

Abstract Abstract
Patients with acute respiratory distress syndrome (ARDS) exhibit elevated levels of interleukin-6 (IL-6), which correlate with increased morbidity and mortality. The exact role of IL-6 in ARDS has proven difficult to study because it exhibits either pro- or anti-inflammatory actions in mouse models of lung injury, depending on the model utilized. In order to improve understanding of the role of this complex cytokine in ARDS, we evaluated IL-6 using the clinically relevant combination of lipopolysaccharide (LPS) and ventilator-induced lung injury (VILI) in IL-6−/− mice. Bronchoalveolar lavage fluid (BAL), whole-lung tissue, and histology were evaluated for inflammatory markers of injury. Transendothelial electrical resistance was used to evaluate the action of IL-6 on endothelial cells in vitro. In wild-type mice, the combination model showed a significant increase in lung injury compared to either LPS or VILI alone. IL-6−/− mice exhibited a statistically significant decrease in BAL cellular inflammation as well as lower histologic scores for lung injury, changes observed only in the combination model. A paradoxical increase in BAL total protein was observed in IL-6−/− mice exposed to LPS, suggesting that IL-6 provides protection from vascular leakage. However, in vitro data showed that IL-6, when combined with its soluble receptor, actually caused a significant increase in endothelial cell permeability, suggesting that the protection seen in vivo was likely due to complex interactions of IL-6 and other inflammatory mediators rather than to direct effects of IL-6. These studies suggest that a dual-injury model exhibits utility in evaluating the pleiotropic effects of IL-6 in ARDS on inflammatory cells and lung endothelium.
Keywords: acute respiratory distress syndrome (ARDS), interleukin-6 (IL-6), lipopolysaccharide (LPS), ventilator-induced lung injury (VILI)
Introduction
Acute respiratory distress syndrome (ARDS) is a clinical syndrome characterized by acute hypoxemic respiratory failure and noncardiogenic pulmonary infiltration.1-3 Pneumonia, sepsis, trauma, and aspiration of gastric contents represent the most common causes of ARDS.2,3 The pathogenesis of ARDS is characterized by both lung microvascular endothelial and epithelial damage that results in accumulation of protein-rich edema in alveoli and cellular infiltration composed of neutrophils, macrophages, and red blood cells.1-3
Interleukin 6 (IL-6) is a cytokine that exhibits both proinflammatory and anti-inflammatory properties and is produced by various cell types, including T cells, B cells, monocytes, fibroblasts, endothelial cells, and synovial cells. Circulating IL-6 levels are elevated in nearly all infectious, traumatic, and inflammatory states,4 including ARDS. Elevated levels of IL-6 are found in the bronchoalveolar lavage fluid (BAL) and plasma of patients with ARDS and patients at risk of ARDS.5-7 Higher levels of IL-6 are also associated with increased risk of mortality.7 However, the exact role of IL-6 in ARDS pathogenesis has not yet been elucidated.
The difficulties in interrogating the role of IL-6 in ARDS arise, in part, from the observed pro- and anti-inflammatory effects of IL-6, which occur in a manner that is dependent on the type of signaling pathway utilized.4,8 Previous studies in mouse models of lung injury have shown model-specific responses related to IL-6, either protective or injurious.9-12 These findings highlight the critical importance of using a preclinical model that most closely mimics human disease. This is particularly true in interrogating a disease with a multifactorial etiology, such as ARDS. In this study, we used a mouse model that closely mimics human ARDS to evaluate the role of IL-6 in ARDS pathobiology, particularly its effects on endothelial damage, inflammation, and vascular leakage. We utilized lipopolysaccharide (LPS), followed 24 hours later by ventilator-induced lung injury (VILI), to produce acute lung injury, thereby creating a combination model that closely mimics the human condition of underlying lung infection exacerbated by mechanical ventilation. Using this inflammatory lung injury model, we evaluated the effect of IL-6 expression on vascular leakage and cellular infiltration in the injured lung.
Methods
Combination model of ARDS
All animal experiments were approved by the Animal Care and Use Committee at the University of Illinois at Chicago. Mice were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited institution in autoclaved microisolator cages with free access to food and water. Experiments used C57Bl/6J mice of similar weights, referred to as wild-type (WT), and IL-6−/− mice on a C57Bl/6J background (B6.129S2-Il6tm1Kopf/J)13 between 10 and 16 weeks of age, with at least 5 animals per group.
For the combination model, mice were anesthetized with ketamine (100 mg/kg) and xylazine (5 mg/kg) given by intraperitoneal injection, intubated with a 20-g catheter, and intratracheally injected with 0.2 mg/kg LPS (Sigma, St. Louis, MO) diluted in phosphate-buffered saline (PBS) for a total volume of 1.6 μL/g. Twenty hours after injection, mice were again anesthetized with ketamine and xylazine and intubated with a 20-g catheter. Mice were then placed on mechanical ventilation for 4 hours (20 mL/kg tidal volume, 90 breaths/min, room air) with additional ketamine/xylazine given as needed at one-quarter of the original dose. At the end of 4 hours, mice were euthanized by exsanguination, and samples were collected.
