation and oxidative stress, which are PAH hallmarks, lead to DNA damage.64,65 In the past few years, literature has described DNA damage in the lungs of PAH patients and the MCT rat model,66,67 and recently, numerous groups have focused their attention on this area of research.68,69 We have shown that DNA damage is increased in distal pulmonary arteries and in PASMCs of PAH patients.59,70,71 Interestingly, extreme DNA damage in healthy cells is classically associated with increased apoptosis;72 however, as described above, PAH cells display strong apoptosis resistance. This apparent paradox could be explained by increased activity and efficiency of cellular DNA repair machinery in PAH cells. In accordance with this hypothesis, we showed that protein expression of PARP-1 (poly [ADP-ribose] polymerase 1), an important actor in single-strand DNA repair, is increased in PAH-PASMC. PARP-1 also regulates cell survival, cell death, and gene expression implicated in neoplasic processes. PARP-1 upregulation could therefore explain why decreased apoptosis is observed in PAH-PASMC despite substantial DNA damage.59,70,71
We have also shown that in vitro PARP-1 inhibition using the chemical inhibitor ABT-888 in PAH-PASMC increases miR-204 expression and thereby decreases NFAT and HIF-1α activation (Fig. 2). Interestingly, these data suggest that activation of DNA repair mechanisms through PARP-1 is strongly implicated in the decreased miR-204 expression observed in PAH-PASMC. Furthermore, our laboratory documented, by in vivo experimentation, that inhibition of PARP-1 improves PAH prognoses in MCT- and Sugen hypoxia-induced PAH rat models.59
Expression of miR-204 has been widely recognized has a key actor in vascular remodeling in PAH distal pulmonary arteries, particularly through PASMC NFAT and HIF-1α activation. Meloche et al.73 suggest that DNA damage and abnormal activation of PARP-1 are the first players in STAT3/NFAT/HIF-1α-induced PAH distal pulmonary artery vascular remodeling.
Systemic angiogenic impairment in PAH
Historically, vascular remodeling was documented only in distal pulmonary arteries of PAH patients, suggesting that PAH has effects on the lungs and surrounding vasculature exclusively. However, increasing data in the literature describe peripheral vascular damage in PAH, suggesting a more systemic impairment in this disease.74,75 Potus et al.76 showed a decrease of capillary density in peripheral skeletal muscles of PAH patients, suggesting a peripheral angiogenesis defect in PAH. Furthermore, they identified the molecular pathway implicated and showed the major role of microRNA in this pathological process. Indeed, in PAH skeletal muscle they showed a decrease of miR-126, which is, through its target SPRED-1 (Sprouty-related, EVH1 domain-containing protein 1), one of the major regulators of the VEGF/ERK (extracellular-signal-regulated kinase) pathway (Fig. 3). Furthermore, it was demonstrated that decreased miR-126 expression and capillary density strongly correlate with the reduced exercise tolerance observed in PAH, suggesting association between systemic angiogenic impairment and the major symptom of PAH patients.
Figure 3.
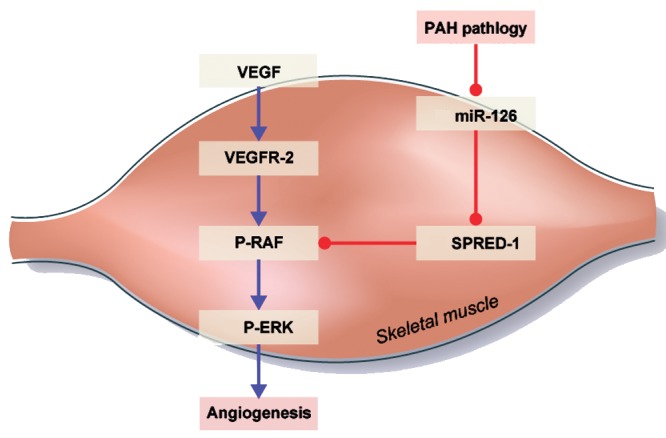
Pulmonary arterial hypertension (PAH) pathology on systemic angiogenesis. Microcirculation impairment in peripheral skeletal muscle and the right ventricle is associated with miR-126 expression downregulation and SPRED-1 upregulation. VEGF: vascular endothelial growth factor; miR: microRNA; VEGFR-2: vascular endothelial growth factor receptor 2; P-RAF: phosphorylated RAF; SPRED-1: Sprouty-related, EVH1 domain–containing protein 1; P-ERK: phosphorylated extracellular signal-regulated kinase. Blue arrows indicate activation and red arrows expression.
