

Abstract Abstract
Circulating levels of asymmetric dimethylarginine (ADMA), a nitric oxide synthase inhibitor, are increased in patients with idiopathic pulmonary hypertension (IPAH). We hypothesized that ADMA abrogates gap junctional communication, required for the coordinated regulation of endothelial barrier function and angiogenesis, and so contributes to pulmonary endothelial dysfunction. The effects of ADMA on expression and function of gap junctional proteins were studied in human pulmonary artery endothelial cells; pulmonary endothelial microvascular cells from mice deficient in an enzyme metabolizing ADMA, dimethylarginine dimethylaminohydrolase I (DDAHI); and blood-derived endothelial-like cells from patients with IPAH. Exogenous and endogenous ADMA inhibited protein expression and membrane localization of connexin 43 (Cx43) in a nitric oxide/soluble guanosine monophosphate/c-jun-dependent manner in pulmonary endothelial cells, resulting in the inhibition of gap junctional communication, increased permeability, and decreased angiogenesis. The effects of ADMA were prevented by overexpression of DDAHI or Cx43 and by treatment with rotigaptide. Blood-derived endothelial-like cells from IPAH patients displayed a distinct disease-related phenotype compared to cells from healthy controls, characterized by reduced DDAHI expression, increased ADMA production, and abnormal angiogenesis. In summary, we show that ADMA induces pulmonary endothelial dysfunction via changes in expression and activity of Cx43. Cells from IPAH patients exhibit abnormal DDAHI/Cx43 signaling as well as differences in gap junctional communication, barrier function, and angiogenesis. Strategies that promote DDAHI/Cx43 signaling may have an endothelium-protective effect and be beneficial in pulmonary vascular disease.
Keywords: nitric oxide, pulmonary hypertension, endothelial cells, angiogenesis
Introduction
Pulmonary hypertension (PH) is characterized by endothelial dysfunction, as shown by imbalance in the levels of vasoconstrictors and vasorelaxants, decreased barrier function, and disordered angiogenesis in plexiform lesions.1 The mechanisms are not fully understood, but reduced nitric oxide signaling is well described,1 as are increased plasma and tissue levels of the naturally occurring nitric oxide synthase (NOS) inhibitor asymmetric dimethylarginine (ADMA).2
Dimethylarginine dimethylaminohydrolase I and II (DDAHI and DDAHII) are responsible for ADMA metabolism.3 DDAHI heterozygous knockout mice have high plasma ADMA levels and show remodeling of small pulmonary arteries, increased pulmonary vascular permeability, and right ventricular pressure.3,4 Increased ADMA levels have also been associated with reduced DDAH expression and activity in lung tissues and pulmonary endothelium of patients with idiopathic pulmonary arterial hypertension (IPAH) and animal models of PH.2,5
ADMA has been shown to induce pulmonary endothelial leakage and inhibit vascular endothelial growth factor (VEGF)–induced angiogenesis in vitro, with these effects being prevented by overexpression of DDAHI.4,6 Exogenous or lysophosphatidylcholine-induced ADMA was shown to decrease connexin (Cx) 43 expression and gap junctional communication7 in human umbilical vein endothelial cells, suggesting a link between elevated ADMA level and progression of atherosclerosis. However, the role of other vascular Cxs or functional implications of ADMA-induced dysregulation of gap junctional communication has not been elucidated.
Gap junctions connect the cytoplasm of adjacent cells and allow exchange of small signaling molecules such as cyclic nucleotides, Ca2+, adenosine triphosphate, and inositol 1,4,5-trisphosphate between cells. They are formed by 2 hemichannels, each composed of 6 Cx subunits. In the vascular system, the core proteins are Cx37, Cx40, and Cx43, and deletion of these genes causes severe cardiovascular abnormalities.8,9 We hypothesized that the effects of ADMA on pulmonary vascular endothelium are propagated by changes in the expression, localization, and function of Cx proteins. We also hypothesized that an accessible endothelial-like cell population (also known as late-outgrowth endothelial progenitor cells or blood-outgrowth endothelial cells) from IPAH patients may exhibit phenotypic changes associated with impaired ADMA metabolism.
We show for the first time that, of the 3 core proteins studied, Cx43 mediated the effects of ADMA/DDAHI activity on pulmonary endothelial gap junctional communication, permeability, and angiogenesis. Overexpression of Cx43 or enhancement of Cx43 function by an antiarrhythmic drug rotigaptide also had an endothelium-protective effect. Furthermore, we present evidence for the dysregulation of ADMA metabolism and Cx43 expression in endothelial-like cells derived from the peripheral blood of patients with IPAH.
Methods
Cell culture
Human pulmonary artery endothelial cells (HPAECs) were cultured in endothelial growth medium-2 (Promocell). Endothelial-like cells were derived from peripheral blood as previously described.10 Venous blood samples were obtained with local ethics committee approval and informed written consent from 6 patients with IPAH and 6 age- and sex-matched healthy volunteers (Table 1). The investigation conformed with the principles outlined in the Declaration of Helsinki and the local research ethics committee (reference no. 09/HO711/4). Mouse pulmonary microvascular cells (PMVECs) were isolated from peripheral parts of the lung of LoxP-DDAHI mice, purified, and cultured as previously described.11 DDAHI gene knockout was induced in cells by adenoviral overexpression of Cre recombinase.
Table 1.
Patient and healthy volunteer data
| Control (n = 6) | IPAH (n = 6) | |
|---|---|---|
| Males/females | 1/5. | 0/6 |
| Age, years | 27.5 (24.0–33.0) | 43.0 (27.0–67.0) |
| Time from diagnosis, months | … | 17.0 (0.1–60.0) |
| mPAP, mmHg | … | 67.0 (50.0–71.0) |
| 6MWD, m | … | 382.0 (0–438.0) |
| WHO class | ||
| 1 | … | 0 |
| 2 | … | 1 |
| 3 | … | 4 |
| 4 | … | 1 |
| Warfarin | … | 4 |
| Treatment naive | 1 | |
| Calcium channel blocker | 1 | |
| Endothelin receptor antagonist | … | 3 |
| PDE5 inhibitor | … | 4 |
| Prostanoid | … | 2 |
| Statin | … | 0 |
Data are presented as median (range). IPAH: idiopathic pulmonary hypertension; mPAP: mean pulmonary artery pressure; 6MWD: 6-minute walk distance; WHO class: World Health Organization functional class; PDE5: phosphodiesterase type 5.