Three different control groups of mice were used. The first group (LPS alone) was anesthetized with ketamine/xylazine and given 0.2 mg/kg LPS diluted in PBS for a total volume of 1.6 μL/g intratracheally. After 24 hours, the animals were euthanized (ketamine/xylazine anesthesia followed by exsanguination), and BAL and lung tissue were collected. The second group (VILI alone) was anesthetized with ketamine/xylazine and given 1.6 μL/g PBS intratracheally. After 20 hours, the animals were anesthetized again with ketamine/xylazine and placed on mechanical ventilation for 4 hours (20 mL/kg tidal volume, 90 breaths/min). At the end of 4 hours, animals were euthanized by exsanguination, and BAL and lung tissue were collected. The final group (PBS alone) was anesthetized with ketamine/xylazine and received 1.6 μL/g PBS intratracheally. After 24 hours, the animals were euthanized (ketamine/xylazine anesthesia followed by exsanguination), and BAL and lung tissue were collected.
Bronchoalveolar lavage
Immediately after euthanasia, the lungs were flushed with 1 mL Hank’s balanced salt solution intratracheally, and the resulting lavage fluid was centrifuged to collect a cell pellet and supernatant, as previously described.14-17 Total cells were counted with a hemacytometer (Bio-Rad, Hercules, CA), and a manual differential was performed, as previously described.14 The supernatant was used to measure total protein (Pierce BCA protein assay; Thermo Scientific, Rockford, IL) and total IL-6 (Mouse IL-6 ELISA Ready-SET-Go!, eBioscience, San Diego, CA).
Lung myeloperoxidase (MPO) assay
PBS was flushed through the pulmonary artery to flush the blood from the vessels in the lung, and then the lungs were removed and frozen in liquid nitrogen. An MPO assay was performed as previously described.18 Lung tissue was homogenized in 50 mM potassium phosphate buffer (pH 6.0) containing 0.5% hexatrimethylammonium bromide, followed by 3 cycles of freezing and thawing and by 40 seconds of full-speed sonication (Virsonic V60; Boston Laboratory Equipment, Woburn, MA) on ice. Samples were centrifuged (10,000 g for 10 minutes), and the supernatant was collected. An assay buffer containing 0.167 mg/mL of o-dianisidine and 0.0005% of H2O2 was prepared. An aliquot (10 μL) of supernatant was added to a 96-well plate, and the reaction was initiated by adding 290 μL of assay buffer. Change in absorbance was read over 1 minute at 460 nm with a kinetic spectrometer (Bio-Rad, Philadelphia, PA).
Histology
Lung tissue samples were fixed in formalin, embedded in paraffin, cut into 10-μm sections, and stained with hematoxylin and eosin (H&E). Photomicrographs were taken at 40× magnification. Histology slides were scanned and evaluated with ImageScope (Aperio, Vista, CA). H&E-stained lung sections (n = 3–6 per condition) were scored by a single individual experienced in scoring lung pathology and blinded to the experiment, using a scoring system similar to one previously described.19 In each lung section, 5 randomly selected fields were scored for interstitial edema, neutrophil infiltration, and alveolar wall damage at 40× magnification, utilizing the digitized slides. Each parameter of lung injury was given a score from 0 to 4, with 0 representing absence of the parameter, 1 that approximately 25% of the field was affected, 2 that approximately 50% of the field was affected, 3 that approximately 75% of the field was affected, and 4 severe and diffuse presence of the parameter throughout the chosen field. The total score for each field is the sum of the scores for each of the three parameters of injury.
Measurement of transendothelial electrical resistance (TER)
The cellular barrier properties of human lung microvascular endothelial cells (HLMVECs) were measured with an electrical cell substrate impedance–sensing system (Applied Biophysics, Troy, NY), as described previously.20 Briefly, cells were grown to a monolayer on small gold electrodes in complete culture medium containing 10% fetal bovine serum and growth factor supplement. Four hours before TER measurement, the culture medium was changed to plain medium with no serum supplement. Total electrical resistance was measured across the monolayer for 10 hours with IL-6 (recombinant human IL-6; R&D Systems, Minneapolis, MN) at 10 ng/mL, IL-6 receptor (recombinant human IL-6 Rα; R&D Systems) at 50 ng/mL, both IL-6 and IL-6 Rα (10 and 50 ng/mL, respectively), or PBS control (vehicle) added to the media after 45 minutes of baseline recording. Resistance was normalized to the initial voltage. TER values from 10 microelectrodes for each condition were pooled at discrete time points with custom-designed Epool software, as described previously.21
Statistics
Statistical analysis was performed with the OriginPro 8.6 software program (OriginLab, Northampton, MA). For BAL total protein, total cell counts, total neutrophils, IL-6 levels, and lung MPO levels, the data were normally distributed, so a one-way ANOVA was performed to compare the combination model to controls. Post hoc analysis was performed with Tukey’s method. For comparing WT mice to IL-6−/− mice within each condition, a Student’s t test was used, with a 4-group Bonferroni correction to the P values (for a result to be significant at the 0.05 level, a P value of <0.0125 was required); data are expressed as mean ± standard error of the mean. For TER analysis, a one-way ANOVA was performed on the normalized data for the 4 groups at each discrete time point; data are expressed as mean ± standard error of the mean. A P value of <0.05 was considered significant.