Interestingly, Potus et al.77 also observed a vascular defect in the free wall of the PAH human right ventricle (RV). In addition, they showed reduced miR-126 expression and increased target protein SPRED-1 expression in the PAH decompensated RV. Reciprocally, they discovered increased microvessel density and miR-126 expression, as well as decreased SPRED-1 protein expression, in compensated RV. On the basis of these data, they proposed that specific miR-126 downregulation and SPRED-1 upregulation will decrease angiogenesis,78,79 reducing O2 and nutrient RV supply and leading to transition from compensated to decompensated RV.
These findings show that similar defects of the VEGF/ERK pathway may lead to angiogenic deficiency in peripheral skeletal muscles and the RV free wall of PAH patients. Potus et al. confirmed, by an in vitro approach on human RV77 and peripheral skeletal muscle (quadriceps)76 endothelial cells, that angiogenic capacity of PAH cells is decreased compared to that in healthy endothelial cells. Furthermore, artificial increase of miR-126 using mimic-126 restored the angiogenic ability of PAH endothelium to similar levels of healthy cells in both quadriceps and RV endothelial cells (Fig. 3). These observations provide new insight on PAH and strongly indicate that systemic vascular impairment is implicated in the etiology and establishment of major symptoms of PAH, such as exercise intolerance and RV decompensation. Moreover, they suggest a systemic angiogenic defect signature in PAH through miR-126/VEGF/ERK pathway impairment, which could be reversed by miR-126 expression modulation.
Therapeutic promise and implications of miRNA
Numerous studies have increased our knowledge of the molecular, cellular, and physiologic bases of PAH in order to reach a better understanding of PAH etiology. Research in the past few years has shown that aberrantly expressed miRNA is implicated in most, if not all, PAH pathological processes, causing regulation defects.80 This review has focused on the implications of miRNA in the vascular remodeling process. Interestingly, recent data suggest that alteration of miRNA expression in PAH not only occurs in the lung but is also implicated in more systemic impairment. Indeed, downregulation of miR-204 in PAH-PASMC leads to proliferation-apoptosis imbalance, contributing to the vascular remodeling of distal pulmonary arteries. In addition, it was shown that miR-126 is abated in the skeletal muscles and RV of PAH patients, leading to angiogenic impairment. Interestingly, each level of miRNA expression alteration is associated with different symptoms: increase of pulmonary artery resistance in the lung, contributing to exercise intolerance in skeletal muscles and to PAH in the RV.