Blood-derived endothelial cells, also known as endothelial colony-forming cells or late outgrowth endothelial progenitor cells, were cultured from peripheral blood. Venous blood samples (∼50 mL) were collected in ethylenediaminetetraacetic acid (EDTA) vacutainers, diluted 1∶1 with phosphate-buffered saline (PBS) containing 0.2% EDTA and 2% fetal bovine serum (FBS) and layered onto Ficoll Plaque PLUS (GE Healthcare, Amersham, UK) for density gradient centrifugation. Peripheral blood mononuclear cells (MNCs) were aspirated, washed in PBS, and resuspended in endothelial growth medium 2 (EGM-2) medium (CC-3156, Lonza Biologics, Slough, UK), supplemented with growth factors (CC-4176, EGM-2 bullet kit, Lonza), 20% FBS (HyClone, Thermo Scientific, South Logan, UT), and 1% antibiotic/antimycotic solution (Gibco, Invitrogen, Paisley, UK). Cells were seeded (3–5 × 107 MNCs in 4 mL of medium per well) in 6-well plates coated with type 1 rat tail collagen (BD Biosciences, Bedford, MA). The medium was replaced daily for the first week (removing nonadherent cells) and every 2 days thereafter. Individual colonies were harvested by attaching a cloning cylinder (8 or 10 mm in diameter; Millipore, Watford, UK) with vacuum grease, removing the cells with warm 0.05% Trypsin-EDTA solution (Invitrogen), resuspending in fresh EGM-2 medium, and seeding (3,000–5,000 cells/cm2) in 1% gelatin precoated culture vessels. The cells were propagated when 70%–80% confluent. Distinct colonies of cells, capable of serial propagation for at least 8 serial passages and displaying a stable population doubling time as well as endothelial phenotype, were used between passages 4 and 7.
The endothelial lineage was confirmed by flow cytometry and immunostaining. For immunostaining, cells were grown on sterile glass coverslips (VWR International, Leicestershire, UK), prewashed with hydrochloric acid, and coated with 1% gelatin. Prior to immunostaining, cells were fixed in 4% paraformaldehyde in PBS (w/v) for 20 minutes at 4°C. After washing twice in PBS, coverslips were immersed in 3% bovine serum albumin in Tris-buffered saline for 30 minutes at room temperature. Cells were then incubated overnight at 4°C with mouse monoclonal antihuman CD31 (JC/70A, Dako, Glostrup, Denmark), antihuman CD144 (sc-9989, Santa Cruz Biotechnology, Santa Cruz, CA), or rabbit polyclonal antihuman von Willebrand factor (A0082, Dako). After washing, the coverslips were incubated for 1 hour at room temperature with Alexa-488 green-conjugated secondary antibodies (Dako), mounted in 4′,6-diamidino-2-phenylindole-containing Vectashield (Vector Laboratories, Peterborough, UK) and observed using fluorescence microscopy.
A 3-color FACSCalibur flow cytometer and CellQuest Pro software (BD Biosciences, Oxford, UK) were used for flow cytometry. Cells were suspended in Fc receptor blocker (Miltenyl Biotec, Bergisch Gladbach, Germany) for 5 minutes before being resuspended for 30 minutes at 4°C with mouse IgG1 antihuman CD31 (555446, phycoerythrin [PE]-labeled, BD Pharmingen, Cowley, UK), VEGFR2/KDR (FAB357A, allophycocyanin [APC]-labeled, R&D Systems, Minneapolis, MN), CD45 (345785, PE-labeled, BD Pharmingen), or CD133 (130-080-801, PE-labeled, Miltenyi Biotec). Fluorochrome matched isotype controls were used at the same dilution factor, as per the manufacturer’s recommendation (BD Pharmingen). The data were analyzed using FCS Express software (De Novo Software, Los Angeles, CA) and presented as the number of antigen-expressing (positive) cells expressed as a proportion of the gated blood-derived endothelial cell population.
Animals
The investigation conformed to directive 2010/63/EU of the European Parliament, and all the experiments involving animals were carried out under a home office licence and conducted according to the Animal Scientific Procedures Act 1986. DDAH−/− mice were generated by the introduction of LoxP sites flanking exon 1 of the mouse DDAHI gene by homologous recombination.12 DDAHIflox/flox mice were crossed with mice expressing Cre recombinase to generate DDAHI +/− mice. Heterozygotes were intercrossed to produce DDAHI−/− mice and wild-type littermates. Following schedule 1 killing using pentobarbital/euthatal (100 mg/animal), mouse lungs were used for cell culture or protein expression studies. Alternatively, the deep-anesthetized mice were used for the lung permeability in vivo studies.
Use of drugs
ADMA (100 μmol/L; Sigma), symmetric methylarginine (SDMA; 100 μmol/L, Sigma), SP600125 (20 μM, Sigma), N (G)-nitro-L-arginine methyl ester (L-NAME; 1 mmol/L, Calbiochem), L-257 (1 μmol/L), or S-nitroso-N-acetyl-d, l-penicillamine (SNAP; 10 μmol/L, Calbiochem) was added to cell cultures for 24 hours. Guanosine 3′,5′-cyclic monophosphate, 8-bromo-, sodium salt (8Br-cGMP, Na; 500 μmol/L; Calbiochem) was added to untreated cells or ADMA-treated cells 1.5 hours before the end of the experiment. Nonspecific gap junction inhibitor 18β-glycyrrhetinic acid (18β-GA, 10 μmol/L, Sigma) was added to cells 1 hour before fluorescence recovery after photobleaching (FRAP) measurement. The Cx43 gap junction blocker Gap26 (VCYDKSFPISHVR, 200 μmol/L, Peptide Protein Research) and the Cx43 gap junction activator rotigaptide (100 nmol/L) were added to cells for 24 hours in cell metabolic activity assay and Matrigel assay and 5 hours before the end of experiment in endothelial permeability and FRAP experiments.
Construction of recombinant adenoviruses
Adenoviruses for overexpression of green fluorescent protein (GFP), DDAHI,4,6 Cx43-GFP (AdGFP, AdDDAHI, AdCx43-GFP, respectively; Welgen, Worcester, MA), and Cre (AdCre; ViraQuest, North Liberty, IA) were generated using Ad5 ΔE1ΔE3 backbone vector. The cDNA of wild-type Cx43 (kind gift of D. Becker, University College London) was cloned into KpnI and XbaI restriction sites of pAdTrackCMV vector. The recombinant adenoviruses were constructed using the Ad-Easy-1 system, where the adenoviral construct is generated in bacteria BJ-5183 cells. The recombinant adenoviral vectors were linearized with PacI and used to infect 911 cells. All adenoviruses were amplified in HEK293 cells and subsequently purified on 2 sequential cesium chloride gradients and then passed through PD10 columns (GE Healthcare) to reduce the salt concentration. To establish numbers of active viral particles, adenoviral titer was measured in plaque assay. The titer of AdGFP was 1 × 1011 plaque-forming units/mL and of AdCx43-GFP was 8.5 × 1010. Adenoviral transient infections of cells using multiplicity of infection 1∶100 lead to a maximal rate of infection efficiency (∼90%–95%) without obvious cytopathic effects. The cells were used for experiments 24 hours postinfection. Protein expression was confirmed by immunofluorescence and Western blotting.
Detergent fractionation
Detergent-soluble and detergent-insoluble cell fractions were obtained as in Tilghman et al.13 Briefly, confluent HPAECs grown on 3-cm dishes were washed once with PBS and incubated on ice with 300 μL of lysis buffer containing 50 mmol/L HEPES, pH 7.4, 150 mmol/L NaCl, 1 mmol/L CaCl2, 1 mmol/L MgCl2, 100 μM phenylmethylsulfonyl fluoride, 10 mg/L leupeptin, 10 mg/L aprotinin, 2 mmol/L Na3VO4, 10 mmol/L NaPO4, 10 mmol/L NaF, and 0.1% Triton-X for 2 minutes. The buffer, containing detergent-soluble components of the cells, was collected and reserved for further analysis. The culture dish with detergent-insoluble (cytoskeletal) components of the cells was washed once with ice-cold PBS, and the contents were scraped and resuspended in 300 μL of detergent-free buffer. Both the detergent-soluble and detergent-insoluble samples were diluted 1∶1 in sodium dodecyl sulfate polyacrylamide gel electrophoresis sample buffer.