For lung injury score analysis, a Kruskal-Wallis ANOVA was used for comparing the combination model to controls. Post hoc analysis was performed with a Mann-Whitney test, with a 4-group Bonferroni correction to the P values (a P value of <0.0125 on the Mann-Whitney test was considered significant at the 0.05 level). For comparing WT mice to IL-6−/− mice within each group, a Mann-Whitney test was used. A P value of <0.05 was considered significant. Data are expressed in box-and-whisker plots, with the box representing the 25th and 75th percentiles and the whiskers representing the fifth and 95th percentiles.
Results
Quantitative measures of combination model lung injury in WT mice
Analysis of BAL from WT mice exposed to the combination model demonstrated significantly greater injury and inflammation compared to that from animals in the control groups (Fig. 1). BAL from mice exposed to the combination model of lung injury showed significantly higher total cell counts (Fig. 1A), total neutrophils (Fig. 1B), and total protein (Fig. 1D) compared to that from mice receiving LPS alone (P = 0.026, 0.013, and 7.3 × 10−4 for total cell count, total neutrophils, and total protein, respectively), VILI alone (P = 1.9 × 10−6, 1.5 × 10−7, and 2.2 × 10−7, respectively), or PBS alone (P = 1.9 × 10−7, 1.06 × 10−7, and <1 × 1010, respectively). MPO activity in the lungs of WT mice exposed to the combination model was significantly higher than that of the WT mice exposed to PBS alone (P = 4.5 × 10−4) or VILI alone (P = 0.0026). However, the difference in MPO activity between the WT mice exposed to the combination model and those that received LPS alone was not significant. BAL levels of IL-6 in animals exposed to the combination model were not significantly increased when compared to IL-6 levels from the LPS-only group (Fig. 1E) but were significantly higher than those in the VILI-only and PBS-only groups (P = 4.4 × 10−4 and 3.1 × 10−4, respectively), exhibiting a more-than-100-fold increase when compared to either group.
Figure 1.
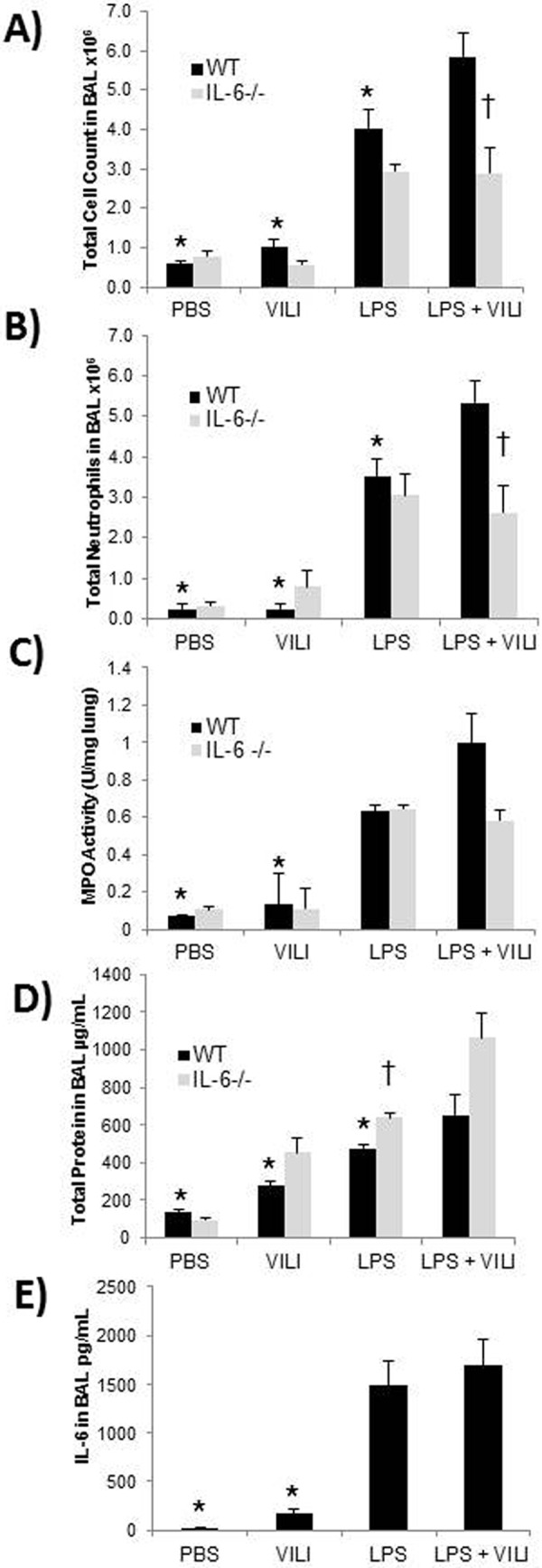
Measurements of lung injury in wild-type (WT) and IL-6−/− mice. An asterisk represents a statistically significant difference between the WT control group so marked and WT mice that received the combination model. A dagger (†) represents a significant difference between the IL-6−/− mice and WT mice exposed to the same condition. Brochoalveolar lavage fluid (BAL) from WT mice exposed to the combination model of lung injury contained significantly higher total cell counts (A), total neutrophils (B), and total protein (D) than all of the WT control groups (P < 0.05 for the combination model vs. control groups). There was no statistical difference between the myeloperoxidase (MPO) levels in WT mice that received the combination model and those in mice that received lipopolysaccharide (LPS) alone, but levels in the combination model were significantly higher than those in mice that received either phosphate-buffered saline (PBS) or ventilator-induced lung injury (VILI) alone (D). Also, IL-6 levels in the BAL of mice that received LPS alone and the combination model were not significantly different, but levels in the combination model were significantly higher than those for either PBS or VILI alone (E). The IL-6−/− mice that received the combination model show significantly lower total cell counts (A) and total neutrophils (B) in the BAL than the WT mice (P < 0.01). IL-6−/− mice that received the combination model did not show a significant difference in total MPO activity in lung tissue compared to the WT mice (C). IL-6−/− mice had higher total protein levels in the BAL than WT mice after receiving LPS alone (D; P < 0.01).