As a first therapeutic miRNA strategy is in clinical trial for various diseases, including cancer81 and hepatitis C infection,82 there is hope for clinical miRNA-based therapies for other diseases as well. Therefore, in addition to elucidating pathological mechanisms of PAH, research regarding miRNA opens a promising new field of investigation for clinical treatment of this disease.62,83
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009;119(16):2250–2294. http://www.ncbi.nlm.nih.gov/pubmed/19332472. Accessed August 5, 2013. [DOI] [PubMed]
- 2.Frost AE, Badesch DB, Barst RJ, Benza RL, Elliott CG, Farber HW, Krichman A, et al. The changing picture of patients with pulmonary arterial hypertension in the United States: how REVEAL differs from historic and non-US contemporary registries. Chest 2011;139(1):128–137. http://www.ncbi.nlm.nih.gov/pubmed/20558556. Accessed August 13, 2013. [DOI] [PubMed]
- 3.Peacock AJ, Murphy NF, McMurray JJV, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 2007;30(1):104–109. http://www.ncbi.nlm.nih.gov/pubmed/17360728. Accessed August 5, 2013. [DOI] [PubMed]
- 4.Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 2011;8(8):443–455. http://www.ncbi.nlm.nih.gov/pubmed/21691314. Accessed August 5, 2013. [DOI] [PMC free article] [PubMed]
- 5.D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med 1991;115(5):343–349. http://www.ncbi.nlm.nih.gov/pubmed/1863023. Accessed October 4, 2012. [DOI] [PubMed]
- 6.Rubin LJ. Primary pulmonary hypertension. N Engl J Med 1997;336(2):111–117. http://www.ncbi.nlm.nih.gov/pubmed/8988890. Accessed August 5, 2013. [DOI] [PubMed]
- 7.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 2004;43(12 suppl.):13S–24S. http://dx.doi.org/10.1016/j.jacc.2004.02.029. Accessed August 5, 2013. [DOI] [PubMed]
- 8.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006;173(9):1023–1030. http://www.ncbi.nlm.nih.gov/pubmed/16456139. Accessed August 5, 2013. [DOI] [PubMed]
- 9.Aslam MI, Taylor K, Pringle JH, Jameson JS. MicroRNAs are novel biomarkers of colorectal cancer. Br J Surg 2009;96(7):702–710. http://www.ncbi.nlm.nih.gov/pubmed/19526617. Accessed August 5, 2013. [DOI] [PubMed]
- 10.Suárez Y, Sessa WC. MicroRNAs as novel regulators of angiogenesis. Circ Res 2009;104(4):442–454. http://circres.ahajournals.org/content/104/4/442.abstract. Accessed August 5, 2013. [DOI] [PMC free article] [PubMed]
- 11.Lee R, Feinbaum R, Ambros V. A short history of a short RNA. Cell 2004;116(2 suppl.):S89–S92. http://www.ncbi.nlm.nih.gov/pubmed/15055592. Accessed August 5, 2013. [DOI] [PubMed]
- 12.Kwak PB, Iwasaki S, Tomari Y. The microRNA pathway and cancer. Cancer Sci 2010;101(11):2309–2315. http://www.ncbi.nlm.nih.gov/pubmed/20726859. Accessed June 7, 2013. [DOI] [PMC free article] [PubMed]
- 13.Banerjee J, Chan YC, Sen CK. MicroRNAs in skin and wound healing. Physiol Genomics 2011;43(10):543–556. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3110888&tool=pmcentrez&rendertype=abstract. Accessed August 5, 2013. [DOI] [PMC free article] [PubMed]
- 14.Donnem T, Fenton CG, Lonvik K, Berg T, Eklo K, Andersen S, Stenvold H, et al. MicroRNA signatures in tumor tissue related to angiogenesis in non-small cell lung cancer. PLoS ONE 2012;7(1):e29671.http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3266266&tool=pmcentrez&rendertype=abstract. Accessed August 5, 2013. [DOI] [PMC free article] [PubMed]
- 15.Gammell P. MicroRNAs: recently discovered key regulators of proliferation and apoptosis in animal cells: identification of miRNAs regulating growth and survival. Cytotechnology 2007;53(1–3):55–63. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2267611&tool=pmcentrez&rendertype=abstract. Accessed August 5, 2013. [DOI] [PMC free article] [PubMed]