C-jun gene knockdown
C-jun protein was depleted using SignalSilence c-jun small interfering RNAs (siRNAs) I (Cell Signaling) at a final concentration of 100 nmol/L. An equal concentration of siGENOME nontargeting siRNA 2 (Thermo Scientific) served as a control. Transfection was carried out using OligofectAMINE (Invitrogen). Cells were harvested 48 hours after transfection.
Measurement of intercellular gap junctional communication
Kinetics of gap junctional communication was analyzed by FRAP. Confluent HPAECs grown on fibronectin-coated 96-well glass-bottom plates (Thermo Scientific) were incubated with acetomethoxy calcein (calcein AM) or its red-orange variant (Molecular Probes) and washed 3 times in PBS. After the last wash, PBS was replaced with fresh culture medium and plates were placed on a heated stage of a confocal laser scanning microscope (Leica TCS SP5, Leica Biosystems, Peterborough, UK) in an atmosphere containing 5% CO2. A 200-μm square of calcein-labeled cell monolayer was bleached at maximum laser power for 150 seconds. Recovery of fluorescence in the bleached area was monitored for 5 minutes at intervals of 10 seconds. An area of 388 μm2 was recorded for prebleach and postbleach scans at low laser power. Analysis was carried out using the Leica LAS-AF Lite 2.0. Fluorescence intensity in selected regions of interest (ROIs) was expressed as a function of time. A 100-μm2 ROI was selected within the uniformly bleached region, covering 4–6 cells. Another ROI was selected in the unbleached region, which served as control for an overall decrease in fluorescence attributable to successive scanning. The relative fluorescence recovery at time t was displayed as the percentage of prebleach levels:
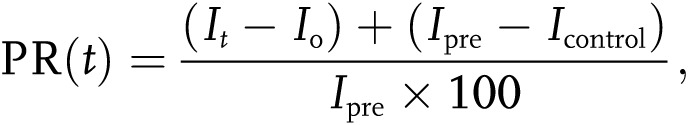 |
where Ipre, Io, and It were the measured fluorescence intensities of the ROI before (prebleach), at the first postbleach scan, and at the time t after the first scan, respectively; Icontrol was measured in unbleached cells, and the fluorescence recovery was corrected for the overall bleaching (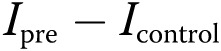 ) attributable to successive scanning.
) attributable to successive scanning.
Western blotting
Following protein electrophoresis and protein transfer, the membranes were probed with primary antibodies rabbit polyclonal anti-Cx40 (AV36635, Sigma), mouse monoclonal anti-Cx43 (C8093, Sigma), mouse monoclonal anti-β-actin (A2228, Sigma), rabbit polyclonal anti-Cx37 (Ab58918, AbCam, Cambridge, UK), rabbit monoclonal anti-c-Jun (60A8, Cell Signaling Technology), and rabbit monoclonal anti-phospho-c-Jun (Ser63; 54B3, Cell Signaling Technology). Goat polyclonal anti-DDAHI was prepared as described in Leiper et al.3 Secondary antibodies horseradish peroxidase (HRP)–labeled goat antirabbit (AG154) and rabbit antigoat (A5420) antibodies were from Sigma, and goat antimouse HRP-labeled antibody (2016–07) was from Dako. Primary antibodies were used at dilution 1∶1,000 and secondary antibodies at dilution 1∶5,000. The relative intensity of the immunoreactive bands was determined by the Quantity One 1-D analysis software, version 4.6.5 (Bio-Rad). The levels of phosphorylated Cx43 were estimated by measuring optical density of the upper band on Western blots. The results were normalized to β-actin levels and expressed as percent of untreated controls. Untreated control samples were included in every experiment, and all experiments were repeated at least 3 times.
Endothelial permeability in vitro and cell morphology
Cells were plated in Transwell-Clear chambers (1-μm pore size, 12-mm diameter; Costar Corning, Costar, High Wycombe, UK) at cell density of 1 × 104 cells/well and grown until confluence. The cells were left untreated or were infected with recombinant adenoviruses to induce overexpression of GFP, DDAHI, Cx43, or Cre. Two hours postinfection, adenovirus was removed and cells were incubated with fresh medium for a further 24 hours. Drugs were added to the upper and lower chambers of Transwell dishes at various time points (see “Use of drugs”). Fluorescein isothiocyanate (FITC)–dextran (MW 42,000, 1 g/L, Sigma) was added to the upper chamber of Transwell dishes. Following 1 hour incubation, the amount of FITC-dextran in the lower compartment of Transwell dishes was determined with the GloMax Multi+ plate reader (Promega, Southampton, UK), using an excitation wavelength of 490 nm and emission at 510–570 nm.4,6
Intracellular localization of vascular endothelial (VE)–cadherin and Cx43 in HPAECs grown on Thermanox coverslips was examined by immunofluorescence. Following fixation with 4% formaldehyde in PBS and permeabilization with 0.1% Triton X-100 in PBS, the cells were incubated with 2% bovine serum albumin in PBS for 1 hour. Next, the cells were incubated with rabbit polyclonal anti-Cx43 antibody (C6219, Sigma) or mouse monoclonal anti-VE-cadherin antibodies (sc-9989, Santa Cruz Biotechnology) at dilution 1∶200. Following the incubation with primary antibodies, the cells were washed 3 times in PBS and incubated with Cy5-conjugated goat antirabbit or tetramethyl rhodamine iso-thiocyanate (TRITC)–goat antimouse secondary antibodies (Jackson ImmunoResearch, West Grove, PA) at dilution 1∶200 for 1 hour. Alternatively, to stain the F-actin, cells were incubated with TRITC-phalloidin (1 μg/mL, Sigma). The cells were then washed 3 times in PBS and mounted in Vectashield (Vector Laboratories). Coverslips were examined under fluorescent confocal microscope (Leica, TCS SP5).
Lung permeability in vivo
All experiments were carried out under a home office license and conducted according to the Animal Scientific Procedures Act 1986. The lungs of 4 anesthetized (Hypnorm [fentanyl and fluanisone 0.25 mL/kg] and midazolam 25 mg/kg intraperitoneus) 10-week-old DDAHI knockout mice and 4 wild-type littermate controls were flushed via a catheter inserted through right ventricle in situ in the open chest with 5 mL PBS at a flow rate of 1.8 mL/minute with a nonpulsatile pump (Masterflex model 7518-00) until it was blood free. The lungs were then perfused with 10 mL of 0.16 g/L Evans Blue (Sigma) in PBS (with or without 100 nmol/L rotigaptide) and with 10 mL PBS afterward. The left lung lobes were dissected, weighed, homogenized, and suspended in 2.5 mL formamide for extraction overnight at room temperature. The homogenates were then centrifuged at 12,000 g, and the concentration of Evans blue in supernatants was quantified by spectrophotometric analysis at 620 nm. The Evans blue concentration was expressed in micrograms per gram wet weight of lungs.4
Study of angiogenic responses
Matrigel tube formation assay was performed to examine angiogenic response of HPAECs.6 HPAECs or blood-derived late outgrowth endothelial cells were left untreated or were transfected with GFP, DDAHI, or Cx43 by adenoviral gene transfer. Twenty-four hours posttransfection, the cells were trypsinized, resuspended in growth factor–deprived media containing 0.5% or 0.1% FBS, and seeded at a density of 7,000 cells/well in 96-well plates coated with 50 μL growth factor–reduced Matrigel (BD Bioscience 354230). Following cell plating, drugs were added to the wells as appropriate, and the cells were incubated for 18 hours prior to fixation in 4% formaldehyde in PBS and microscopic examination. Phase-contrast images were captured from each well, and total tube length was determined using Image J software.