Effect of IL-6 deletion on quantitative measures in a combination model of lung injury
The IL-6−/− animals subjected to the combination model of lung injury exhibited significantly less cellular inflammation than the WT animals with similar exposure (Fig. 1). Compared to WT mice, IL-6−/− mice demonstrated significant reductions in BAL total cell counts (P = 0.0068; Fig. 1A) and total BAL neutrophils (P = 0.0089; Fig. 1B). Lung MPO activity was lower in IL-6−/− mice subjected to the combination model than in WT mice, although the results were not significant (P = 0.042; significance requires P < 0.0125 with the Bonferroni correction; Fig. 1C). In contrast to the apparent protective effect of IL-6 deletion on cellular markers of inflammatory lung injury, when compared to similarly exposed WT animals IL-6−/− mice showed increased BAL total protein influx in response to either the combination model, LPS alone, or VILI alone (Fig. 1D). However, this increase in protein was significant only in the LPS-alone model (P = 0.00135), although the difference approached significance in the combination model (P = 0.022; Bonferroni correction). The differences in total protein between IL-6−/− and WT animals in the PBS control and VILI-alone groups were not significant.
Histologic evaluation of the combination model of lung injury
Neutrophilic infiltration, interstitial edema, and alveolar wall damage were greatest in the histologic sections of lungs from WT mice that received the combination model, with decreasing amounts of inflammation, edema, and alveolar damage seen in lungs from mice exposed to LPS, VILI, or PBS alone (Fig. 2A). Significant differences were observed in total lung injury scores among the 4 groups in WT animals (P = 2.75 × 10−9; Fig. 2B). The total scores for the combination model in WT animals were significantly higher than those in the PBS-only (P = 1.3 × 10−10) and VILI-only (P = 5.1 × 10−6) groups. However, the total scores for the combination model in WT animals were not significantly higher than the scores for LPS alone, although these values trended higher, with a P value approaching significance (P = 0.016; Bonferroni correction). Neutrophilic infiltration, interstitial edema, and alveolar wall damage seen in the histologic sections of lungs from IL-6−/− animals that received the combination model were less than those seen in WT animals (Fig. 2A). Total lung injury scores from IL-6−/− mice were significantly lower than those from the WT mice only in the combination model (P = 2.6 × 10−7; Fig. 2B).
Figure 2.
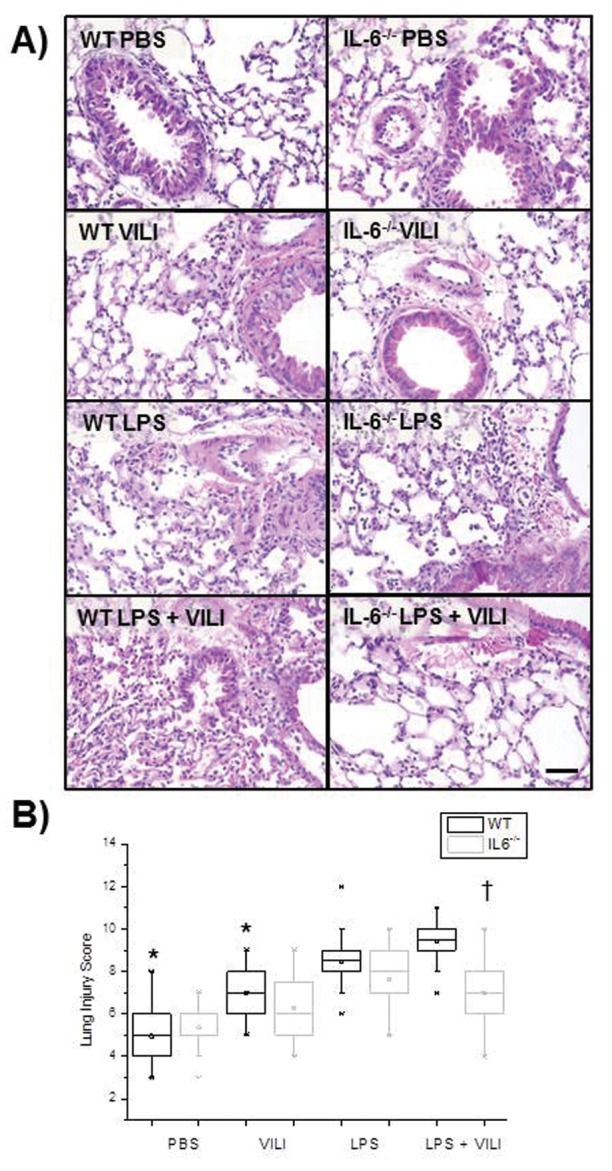
Histologic evidence of lung injury in wild-type (WT) and IL-6−/− mice. A, Representative hematoxylin-and-eosin stain slides at 40× magnification. In WT mice, the greatest lung injury is seen in the combination model, with decreasing injury observed in mice exposed to lipopolysaccharide (LPS), ventilator-induced lung injury (VILI), or phosphate-buffered saline (PBS) alone. Histology from the combination model–exposed IL-6−/− mice shows less inflammatory infiltration when compared to similar exposure in WT mice. Scale bar: 50 μm. B, Box-and-whisker plot showing the total lung injury scores from each experimental group. Asterisks represent statistically significant difference between the WT control group so marked and WT mice that received the combination model. The differences between the groups in WT animals were significant (P < 0.001), with the combination-injury model having significantly higher total lung injury scores than treatment with PBS or VILI alone (P < 0.001) but not significantly higher total scores than treatment with LPS alone. The difference between the scores of the IL-6−/− and WT mice was significant only in the LPS+VILI model (dagger [†]; P < 0.001).