- 16.Paulin R, Courboulin A, Meloche J, Mainguy V, de la Roque ED, Saksouk N, Côté J, Provencher S, Sussman MA, Bonnet S. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 2011;123(11):1205–1215.http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3545712&tool=pmcentrez&rendertype=abstract. Accessed August 1, 2013. [DOI] [PMC free article] [PubMed]
- 17.Chang Y-M, Kung H-J, Evans CP. Nonreceptor tyrosine kinases in prostate cancer. Neoplasia 2007;9(2):90–100. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1813931&tool=pmcentrez&rendertype=abstract. Accessed August 11, 2013. [DOI] [PMC free article] [PubMed]
- 18.Johnston PA, Grandis JR. STAT3 signaling: anticancer strategies and challenges. Mol Interventions 2011;11(1):18–26. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3063716&tool=pmcentrez&rendertype=abstract. Accessed August 11, 2013. [DOI] [PMC free article] [PubMed]
- 19.Lim CP, Fu X-Y. Multiple roles of STAT3 in cardiovascular inflammatory responses. Prog Mol Biol Transl Sci 2012;106:63–73. http://www.ncbi.nlm.nih.gov/pubmed/22340714. Accessed August 11, 2013. [DOI] [PubMed]
- 20.Darnell JE Jr. STATs and gene regulation. Science 1997;277(5332):1630–1635. http://www.ncbi.nlm.nih.gov/pubmed/9287210. Accessed August 1, 2013. [DOI] [PubMed]
- 21.Banes-Berceli AKL, Ketsawatsomkron P, Ogbi S, Patel B, Pollock DM, Marrero MB. Angiotensin II and endothelin-1 augment the vascular complications of diabetes via JAK2 activation. Am J Physiol Heart Circ Physiol 2007;293(2):H1291–H1299. http://www.ncbi.nlm.nih.gov/pubmed/17526654. Accessed August 1, 2013. [DOI] [PubMed]
- 22.Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer 2003;3(1):46–54. http://www.ncbi.nlm.nih.gov/pubmed/12509766. Accessed August 1, 2013. [DOI] [PubMed]
- 23.McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Bonnet S, Puttagunta L, Michelakis ED. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest 2005;115(6):1479–1491.http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1136986&tool=pmcentrez&rendertype=abstract. Accessed August 1, 2013. [DOI] [PMC free article] [PubMed]
- 24.Courboulin A, Tremblay VL, Barrier M, Meloche J, Jacob MH, Chapolard M, Bisserier M, et al. Krüppel-like factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir Res 2011;12:128. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3193170&tool=pmcentrez&rendertype=abstract. Accessed August 1, 2013. [DOI] [PMC free article] [PubMed]
- 25.Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res 2009;104(10):1184–1191. http://www.ncbi.nlm.nih.gov/pubmed/19390056. Accessed August 1, 2013. [DOI] [PubMed]
- 26.Meloche J, Courchesne A, Barrier M, Carter S, Bisserier M, Paulin R, Lauzon-Joset J-F, et al. Critical role for the advanced glycation end-products receptor in pulmonary arterial hypertension etiology. J Am Heart Assoc 2013;2(1):e005157. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3603259&tool=pmcentrez&rendertype=abstract. Accessed August 1, 2013. [DOI] [PMC free article] [PubMed]
- 27.Wong WKP, Knowles JA, Morse JH. Bone morphogenetic protein receptor type II C-terminus interacts with c-Src: implication for a role in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2005;33(5):438–446. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2715351&tool=pmcentrez&rendertype=abstract. Accessed November 20, 2013. [DOI] [PMC free article] [PubMed]
- 28.Zhang S, Fantozzi I, Tigno DD, Yi ES, Platoshyn O, Thistlethwaite PA, Kriett JM, Yung G, Rubin LJ, Yuan JX. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2003;285(3):L740–L754. http://www.ncbi.nlm.nih.gov/pubmed/12740218. Accessed August 1, 2013. [DOI] [PubMed]