Angiogenic response of pulmonary microvascular endothelial cells from the lungs of LoxP DDAHI mice was studied in 24-well plates, each well coated with 300 μL collagen I gel solution, made of 2.1 g/L PureCol collagen I from Advanced BioMatrix (San Diego, CA) in M199 medium containing 10 mmol/L NaOH. Cells were transfected with GFP, Cre, DDAHI, or Cx43 by adenoviral gene transfer 24 hours prior to the experiment. Cells were seeded on to collagen I gel and cultured in medium containing 0.5% FBS. Tubes were fixed in 4% formaldehyde after 18 hours of incubation. Two phase-contrast images were captured from each well, and total tube length and the number of blind tubes (not connected with the network at both ends) in each image were determined using Image J software.
Intracellular calcium levels
HPAECs grown on optical (glass-bottom) 96-well plates (Thermo Scientific) were incubated with L-NAME, ADMA, L-257, or Gap26 for 24 hours, while rotigaptide was added to the cells for 15 minutes, 1 hour, or 5 hours before the end of experiment. Ionomycin (0.5 μmol/L, 15-minute incubation) was used as a positive control. Fluorescent Ca2+ indicator Fluo-4 AM (10 μmol/L; Invitrogen) was incubated with the cells for 45 minutes, and fluorescence was measured at excitation/emission 490/510–570 nm with the GloMax Multi+ plate reader (Promega).
Apoptosis
HPAECs grown on optical (glass-bottom) 96-well plates (Thermo Scientific) were incubated with L-NAME, ADMA, L-257, Gap26, or rotigaptide for 24 hours. Menadione (20 μmol/L, 6-hour incubation) served as a positive control. Apoptosis was measured by 3,3-Dihexyloxacarbocyanine iodide (Sigma) staining.14 Fluorescence was measured at excitation/emission 490/510–570 nm with the GloMax Multi+ plate reader (Promega).
ADMA measurement
Levels of ADMA were quantified by HPLC in a conditioned medium as previously described.3
Statistical analysis
All the experiments were performed at least in triplicate (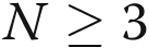 ). Data are presented as means ± standard error of the mean. Comparisons between 2 groups were carried out with 2-tailed Student’s t test, while comparisons between >2 groups were made with 1-way ANOVA followed by Newman-Keuls posttest using GraphPad Prism 4. Statistical significance was accepted for
). Data are presented as means ± standard error of the mean. Comparisons between 2 groups were carried out with 2-tailed Student’s t test, while comparisons between >2 groups were made with 1-way ANOVA followed by Newman-Keuls posttest using GraphPad Prism 4. Statistical significance was accepted for 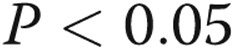 .
.
Results
ADMA/DDAHI regulates Cx43 expression and localization in pulmonary artery endothelial cells
ADMA reduced Cx43 protein expression and phosphorylation in cultured HPAECs (Fig. 1A–1C) and human pulmonary microvascular cells (HPMVCs; data not shown), while the protein levels of Cx37 and Cx40 remained unchanged. The effects of ADMA were concentration dependent, a maximal response being seen with 50–100 μmol/L ADMA (up to 2-fold; 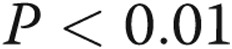 ), and less pronounced effects were observed at lower concentrations (Fig. 1D). ADMA reduced the levels of detergent-insoluble Cx43 associated with cytoskeletal fraction (Fig. 1E, 1F) and induced dispersion of Cx43 and the endothelial adherens junctions marker VE-cadherin from intercellular junctions (Fig. 1G). Overexpression of DDAHI increased Cx43 expression and restored junctional localization of Cx43 and its association with cytoskeletal fraction (Fig. 1A, 1B, 1E, 1G).
), and less pronounced effects were observed at lower concentrations (Fig. 1D). ADMA reduced the levels of detergent-insoluble Cx43 associated with cytoskeletal fraction (Fig. 1E, 1F) and induced dispersion of Cx43 and the endothelial adherens junctions marker VE-cadherin from intercellular junctions (Fig. 1G). Overexpression of DDAHI increased Cx43 expression and restored junctional localization of Cx43 and its association with cytoskeletal fraction (Fig. 1A, 1B, 1E, 1G).
Figure 1.

Asymmetric dimethylarginine (ADMA) and dimethylarginine dimethylaminohydrolase I (DDAHI) selectively regulate connexin (Cx) 43 expression. A, B, Effects of ADMA and DDAHI on Cx43 protein expression and phosphorylation in human pulmonary artery endothelial cells (HPAECs). C, Corresponding representative Western blots showing expression of Cx37, Cx40, and Cx43. D, Concentration-dependent reduction of Cx43 expression in HPAECs. E, F, ADMA reduces the levels of cytoskeleton-bound Cx43, and DDAHI prevents this effect. G, Intracellular distribution of Cx43 and vascular endothelial–cadherin in HPAECs treated as indicated. Confocal microscopy. Scale bar = 10 μm. H, Expression levels of DDAHI in cells 24 hours postinfection with adenoviruses for overexpression of DDAHI. One asterisk: 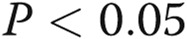 , comparison with controls; Delta:
, comparison with controls; Delta: 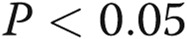 ; two Deltas:
; two Deltas: 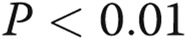 , comparison with ADMA.
, comparison with ADMA. 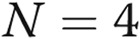 . VE: vascular endothelial; AdGFP: adenoviruses for overexpression of green fluorescent protein.
. VE: vascular endothelial; AdGFP: adenoviruses for overexpression of green fluorescent protein.
Consistent with our observations in cultured human cells, Cx43 protein levels were significantly reduced in the lungs of homozygous DDAHI−/− mice, while the levels of Cx37 and Cx40 remained unchanged (Fig. 2A, 2C). The DDAHI gene knockout in murine pulmonary microvascular endothelial cells (PMVECs) also resulted in a significant reduction in Cx43 expression with no effect on Cx37 or Cx40 protein levels (Fig. 2B, 2C).
Figure 2.
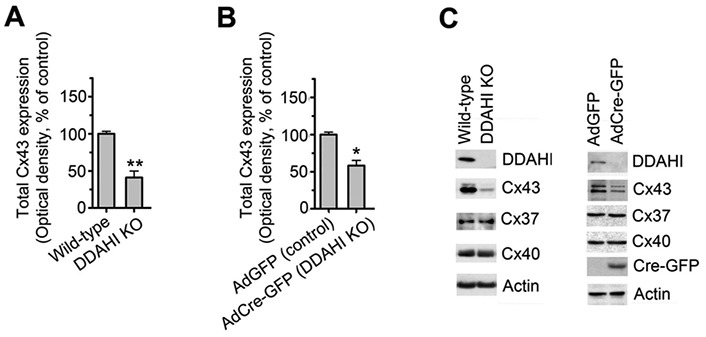
Connexin (Cx) 43 expression in lungs from dimethylarginine dimethylaminohydrolase I (DDAHI) knockout (KO) mice (A) and in DDAHI-deficient pulmonary microvascular cells (PMVECs; B). DDAHI gene KO was induced in PMVECs isolated from lungs of DDAHI-LoxP mice by adenoviral overexpression of Cre-GFP (AdCre-GFP). C, Representative Western blots showing expression of Cx43, Cx37, and Cx40, corresponding to A and B. One asterisk: 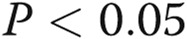 ; two asterisks:
; two asterisks: 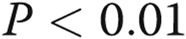 , comparisons with untreated controls.