TER of HLMVECs exposed to IL-6 and IL-6 Rα
After IL-6 and IL-6 Rα were added to HLMVECs, there was a significant decrease in TER compared to that in the control group (P < 0.05), starting at 1.2 hours and continuing until 9 hours (Fig. 3). HLMVECs exposed to both IL-6 and IL-6 Rα had a significantly lower TER than HLMVECs exposed to IL-6 only at the 1.2-, 1.95-, 2.2-, and 2.45-hour time points (P = 0.025, 0.027, 0.015, and 0.033, respectively). HLMVECs exposed to both IL-6 and IL-6 Rα had a significantly lower TER than HLMVECs exposed to IL-6 Rα only at all time points from 1.2 through 2.45 hours (P < 0.05). At 1.95 hours, the TER change from baseline with IL-6+IL6Rα was significantly greater than that with the vehicle (P = 7.58 × 10−4), IL-6 alone (P = 0.028), or IL-6 Rα alone (P = 0.023; Fig. 3B). HLMVECs exposed to either IL-6 alone or IL-6 Rα alone exhibited TER values significantly lower than control values for the entire time period.
Figure 3.

Transendothelial electrical resistance (TER) of human lung microvascular endothelial cells (HLMVECs) exposed to interleukin-6 (IL-6). A, IL6 combined with the IL-6 receptor (IL-6 Rα) has an additive effect on HLMVEC permeability, shown as a small but significant decrease in resistance compared to vehicle (control) or treatment with IL-6 or IL-6 Rα alone between 1.2 and 2.45 hours (P < 0.05). IL-6 combined with IL-6 Rα has significantly less resistance than the vehicle from 1.2 to 9 hours. B, At 1.95 hours, the percent change from baseline in the TER for IL-6+IL6Rα is significantly greater than that for the vehicle (P = 7.58 × 10−4), IL-6 alone (P = 0.028), or IL-6 Rα alone (P = 0.023).
Discussion
To our knowledge, this study is unique in evaluating the role of IL-6 in preclinical ARDS by utilizing an LPS-and-VILI combination model. Because of the pleiotropic nature of this cytokine and the contradictory results produced from previous in vivo lung injury models, we chose a dual-injury model more closely mimicking the clinical scenario, to better determine the role of IL-6 in ARDS. Our results demonstrate that the combination of dual injury stimuli, LPS and VILI, provides a clinically relevant in vivo murine model of ARDS and produced results that were not observed in the single-injury controls. The clinical ARDS scenario often involves infection that is present before the onset of mechanical ventilation, with cytokine expression at 24 hours differing from that in the initial hours after exposure to infection,5,6,22,23 making this model, with a delay between LPS and VILI lung injury, a relevant design. The BAL characteristics from human patients with ARDS include an acute neutrophilic inflammatory response with proteinaceous exudate containing increased cytokine levels.1-3,24 The combination lung injury model in WT mice mimicked the human condition by producing multifactorial injury with neutrophilic inflammation and proteinaceous edema and evoked greater lung injury than exposure to either LPS or mechanical ventilation alone, reflected by higher levels of cellular inflammation and vascular leakage (Fig. 1). Tidal volumes commonly utilized for mechanical ventilation in normal lungs have been shown to damage the injured lung, especially in cases of ARDS.1 In this study, the tidal volume used for VILI was low and similar to what would be used for ventilation of a healthy lung. VILI alone predictably produced minimal increases in lung injury parameters, compared to PBS alone. However, the addition of VILI to the previously LPS-injured lung produced the greatest level of injury. The combination model did not exhibit significantly higher BAL IL-6 levels than LPS alone (Fig. 1E); however, levels in both LPS and VILI groups demonstrated extremely elevated rates of IL-6 production.