- 29.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci USA 2007;104(27):11418–11423. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1903339&tool=pmcentrez&rendertype=abstract. Accessed August 1, 2013. [DOI] [PMC free article] [PubMed]
- 30.Bonnet S, Paulin R, Sutendra G, Dromparis P, Roy M, Watson KO, Nagendran J, Haromy A, Dyck JR, Michelakis ED. Dehydroepiandrosterone reverses systemic vascular remodeling through the inhibition of the Akt/GSK3-β/NFAT axis. Circulation 2009;120(13):1231–1240. http://www.ncbi.nlm.nih.gov/pubmed/19752325. Accessed August 1, 2013. [DOI] [PubMed]
- 31.Courboulin A, Barrier M, Perreault T, Bonnet P, Tremblay VL, Paulin R, Tremblay E, et al. Plumbagin reverses proliferation and resistance to apoptosis in experimental PAH. Eur Respir J 2012;40(3):618–629. http://www.ncbi.nlm.nih.gov/pubmed/22496325. Accessed August 1, 2013. [DOI] [PubMed]
- 32.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11(1):37–51. http://www.ncbi.nlm.nih.gov/pubmed/17222789. Accessed August 1, 2013. [DOI] [PubMed]
- 33.Remillard CV, Yuan JX. Activation of K+ channels: an essential pathway in programmed cell death. Am J Physiol Lung Cell Mol Physiol 2004;286(1):L49–L67. [DOI] [PubMed]
- 34.Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, Seiden JE, Rubin LJ, Yuan JX. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 2000;279(5):C1540–C1549. http://www.ncbi.nlm.nih.gov/pubmed/11029301. Accessed August 1, 2013. [DOI] [PubMed]
- 35.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 2005;5(6):472–484. http://www.ncbi.nlm.nih.gov/pubmed/15928679. Accessed August 1, 2013. [DOI] [PubMed]
- 36.Niu G, Briggs J, Deng J, Ma Y, Lee H, Kortylewski M, Kujawski M, et al. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1α RNA expression in both tumor cells and tumor-associated myeloid cells. Mol Cancer Res 2008;6(7):1099–1105. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2775817&tool=pmcentrez&rendertype=abstract. Accessed August 1, 2013. [DOI] [PMC free article] [PubMed]
- 37.Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS, Gallick GE. HIF-1α, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene 2005;24(19):3110–3120. http://www.ncbi.nlm.nih.gov/pubmed/15735682. Accessed August 5, 2013. [DOI] [PubMed]
- 38.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, et al. An abnormal mitochondrial–hypoxia inducible factor-1α–Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 2006;113(22):2630–2641. http://circ.ahajournals.org/cgi/content/abstract/113/22/2630. Accessed March 29, 2012. [DOI] [PubMed]
- 39.Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med 2001;7(8):345–350. http://www.ncbi.nlm.nih.gov/pubmed/11516994. Accessed August 1, 2013. [DOI] [PubMed]
- 40.Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem 2001;276(46):43407–43412. http://www.ncbi.nlm.nih.gov/pubmed/11557773. Accessed August 1, 2013. [DOI] [PubMed]
- 41.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3(3):177–185. http://www.ncbi.nlm.nih.gov/pubmed/16517405. Accessed August 1, 2013. [DOI] [PubMed]
- 42.Lambert CM, Roy M, Robitaille GA, Richard DE, Bonnet S. HIF-1 inhibition decreases systemic vascular remodelling diseases by promoting apoptosis through a hexokinase 2-dependent mechanism. Cardiovasc Res 2010;88(1):196–204. http://www.ncbi.nlm.nih.gov/pubmed/20498255. Accessed August 1, 2013. [DOI] [PubMed]
- 43.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol 2001;2(1):67–71. http://www.ncbi.nlm.nih.gov/pubmed/11413468. Accessed August 1, 2013. [DOI] [PubMed]
- 44.McMurtry MS, Bonnet S, Wu X, Dyck JRB, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 2004;95(8):830–840. http://circres.ahajournals.org/content/95/8/830.full. Accessed August 1, 2013. [DOI] [PubMed]