, comparisons with untreated controls. 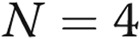 .
.
Effects of ADMA on Cx43 expression are mediated by NO–cyclic guanosine monophosphate (cGMP) pathway
ADMA is an endogenous NOS inhibitor, and therefore, we studied the role of NO and cGMP as potential mediators of ADMA/DDAH effects. In contrast to ADMA, SDMA, an inactive isomer that does not inhibit NOS or represent a substrate for DDAH,3 had no effect on either Cx43 expression or phosphorylation (Fig. 3). The NOS inhibitor L-NAME mimicked the effects of ADMA on Cx43 expression/phosphorylation, while an NO donor (SNAP) and stable cGMP analogue (8-Br-cGMP) prevented the effects of ADMA (Fig. 3). These data reinforce previous findings attributing ADMA-induced endothelial dysfunction to deficiency in NO signaling.4,6
Figure 3.
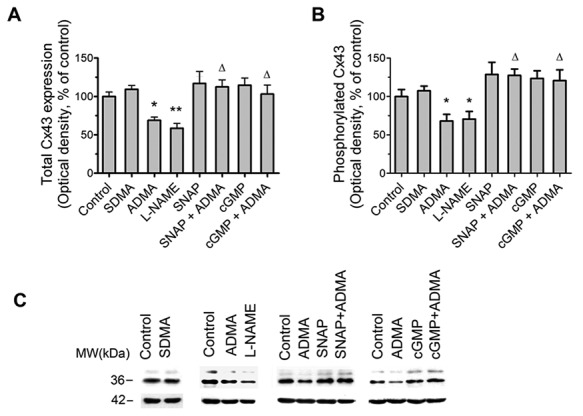
Effects of asymmetric dimethylarginine (ADMA) on connexin 43 (Cx43) expression are mediated by the nitric oxide (NO)–cyclic guanosine monophosphate (cGMP) pathway. A, Effects of symmetric methylarginine (SDMA) and NO pathway inhibitors ADMA and N (G)-nitro-L-arginine methyl ester (L-NAME) and NO pathway activators S-nitroso-N-acetyl-d, l-penicillamine (SNAP) and 8-Br-cGMP (cGMP) on Cx43 expression and Cx43 phosphorylation (B). C, Representative examples of Western blots corresponding to A and B. One asterisk: 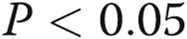 ; two asterisks:
; two asterisks: 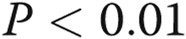 , compared to controls; Delta:
, compared to controls; Delta: 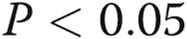 , comparisons with ADMA.
, comparisons with ADMA.  .
.
Transcription factors such as the activator protein 1 (AP-1; Jun/Fos) family are activated by NO and cGMP signaling.15 ADMA significantly inhibited c-jun protein expression and phosphorylation in HPAECs (Fig. 4) and siRNA-mediated knockdown of c-jun inhibited Cx43 expression and abolished the protective effect of DDAHI on Cx43 expression (Fig. 4C, 4D), whereas expression levels of Cx37 or Cx40 were unaffected. Inhibition of c-jun N-terminal kinase (JNK) with SP600125 resulted in a similar inhibition of Cx43 expression in HPAECs (Fig. 4C, 4D).
Figure 4.
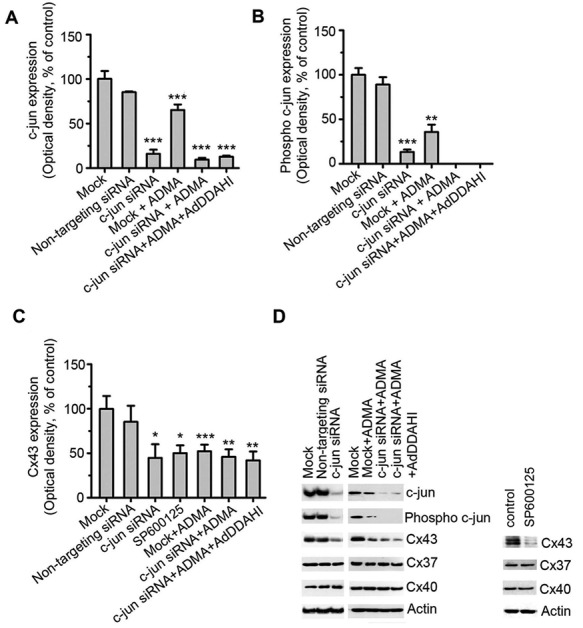
Asymmetric dimethylarginine (ADMA) regulates c-jun expression and phosphorylation. C-jun expression (A), c-jun phosphorylation (B), and connexin 43 (Cx43; C) expression in untreated human pulmonary artery endothelial cells (HPAECs) and HPAECs treated with ADMA, SP600125, mock transfected (OligofectAMINE only), transfected with nontargeting small interfering RNA (siRNA) or c-jun siRNA, or overexpressing DDAHI (AdDDAHI), as indicated. D, Representative Western blots corresponding to A–C. One asterisk: 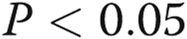 ; two asterisks:
; two asterisks: 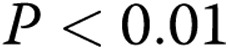 ; three asterisks:
; three asterisks: 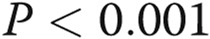 , comparisons with transfection controls.
, comparisons with transfection controls.
Cx43 mediates the effects of ADMA/DDAHI on gap junctional communication, barrier function, and angiogenesis
ADMA reduced the rate of gap junctional communication (40% decrease; 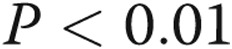 ), studied by FRAP7 (Fig. 5A, 5B). The effect was mimicked by the Cx43-blocking peptide, Gap26, and DDAHI-selective inhibitor L-257 and prevented by overexpression of DDAHI (Fig. 5A). The nonspecific gap junction blocking peptide 18β-GA served as a positive control. Interestingly, DDAHI overexpression also increased gap junctional communication in untreated cells (∼40% increase;
), studied by FRAP7 (Fig. 5A, 5B). The effect was mimicked by the Cx43-blocking peptide, Gap26, and DDAHI-selective inhibitor L-257 and prevented by overexpression of DDAHI (Fig. 5A). The nonspecific gap junction blocking peptide 18β-GA served as a positive control. Interestingly, DDAHI overexpression also increased gap junctional communication in untreated cells (∼40% increase; 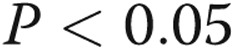 ) and reduced ADMA levels in HPAEC-conditioned medium from
) and reduced ADMA levels in HPAEC-conditioned medium from 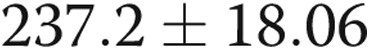 to
to 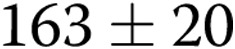 nmol/L (
nmol/L (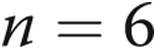 ,
, 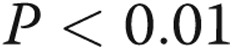 ), suggesting that endogenous ADMA regulates gap junctional communication under basal conditions. Overexpression of Cx43 or treatment with rotigaptide prevented the detrimental effects of ADMA (Fig. 5A). Prolonged (>5-hour) treatment of cells with rotigaptide or Gap26 did not affect Cx43 expression (data not shown).