Studies examining the role of IL-6 in mouse models of acute lung injury have yielded conflicting results. For instance, IL-6 was found to be proinflammatory in lipoteichoic acid–induced lung injury in mice while exhibiting an anti-inflammatory role in peptidoglycan-induced acute lung injury.25 IL-6 was proinflammatory in a hydrochloric acid and mechanical ventilation lung injury model10 and in a model of lung injury induced by acute kidney injury26 but was protective in a hyperoxia-induced lung injury model9 and two models of LPS-induced lung injury.27,28 Since IL-6 is a complex cytokine with both pro- and anti-inflammatory properties, it is likely that interactions of many variables in a complex disease such as ARDS will determine its ultimate effects. Thus, an animal model that mimics the human condition in ARDS in both etiology and major circumstances associated with treatment, such as mechanical ventilation, is of exceptional utility. Our data support the importance of the model by demonstrating that IL-6 had a significant effect on several lung injury parameters, including cellular inflammation (Fig. 1) and lung injury score (Fig. 2), only when the combination model was utilized. Other cytokines also exhibit pleiotropic effects,29 particularly those in the IL-6 family,30,31 and may also show unique effects when examined in a dual-injury animal model of lung injury.
Using the combination model, the results of this study show that mice that do not produce IL-6 exhibit a significant reduction in lung inflammatory indices, particularly neutrophilic inflammation in alveoli and airways (Fig. 1A, 1B), and a nonsignificant reduction in lung tissue, as shown by total MPO levels (Fig. 1C). These data suggest that IL-6 plays an important role in the recruitment of immune effector cells into the lung in the acute stages of inflammation. Given that many steps are involved in the recruitment of neutrophils to a site of inflammation and that many different chemoattractants recruit neutrophils to sites of inflammation, there may be unique temporal or spatial patterns of expression that make certain cytokines critical to neutrophil accumulation.23 The major steps in neutrophil recruitment include mobilization from the bone marrow, adhesion and migration through the endothelium, and moving to the site of infection once inside the tissue.23 Since our data indicate significantly lower BAL neutrophil levels but a nonsignificant change in total lung neutrophil levels in IL-6−/− mice compared to WT mice, IL-6 appears to be most important in the process of neutrophil migration into alveoli. IL-6 has been shown to increase migration of neutrophils to activated lung epithelial cells,32 and it has been shown to augment the expression of adhesion molecules, such as vascular cell adhesion molecule (VCAM-1) and intercellular adhesion molecule (ICAM-1), in inflamed areas and to stimulate production of chemokines.33 Alternatively, it is possible that similar numbers of neutrophils infiltrated inflamed IL-6−/− and WT alveoli but that neutrophil death was higher because of greater apoptosis in IL-6−/− mice, since IL-6 delays neutrophil apoptosis in vitro.34
In contrast to the cellular data, the total BAL protein levels in IL-6−/− mice were higher than those in WT mice in all lung injury groups (Fig. 1D). This change was significant only in the LPS-alone group, although it trended toward significance in the combination model. Protein leakage in the BAL is considered an indicator of vascular leakage in the pulmonary capillaries, and protein-rich alveolar edema is seen in human ARDS patients.2 Our results from the BAL protein suggest that IL-6 is protective against vascular injury and increased permeability. In an attempt to further characterize the role of IL-6 in endothelial permeability, the TER of HLMVECs exposed to IL-6 was measured (Fig. 3). However, these data showed that IL-6, particularly in association with its soluble receptor, increased endothelial permeability, a finding that appears to oppose the BAL protein data.
IL-6 produces pro- or anti-inflammatory effects, depending on the type of receptor that is activated.35,36 Only a few cell types (macrophages, neutrophils, T cells, and hepatocytes) normally express the IL-6 receptor (classic signaling), which is the method through which IL-6 produces anti-inflammatory effects. Signaling through the soluble IL-6 receptor is called trans-signaling and produces mostly proinflammatory effects. Since endothelial cells do not express IL-6 receptors and respond via trans-signaling, IL-6 would be expected to induce vascular leakage,36 which has been demonstrated in vitro in this study and others.37,38 However, IL-6 exhibits regenerative effects critical to the resolution of inflammatory processes,8 and a component of IL-6 anti-inflammatory activity is to signal mononuclear cells to resolve neutrophilic inflammation.36 The absence of this function in IL-6−/− mice may be related to decreased ability to resolve neutrophilic damage in the alveoli, which resulted in higher protein levels in the BAL of these mice, resulting in an apparent contradiction about the action of IL-6 on the pulmonary endothelium between the in vitro and in vivo data. It is also possible that the increase in BAL protein seen in IL-6−/− mice was not due to direct effects of IL-6 at all but rather to increases in other cytokines or inflammatory mediators that were the result of not having IL-6 present. Taken together, these contradictory data on the role of IL-6 in vascular leakage and the significant decrease in neutrophil migration in IL-6−/− mice suggest that the role of IL-6 in recruitment and resolution of inflammatory cells is more important to the overall level of injury seen in the lungs in this model of acute lung injury.
This combination ARDS model provides a valuable tool for future studies of the clinical relevance of IL-6, since it elucidates the role of IL-6 in cellular inflammation better than single-injury models. IL-6 single-nucleotide polymorphisms (SNPs) and genetic haplotypes are associated with increased risk of ARDS.39-41 Potentially, the combination model allows for the assessment of the clinical relevance of IL-6 SNPs to ARDS severity and could be utilized to evaluate IL-6-specific treatments, such as IL-6 antibodies or IL-6 receptor antibodies (e.g., tocilizumab), in acute lung injury.42 However, limitations with this model include that ARDS can be caused by etiologies other than infection, such as major trauma, and that it may have limited use for directly evaluating mechanisms of action of IL-6, since many factors influence IL-6 expression in this model and IL-6 may play different roles at different times over the 24-hour time period. Also, there are differences in the profiles of inflammatory mediators between mice and humans,43 and the dynamics between IL-6 and other cytokines may be important for lung responses.