- 45.Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 1998;101(11):2319–2330. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=508821&tool=pmcentrez&rendertype=abstract. Accessed August 1, 2013. [DOI] [PMC free article] [PubMed]
- 46.Li S, Ran Y, Zhang D, Chen J, Li S, Zhu D. MicroRNA-138 plays a role in hypoxic pulmonary vascular remodelling by targeting Mst1. Biochem J 2013;452(2):281–291. http://www.ncbi.nlm.nih.gov/pubmed/23485012. Accessed August 12, 2013. [DOI] [PubMed]
- 47.Caruso P, Dempsie Y, Stevens HC, McDonald RA, Long L, Lu R, White K, et al. A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples. Circ Res 2012;111(3):290–300. http://www.ncbi.nlm.nih.gov/pubmed/22715469. Accessed August 12, 2013. [DOI] [PubMed]
- 48.Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 2011;208(3):535–548. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3058572&tool=pmcentrez&rendertype=abstract. Accessed March 24, 2012. [DOI] [PMC free article] [PubMed]
- 49.Bockmeyer CL, Maegel L, Janciauskiene S, Rische J, Lehmann U, Maus UA, Nickel, N, et al. Plexiform vasculopathy of severe pulmonary arterial hypertension and microRNA expression. J Heart Lung Transplant 2012;31(7)764–772. http://dx.doi.org/10.1016/j.healun.2012.03.010. Accessed April 29, 2012. [DOI] [PubMed]
- 50.Parikh VN, Jin RC, Rabello S, Gulbahce N, White K, Hale A, Cottrill KA, et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 2012;125(12):1520–1532. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3353408&tool=pmcentrez&rendertype=abstract. Accessed August 11, 2013. [DOI] [PMC free article] [PubMed]
- 51.Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, Sdrimas K, Fernandez-Gonzalez A, Kourembanas S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 2012;126(22):2601–2611. http://www.ncbi.nlm.nih.gov/pubmed/23114789. Accessed August 10, 2013. [DOI] [PMC free article] [PubMed]
- 52.Li G, Luna C, Qiu J, Epstein DL, Gonzalez P. Role of miR-204 in the regulation of apoptosis, endoplasmic reticulum stress response, and inflammation in human trabecular meshwork cells. Invest Ophthalmol Vis Sci 2011;52(6):2999–3007. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3109013&tool=pmcentrez&rendertype=abstract. Accessed August 5, 2013. [DOI] [PMC free article] [PubMed]
- 53.Wang FE, Zhang C, Maminishkis A, Dong L, Zhi C, Li R, Zhao J, et al. MicroRNA-204/211 alters epithelial physiology. FASEB J 2010;24(5):1552–1571. http://www.fasebj.org/content/24/5/1552.full. Accessed August 5, 2013. [DOI] [PMC free article] [PubMed]
- 54.Lee Y, Yang X, Huang Y, Fan H, Zhang Q, Wu Y, Li J, et al. Network modeling identifies molecular functions targeted by miR-204 to suppress head and neck tumor metastasis. PLoS Comput Biol 2010;6(4):e1000730. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2848541&tool=pmcentrez&rendertype=abstract. Accessed April 21, 2012. [DOI] [PMC free article] [PubMed]
- 55.Joshi SR, McLendon JM, Comer BS, Gerthoffer WT. MicroRNAs-control of essential genes: implications for pulmonary vascular disease. Pulm Circ 2011;1(3):357–364. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3224427&tool=pmcentrez&rendertype=abstract. Accessed October 29, 2012. [DOI] [PMC free article] [PubMed]
- 56.Margaillan G, Courboulin A, Potus F, Courchesne A, Couture C, Bonnet P, Provencher S, Bonnet S. RUNX2 regulates the normoxic activation of the hypoxic inducible factor (HIF-1) under the influence of miR-204 in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;187:A2094. http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2013.187.1_MeetingAbstracts.A2094. Accessed July 30, 2013.