), suggesting that endogenous ADMA regulates gap junctional communication under basal conditions. Overexpression of Cx43 or treatment with rotigaptide prevented the detrimental effects of ADMA (Fig. 5A). Prolonged (>5-hour) treatment of cells with rotigaptide or Gap26 did not affect Cx43 expression (data not shown).
Figure 5.
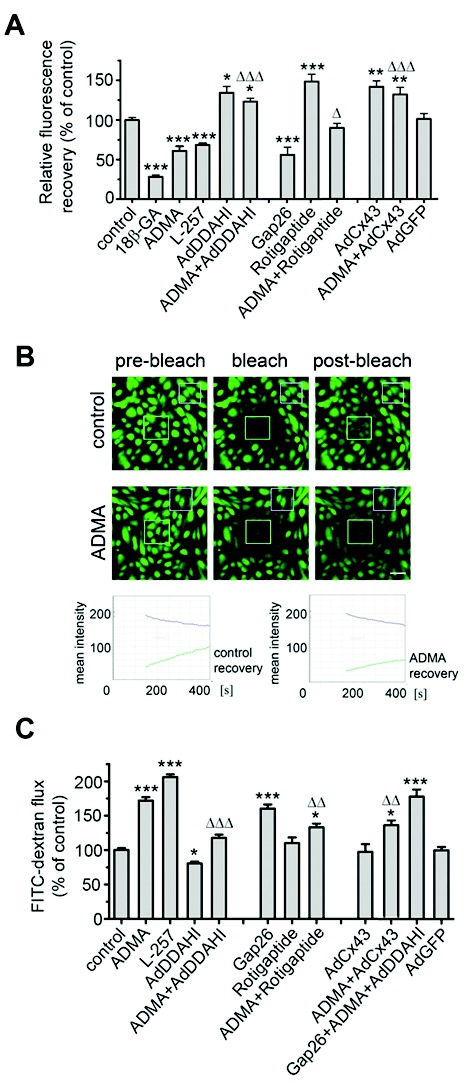
Effects of asymmetric dimethylarginine (ADMA) on gap junctional communication (A, B) and endothelial barrier function (C) in human pulmonary artery endothelial cells (HPAECs) treated, as indicated. B, Example of fluorescence recovery after photobleaching (FRAP) analysis in control and ADMA-treated cells. The lower trace in graphs shows fluorescence recovery in the bleached region marked with a square in the center, while the upper trace shows fluorescence in the reference region. Scale bar = 50 μm. One asterisk: 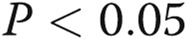 ; two asterisks:
; two asterisks: 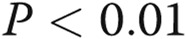 ; three asterisks:
; three asterisks: 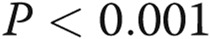 , comparisons with untreated controls; one Delta:
, comparisons with untreated controls; one Delta: 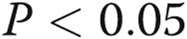 ; two Deltas:
; two Deltas: 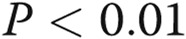 ; three Deltas:
; three Deltas: 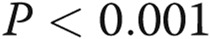 , comparisons between ADMA + treatment and ADMA groups.
, comparisons between ADMA + treatment and ADMA groups. 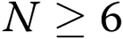 . AdDDAHI: adenoviruses for overexpression of dimethylarginine dimethylaminohydrolaseI; AdGFP: adenoviruses for overexpression of green fluorescent protein. A color version of this figure is available online.
. AdDDAHI: adenoviruses for overexpression of dimethylarginine dimethylaminohydrolaseI; AdGFP: adenoviruses for overexpression of green fluorescent protein. A color version of this figure is available online.
Exogenous ADMA and L-257 increased endothelial permeability (Fig. 5C) and inhibited tube formation in HPAECs (Fig. 6A, 6B), consistent with previously published data.4,6 Overexpression of Cx43 or treatment with rotigaptide prevented the effects of ADMA on HPAEC barrier function and angiogenesis (Figs. 5C, 6), suggesting that Cx43 is important in the maintenance of endothelial function in conditions where ADMA may limit NO supply. None of the treatments affected intracellular calcium levels or cell apoptosis (data not shown).
Figure 6.
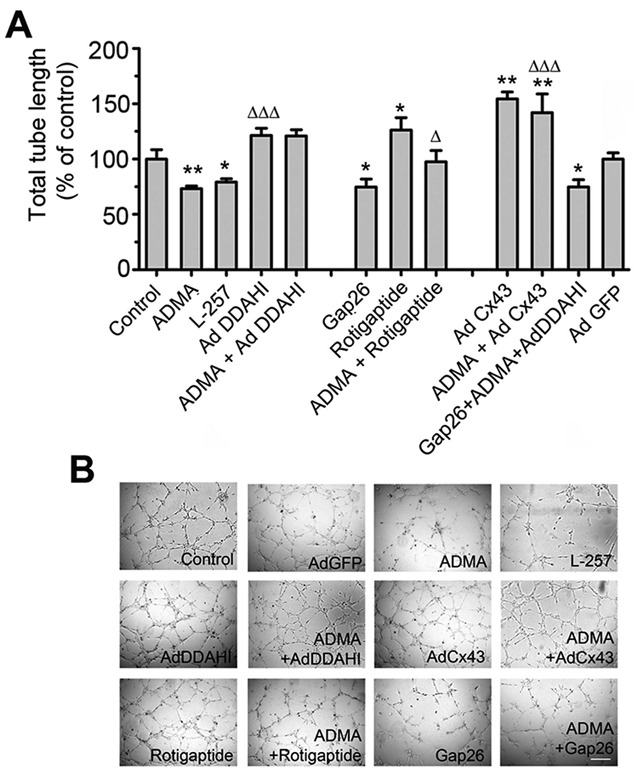
Asymmetric dimethylarginine (ADMA) decreases endothelial tube formation (A, B) in human pulmonary artery endothelial cells (HPAECs). HPAECs were treated with 18β-GA, ADMA, L-257, or rotigaptide or were overexpressing AdDDAHI, AdCx43, or AdGFP, as indicated. B shows representative images of tube formation in HPAECs treated, as indicated. Scale bar = 100 μm. One asterisk: 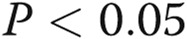 ; two asterisks:
; two asterisks: 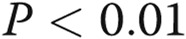 , comparison with control; one Delta:
, comparison with control; one Delta: 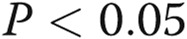 ; two Deltas:
; two Deltas: 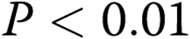 ; three Deltas:
; three Deltas: 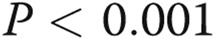 , comparisons between ADMA + treatment and ADMA groups.
, comparisons between ADMA + treatment and ADMA groups. 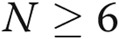 .
.
Consistent with the effects of exogenous ADMA in cultured HPAECs, DDAHI gene knockout inhibited gap junctional communication, increased permeability, and inhibited angiogenesis in murine pulmonary endothelial cells (Fig. 7A, 7C). DDAH−/− mice showed a decrease in pulmonary endothelial barrier function in vivo that was prevented by rotigaptide (Fig. 7D).