In summary, a preclinical combination-injury mouse model of ARDS, likely to more accurately reflect human ARDS than single-stimulus preclinical models, was useful for the evaluation of the vexing effects of the cytokine IL-6 on cellular inflammation. Our study demonstrated an overall proinflammatory role of IL-6 in acute lung injury, with a paradoxical protective role against vascular leakage, and suggested that the influence of IL-6 on inflammatory cells may represent the primary mechanism by which IL-6 contributes to acute lung injury. Further research will have to be performed to elucidate the mechanisms of action of IL-6 in inflammation associated with ARDS as well as the role of IL-6 in other aspects of ARDS pathology, such as epithelial damage and damage to other organ systems.
Acknowledgment
This work was presented at the meeting of the Central Society for Clinical and Translational Research in Chicago, Illinois, April 25, 2013.
Source of Support: This work was supported by the National Institutes of Health/National Heart, Lung, and Blood Institute grants R01 HL058064 (to JGNG) and 1R25RR032021.
Conflict of Interest: None declared.
References
- 1.Dushianthan A, Grocott MP, Postle AD, Cusack R. Acute respiratory distress syndrome and acute lung injury. Postgrad Med J 2011;87(1031):612–622. [DOI] [PubMed]
- 2.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 2012;122(8):2731–2740. [DOI] [PMC free article] [PubMed]
- 3.Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol 2011;6:147–163. [DOI] [PMC free article] [PubMed]
- 4.Jawa RS, Anillo S, Huntoon K, Baumann H, Kulaylat M. Analytic review: interleukin-6 in surgery, trauma, and critical care: part I: basic science. J Intensive Care Med 2011;26(1):3–12. [DOI] [PMC free article] [PubMed]
- 5.Takala A, Jousela I, Takkunen O, Kautiainen H, Jansson S-E, Orpana A, Karonen SL, Repo H. A prospective study of inflammation markers in patients at risk of indirect acute lung injury. Shock 2002;17(4):252–257. [DOI] [PubMed]
- 6.Park WY, Goodman RB, Steinberg KP, Ruzinski JT, Radella F, Park DR, Pugin J, Skerrett SJ, Hudson LD, Martin TR. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;164(10):1896–1903. [DOI] [PubMed]
- 7.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, Leeper K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS: plasma IL-1β and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 1995;107(4):1062–1073. [DOI] [PubMed]
- 8.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 2011;1813(5):878–888. [DOI] [PubMed]
- 9.Ward NS, Waxman AB, Homer RJ, Mantell LL, Einarsson O, Du YF, Elias JA. Interleukin-6-induced protection in hyperoxic acute lung injury. Am J Respir Cell Mol Biol 2000;22(5):535–542. [DOI] [PubMed]
- 10.Gurkan OU, He C, Zielinski R, Rabb H, King LS, Dodd-o JM, D’Alessio FR, Aggarwal N, Pearse D, Becker PM. Interleukin-6 mediates pulmonary vascular permeability in a two-hit model of ventilator-associated lung injury. Exp Lung Res 2011;37(10):575–584. [DOI] [PMC free article] [PubMed]
- 11.Saito F, Tasaka S, Inoue K, Miyamoto K, Nakano Y, Ogawa Y, Yamada W, et al. Role of interleukin-6 in bleomycin-induced lung inflammatory changes in mice. Am J Respir Cell Mol Biol 2008;38(5):566–571. [DOI] [PubMed]
- 12.Wolters PJ, Wray C, Sutherland RE, Kim SS, Koff J, Mao Y, Frank JA. Neutrophil-derived IL-6 limits alveolar barrier disruption in experimental ventilator-induced lung injury. J Immunol 2009;182(12):8056–8062. [DOI] [PMC free article] [PubMed]
- 13.Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Köhler G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 1994;368(6469):339–342. [DOI] [PubMed]
- 14.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JGN. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med 2004;169(11):1245–1251. [DOI] [PubMed]
- 15.Jacobson JR, Barnard JW, Grigoryev DN, Ma SF, Tuder RM, Garcia JG. Simvastatin attenuates vascular leak and inflammation in murine inflammatory lung injury. Am J Physiol Lung Cell Mol Physiol 2005;288(6):L1026–L1032. [DOI] [PubMed]
- 16.Moitra J, Sammani S, Garcia JG. Re-evaluation of Evans Blue dye as a marker of albumin clearance in murine models of acute lung injury. Transl Res 2007;150(4):253–265. [DOI] [PubMed]
- 17.Nonas SA, Moreno-Vinasco L, Ma SF, Jacobson JR, Desai AA, Dudek SM, Flores C, et al. Use of consomic rats for genomic insights into ventilator-associated lung injury. Am J Physiol Lung Cell Mol Physiol 2007;293(2):L292–L302. [DOI] [PMC free article] [PubMed]
- 18.Moreno-Vinasco L, Jacobson J, Bonde P, Sammani S, Mirzapoiazova, Vigneswaran WT, Garcia JGN. Attenuation of rodent lung ischemia-reperfusion injury by sphingosine 1-phosphate. J Organ Dysfunct 2008;4(2):106–114.