- 57.Lee SH, Che X, Jeong J-H, Choi JY, Lee YJ, Lee YH, Bae SC, Lee YM. Runx2 stabilizes hypoxia-inducible factor-1α through competition with pVHL and stimulates angiogenesis in growth plate hypertrophic chondrocytes. J Biol Chem 2012;287(18):14760–14771. http://www.ncbi.nlm.nih.gov/pubmed/22351759. Accessed March 13, 2012. [DOI] [PMC free article] [PubMed]
- 58.Kwon T-G, Zhao X, Yang Q, Li Y, Ge C, Zhao G, Franceschi RT. Physical and functional interactions between Runx2 and HIF-1α induce vascular endothelial growth factor gene expression. J Cell Biochem 2011;112(12):3582–3593. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3202060&tool=pmcentrez&rendertype=abstract. Accessed July 30, 2013. [DOI] [PMC free article] [PubMed]
- 59.von Gise A, Archer SL, MacLean MR, Hansmann G. Full conference report: the first Keystone Symposia Conference on pulmonary vascular disease and right ventricular dysfunction: current concepts and future therapies. Pulm Circ 2013;3(2):275–277. [DOI] [PMC free article] [PubMed]
- 60.Avellino R, Carrella S, Pirozzi M, Risolino M, Salierno FG, Franco P, Stoppelli P, Verde P, Banfi S, Conte I. miR-204 targeting of Ankrd13A controls both mesenchymal neural crest and lens cell migration. PLoS ONE 2013;8(4):e61099. http://dx.plos.org/10.1371/journal.pone.0061099. Accessed August 5, 2013. [DOI] [PMC free article] [PubMed]
- 61.Cui R-R, Li S-J, Liu L-J, Yi L, Liang QH, Zhu X, Liu GY, et al. MicroRNA-204 regulates vascular smooth muscle cell calcification in vitro and in vivo. Cardiovasc Res 2012;96(2):320–329. http://www.ncbi.nlm.nih.gov/pubmed/22871591. Accessed August 12, 2013. [DOI] [PubMed]
- 62.Paulin R, Courboulin A, Barrier M, Bonnet S. From oncoproteins/tumor suppressors to microRNAs, the newest therapeutic targets for pulmonary arterial hypertension. J Mol Med (Berl) 2011;89(11):1089–1101. http://www.ncbi.nlm.nih.gov/pubmed/21761156. Accessed March 24, 2012. [DOI] [PubMed]
- 63.Courboulin A, Barrier M. Abstract 18277: role of the miR-204/miR-145 communication in the etiology of pulmonary arterial hypertension. Circulation 2011;124(21 suppl.):A18277. http://circ.ahajournals.org/cgi/content/meeting_abstract/124/21_MeetingAbstracts/A18277.
- 64.Danielsen PH, Møller P, Jensen KA, Sharma AK, Wallin H, Bossi R, Autrup H, et al. Oxidative stress, DNA damage, and inflammation induced by ambient air and wood smoke particulate matter in human A549 and THP-1 cell lines. Chem Res Toxicol 2011;24(2):168–184. http://www.ncbi.nlm.nih.gov/pubmed/21235221. Accessed August 5, 2013. [DOI] [PubMed]
- 65.Danielsen PH, Loft S, Jacobsen NR, Jensen KA, Autrup H, Ravanat JL, Wallin H, Møller P. Oxidative stress, inflammation, and DNA damage in rats after intratracheal instillation or oral exposure to ambient air and wood smoke particulate matter. Toxicol Sci 2010;118(2):574–585. http://www.ncbi.nlm.nih.gov/pubmed/20864625. Accessed August 5, 2013. [DOI] [PubMed]
- 66.Wang Y, Jing L, Zhao X-M, Han J-J, Xia Z-L, Qin S-C, Wu Y-P, Sun X-J. Protective effects of hydrogen-rich saline on monocrotaline-induced pulmonary hypertension in a rat model. Respir Res 2011;12:26. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3065415&tool=pmcentrez&rendertype=abstract. Accessed August 5, 2013. [DOI] [PMC free article] [PubMed]
- 67.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med 2004;169(6):764–769. http://www.ncbi.nlm.nih.gov/pubmed/14701708. Accessed August 5, 2013. [DOI] [PubMed]
- 68.Federici C, Aldred MA. Elevated levels of DNA damage in patients with pulmonary arterial hypertension measured by cytokinesis-block micronucleus assay. Am J Respir Crit Care Med 2013;187:A5407. http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2013.187.1_MeetingAbstracts.A5407. Accessed August 12, 2013.
- 69.Li M, Vattulainen S, Myllykangas S, Koskenvuo J, Alastalo T-P. Abstract 16996: Meta-analysis of DNA microarrays reveals dysregulation of DNA repair genes downstream of BMPR2 in pulmonary arterial hypertension. Circulation 2012;126(21 suppl.):A16996. http://circ.ahajournals.org/cgi/content/meeting_abstract/126/21_MeetingAbstracts/A16996. Accessed August 12, 2013.