Figure 7.
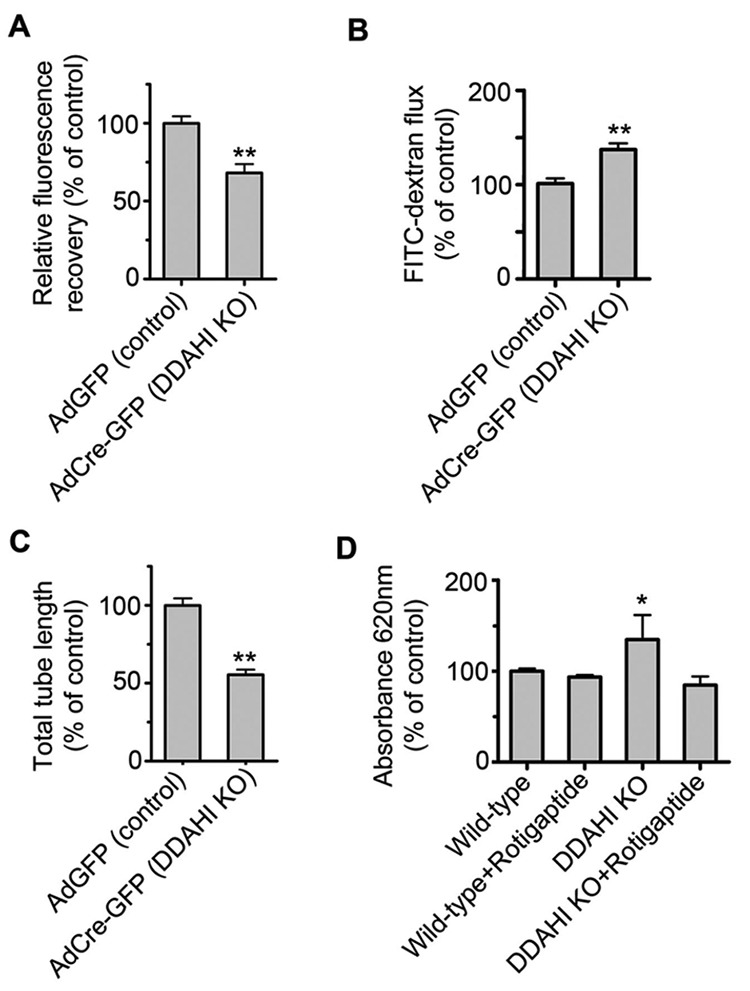
Dimethylarginine dimethylaminohydrolase I gene knockout (DDAHI KO) induces pulmonary endothelial dysfunction in vitro and in vivo. DDAHI KO in murine pulmonary microvascular cells (PMVECs) results in inhibition of gap junctional communication (A), increase in endothelial permeability (B), and inhibition of endothelial tube formation (C). D, Rotigaptide attenuates pulmonary endothelial permeability in lungs of DDAHI KO mice. One asterisk: 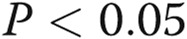 ; two asterisks:
; two asterisks: 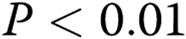 , comparisons with controls.
, comparisons with controls. 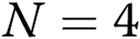 . AdGFP: adenoviruses for overexpression of green fluorescent protein.
. AdGFP: adenoviruses for overexpression of green fluorescent protein.
Blood-derived endothelial-like cells from IPAH patients show reduced expression of DDAHI and Cx43 and increased angiogenesis
The endothelial cell lineage of blood-derived cells was confirmed by immunostaining (data not shown) and flow cytometry, the cells being positive for the endothelial markers CD31 (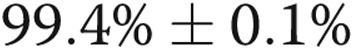 ) and VEGFR-2 (
) and VEGFR-2 (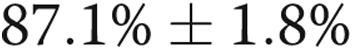 ) but negative (<0.1%) for the hematopoietic/monocytic markers CD45 and CD133. Most importantly, endothelial-like cells from IPAH patients showed reduced expression of DDAHI and significantly increased ADMA levels compared with controls (Fig. 8A, 8B, 8E). Cx43 protein expression was reduced in IPAH cells (∼2.3-fold reduction;
) but negative (<0.1%) for the hematopoietic/monocytic markers CD45 and CD133. Most importantly, endothelial-like cells from IPAH patients showed reduced expression of DDAHI and significantly increased ADMA levels compared with controls (Fig. 8A, 8B, 8E). Cx43 protein expression was reduced in IPAH cells (∼2.3-fold reduction; 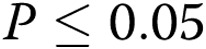 ), whereas Cx40 expression was similar to that in control cells (Fig. 8C, 8E). DDAHII and Cx37 expression were not detectable in either cell population. The reduction in Cx43 expression was accompanied by a significant decrease in gap junctional communication (Fig. 8D). IPAH cells formed leaky monolayers that exhibited 4-fold higher endothelial permeability compared with control cells (Fig. 8F). IPAH cells also showed abnormal angiogenesis in basal conditions in vitro manifested by the formation of numerous poorly developed tubes (not connected with the network at both ends) and cell aggregates (Fig. 8G–8I). IPAH cell phenotype was attenuated by overexpression of DDAHI or Cx43 or treatment with rotigaptide (Fig. 8F–8I). Conversely, inhibition of DDAHI with L-257 significantly increased endothelial permeability and abrogated network formation in control cells, resembling the pattern of responses seen in IPAH cells (Fig. 8G–8I).
), whereas Cx40 expression was similar to that in control cells (Fig. 8C, 8E). DDAHII and Cx37 expression were not detectable in either cell population. The reduction in Cx43 expression was accompanied by a significant decrease in gap junctional communication (Fig. 8D). IPAH cells formed leaky monolayers that exhibited 4-fold higher endothelial permeability compared with control cells (Fig. 8F). IPAH cells also showed abnormal angiogenesis in basal conditions in vitro manifested by the formation of numerous poorly developed tubes (not connected with the network at both ends) and cell aggregates (Fig. 8G–8I). IPAH cell phenotype was attenuated by overexpression of DDAHI or Cx43 or treatment with rotigaptide (Fig. 8F–8I). Conversely, inhibition of DDAHI with L-257 significantly increased endothelial permeability and abrogated network formation in control cells, resembling the pattern of responses seen in IPAH cells (Fig. 8G–8I).
Figure 8.