- 19.Makena PS, Gorantla VK, Ghosh MC, Bezawada L, Balazs L, Luellen C, Parthasarathi K, Waters CM, Sinclair SE. Lung injury caused by high tidal volume mechanical ventilation and hyperoxia is dependent on oxidant-mediated c-Jun NH2-terminal kinase activation. J Appl Physiol 2011;111(5):1467–1476. [DOI] [PMC free article] [PubMed]
- 20.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 2001;108(5):689–701. [DOI] [PMC free article] [PubMed]
- 21.Schaphorst KL, Chiang E, Jacobs KN, Zaiman A, Natarajan V, Wigley F, Garcia JGN. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am J Physiol Lung Cell Mol Physiol 2003;285(1):L258–L267. [DOI] [PubMed]
- 22.Liberati TA, Trammell RA, Randle M, Barrett S, Toth LA. Cytokine and chemokine responses of lung exposed to surrogate viral and bacterial infections. Comp Med 2013;63(2):114–126. [PMC free article] [PubMed]
- 23.Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol 2011;32(10):452–460. [DOI] [PMC free article] [PubMed]
- 24.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2008;295(3):L379–L399. [DOI] [PMC free article] [PubMed]
- 25.Leemans JC, Vervoordeldonk MJ, Florquin S, van Kessel KP, van der Poll T. Differential role of interleukin-6 in lung inflammation induced by lipoteichoic acid and peptidoglycan from Staphylococcus aureus. Am J Respir Crit Care Med 2002;165(10):1445–1450. [DOI] [PubMed]
- 26.Klein CL, Hoke TS, Fang WF, Altmann CJ, Douglas IS, Faubel S. Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int 2008;74(7):901–909. [DOI] [PubMed]
- 27.Bhargave R, Janssen W, Altmann C, Andrés-Hernando A, Okamura K, Vandivier RW, Ahuja N, Faubel S. Intratracheal IL-6 protects against lung inflammation in direct, but not indirect, causes of acute lung injury in mice. PLoS ONE 2013;8:e61405. [DOI] [PMC free article] [PubMed]
- 28.Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, Achong MK. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest 1998;101(2):311–320. [DOI] [PMC free article] [PubMed]
- 29.Ozaki K, Leonard WJ. Cytokine and cytokine receptor pleiotropy and redundancy. J Biol Chem 2002;277(33):29355–29358. [DOI] [PubMed]
- 30.Brasier A. The nuclear factor-κB–interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res 2010;86(2):211–218. [DOI] [PMC free article] [PubMed]
- 31.Garbers C, Hermanns HM, Schaper F, Müller-Newen G, Grötzinger J, Rose-John S, Scheller J. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev 2012;23(3):85–97. [DOI] [PubMed]
- 32.Mul FP, Zuurbier AE, Janssen H, Calafat J, van Wetering S, Hiemstra PS, Roos D, Hordijk PL. Sequential migration of neutrophils across monolayers of endothelial and epithelial cells. J Leukoc Biol 2000;68(4):529–537. [PubMed]
- 33.Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond) 2012;122(4):143–159. [DOI] [PubMed]
- 34.Biffl W, Moore E, Moore F, Barnett C. Interleukin-6 delays neutrophil apoptosis via a mechanism involving platelet activating factor. J Trauma Inj Infect Crit Care 1996;40(4):575–579. [DOI] [PubMed]
- 35.Eulenfeld R, Dittrich A, Khouri C, Müller PJ, Mütze B, Wolf A, Schaper F. Interleukin-6 signalling: more than Jaks and STATs. Eur J Cell Biol 2012;91(6–7):486–495. [DOI] [PubMed]
- 36.Kruttgen A, Rose-John S. Interleukin-6 in sepsis and capillary leakage syndrome. J Interferon Cytokine Res 2012;32(2):60–65. [DOI] [PubMed]
- 37.Maruo N, Morita I, Shirao M, Murota S. IL-6 increases endothelial permeability in vitro. Endocrinology 1992;131(2):710–714. [DOI] [PubMed]
- 38.Desai TR, Leeper NJ, Hynes KL, Gewertz BL. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J Surg Res 2002;104(2):118–123. [DOI] [PubMed]
- 39.Flores C, Ma SF, Maresso K, Wade MS, Villar J, Garcia JG. IL6 gene-wide haplotype is associated with susceptibility to acute lung injury. Transl Res 2008;152(1):11–17. [DOI] [PubMed]
- 40.Meyer NJ, Daye ZJ, Rushefski M, Aplenc R, Lanken PN, Shashaty MGS, Christie JD, Feng R. SNP-set analysis replicates acute lung injury genetic risk factors. BMC Med Genet 2012;13:52. [DOI] [PMC free article] [PubMed]
- 41.Meyer NJ, Garcia JG. Wading into the genomic pool to unravel acute lung injury genetics. Proc Am Thorac Soc 2007;4(1):69–76. [DOI] [PubMed]
- 42.Yanagawa Y, Hirano Y, Kato H, Iba T. The absence of typical pneumonia symptoms in a patient with rheumatoid arthritis during tocilizumab and steroid treatment. BMJ Case Rep 2012(23 May). [DOI] [PMC free article] [PubMed]
- 43.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol 2004;172(5):2731–2738. [DOI] [PubMed]