- 70.Courboulin A, Meloche J, Potus F, Margaillan G, Provencher S, Bonnet S. Role for DNA repair machinery in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;187:A1733. http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2013.187.1_MeetingAbstracts.A1733. Accessed August 5, 2013.
- 71.Courboulin A, Krietsch J, Coulombe Y, Poirier G, Masson JY, Bonnet S. Abstract 16424: a critical role of poly-ADP ribose polymerase 1 (PARP-1) in human pulmonary arterial hypertension. Circulation 2011;124(21 suppl.):A16424. http://circ.ahajournals.org/cgi/content/meeting_abstract/124/21_MeetingAbstracts/A16424. Accessed August 5, 2013.
- 72.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007;35(4):495–516. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2117903&tool=pmcentrez&rendertype=abstract. Accessed July 31, 2013. [DOI] [PMC free article] [PubMed]
- 73.Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, Zervopoulos S, Graydon C, et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014;129(7):786–797. http://www.ncbi.nlm.nih.gov/pubmed/24270264. Accessed March 26, 2014. [DOI] [PubMed]
- 74.Badesch DB, Champion HC, Sanchez MAG, Hoeper MM, Loyd JE, Manes A, McGoon M, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2009;54(1 suppl.):S55–S66. http://www.ncbi.nlm.nih.gov/pubmed/19555859. Accessed May 24, 2013. [DOI] [PubMed]
- 75.de Man FS, Handoko ML, Groepenhoff H, van’t Hul AJ, Abbink J, Koppers RJ, Grotjohan HP, et al. Effects of exercise training in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2009;34(3):669–675. http://www.ncbi.nlm.nih.gov/pubmed/19720810. Accessed May 24, 2013. [DOI] [PubMed]
- 76.Potus F, Malenfant S, Breuils-Bonnet S, Margaillant G, Bonnet S, Provencher S. miR-126, angiogenesis and exercise intolerance in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;187:A5913. http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2013.187.1_MeetingAbstracts.A5913. Accessed August 5, 2013.
- 77.Potus F, Paulin R, Breuils-Bonnet S, Tremblay E, Couture C, Michelakis ED, Provencher S, Bonnet S. Downregulation of the angiomiR-126 contributes to the failing right ventricle in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;187:A5926. http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2013.187.1_MeetingAbstracts.A5926. Accessed August 5, 2013.
- 78.Fish JE, Santoro MM, Morton SU, Yu S, Yeh R-F, Wythe JD, Bruneau BG, Stainier DYR, Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell 2008;15(2):272–284. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2604134&tool=pmcentrez&rendertype=abstract. Accessed March 13, 2012. [DOI] [PMC free article] [PubMed]
- 79.Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell 2008;15(2):261–271. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2685763&tool=pmcentrez&rendertype=abstract. Accessed March 6, 2012. [DOI] [PMC free article] [PubMed]
- 80.Grant JS, White K, Maclean MR, Baker AH. MicroRNAs in pulmonary arterial remodeling. Cell Mol Life Sci 2013;70(23):4479–4494. http://www.ncbi.nlm.nih.gov/pubmed/23739951. Accessed August 5, 2013. [DOI] [PMC free article] [PubMed]
- 81.Brenner A, Mirna Therapeutics. A multicenter phase I study of MRX34, microRNA miR-RX34 liposome injectable suspension. ClinicalTrials.gov identifier: NCT01829971. http://www.clinicaltrials.gov/ct2/show/NCT01829971?term=phase&rcv_d=14.
- 82.Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med 2013;368(18):1685–1694. http://www.nejm.org/doi/full/10.1056/NEJMoa1209026. Accessed July 15, 2013. [DOI] [PubMed]
- 83.Meloche J, Paulin R, Provencher S, Bonnet S. Therapeutic potential of microRNA modulation in pulmonary arterial hypertension. Curr Vasc Pharmacol. http://www.ncbi.nlm.nih.gov/pubmed/23713859. Published May 13, 2013. Accessed August 5, 2013. [DOI] [PubMed]