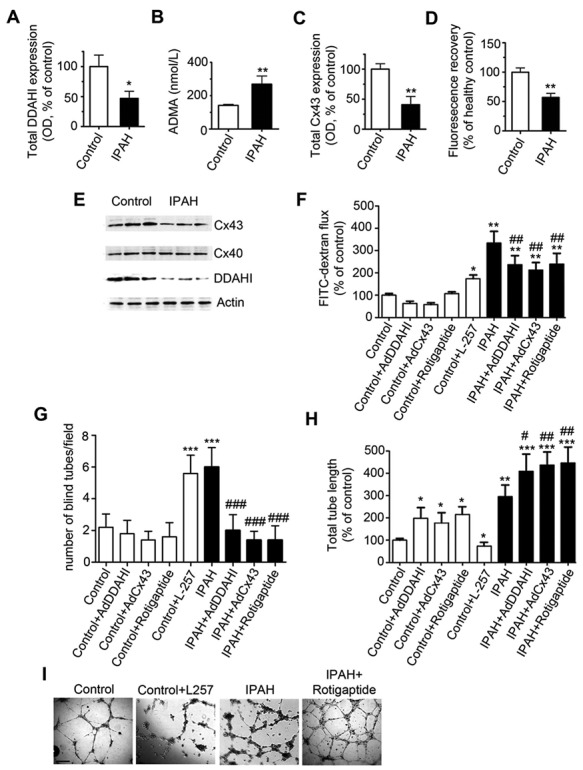
Dimethylarginine dimethylaminohydrolase I (DDAHI) and connexin 43 (Cx43) expression and signaling in cells from idiopathic pulmonary hypertension (IPAH) patients. DDAHI expression (A), asymmetric dimethylarginine (ADMA) production (B), Cx43 expression (C), and gap junctional communication (D) in IPAH cells and healthy controls. E, Representative blot showing expression of DDAHI and Cx43. F, Endothelial permeability. G, Number of blind tubes in IPAH cells and controls. H, Overall tube length in IPAH cells and controls. I, Selected examples of tube formation in cells treated, as indicated. Scale bar = 100 μm. One asterisk: 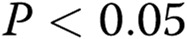 ; two asterisks:
; two asterisks: 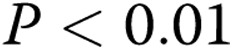 ; three asterisks:
; three asterisks: 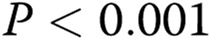 , compared with healthy controls; one hash sign:
, compared with healthy controls; one hash sign: 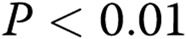 ; two hash signs:
; two hash signs: 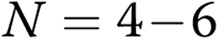 ; three hash signs:
; three hash signs: 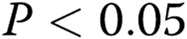 , compared with cells from IPAH patients.
, compared with cells from IPAH patients. 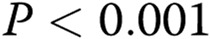 .
.
Figure 9.
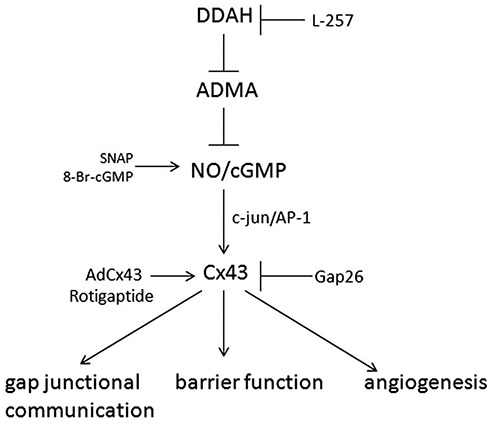
Proposed role of asymmetric dimethylarginine (ADMA)/dimethylarginine dimethylaminohydrolase I (DDAHI) in the regulation of pulmonary endothelial function. ADMA inhibits nitric oxide (NO)–cGMP signaling, leading to a decrease in c-jun expression and phosphorylation, connexin 43 (Cx43) expression, and gap junctional communication. Loss of Cx43 destabilizes intercellular junctions, contributing to the breakdown of endothelial barrier function and inhibition of angiogenesis. The DDAHI-selective inhibitor L-257 and Cx43 inhibitor Gap 26 mimic the effects of ADMA, while overexpression of DDAHI, Cx43, or treatment with rotigaptide has a protective effect. SNAP: S-nitroso-N-acetyl-d, l-penicillamine; NO: nitric oxide; cGMP: cyclic guanosine monophosphate; AP-1: activator protein 1.
Discussion
Using pharmacological and genetic tools, we show that DDAHI activity and ADMA metabolism influence pulmonary endothelial permeability and angiogenesis by regulating the expression, activation, and membrane localization of Cx43 (Fig. 9). Rotigaptide, a Cx43-activating drug, has protective effects on pulmonary endothelial barrier function in vitro and in vivo and in conditions of reduced NO production. We also provide evidence of reduced DDAHI and Cx43 expression/signaling, increased ADMA production, and resulting dysregulation of endothelial barrier function and angiogenic responses in endothelial-like cells derived from IPAH patients.
The ADMA-induced reduction of Cx43 expression in pulmonary endothelial cells resulted, at least in part, from decreased expression and phosphorylation of c-jun, a protein required for activation of the transcription factor AP-1.15 Inhibition of JNK, an upstream activator of c-jun, also decreased Cx43 expression in HPAECs, indicating that functional JNK/c-jun signaling is important in the maintenance of Cx43 expression in basal conditions. In keeping with our observations, c-jun/AP-1 has also been implicated in controlling Cx43 expression in myometrial cells.16 Interestingly, a downregulation of Cx43 activates human aortic endothelial cells to pathological status and increases the activities of JNK and c-jun, suggesting the existence of a regulatory feedback mechanism.17 Nonetheless, the precise regulatory mechanisms will need to be established as a variety of kinases and signaling pathways modulate Cx43 expression and phosphorylation.18
Cx43 augments several processes important in the maintenance of endothelial structure and function, such as cell-cell adhesion, cell migration, and expression of genes in VEGF- and TGF-β-signaling pathways.18,19 The interaction of Cx43 with submembrane actin cytoskeleton helps coordinate formation of protein scaffolds containing proteins such as ZO-1, cadherins, and integrins and is important for the formation and maintenance of intercellular contacts.18 Consistent with this, we observed that destabilization of endothelial junctions by ADMA was accompanied by a loss of cytoskeleton-bound Cx43. In addition to mediating endothelial intercellular communication, Cx43 is also considered important in the cross talk between endothelial and smooth muscle cells, including those in the pulmonary vasculature.20,21
We show for the first time that rotigaptide exerts beneficial effects on pulmonary endothelial function in vitro and in vivo, opposing the negative effects of ADMA. Although its efficacy as an antiarrhythmic peptide in humans is unclear,22 rotigaptide has been safely administered to healthy volunteers and used as a tool to specifically augment Cx43-mediated signaling.23,24 Clinical studies indicate that augmentation of Cx43 gap junction communication with rotigaptide does not affect basal forearm blood flow, agonist-induced vasodilatation, or release of the endogenous fibrinolytic factor tissue-plasminogen activator.23 However, preliminary data suggest that it prevents endothelial dysfunction caused by ischemia-reperfusion injury and reduces myocardial infarct size following coronary artery occlusion.25 These findings are in keeping with the identification of ischemia-regulated phosphorylation sites in Cx43, the dephosphorylation of which is suppressed by rotigaptide,26 and increasing evidence indicating that the phosphorylation state of Cx43 reflects changes in the function and resistance to injury in the endothelium as well as other cell types18,27
Therapeutic targeting of the DDAH/Cx43 pathway in pulmonary arterial hypertension will, however, require careful consideration. While activation of this pathway might reduce endothelial dysfunction, the augmentation of Cx43 expression could promote pulmonary arterial smooth muscle differentiation and hypertensive pulmonary vascular remodeling19,20 or enhance proinflammatory signaling in the pulmonary vasculature.28 The effective dose of ADMA used in our in vitro studies was relatively high (100 μmol/L), as compared to the plasma concentration of ADMA in mice with DDAHI gene deletion or patients with multiple cardiovascular risk factors (0.2–0.6 μmol/L).3 However, we observed that the intracellular ADMA levels in HPAECs following overnight treatment with 10 and 100 μmol/L were only 0.005 and 0.012 μmol/L, respectively. This suggests that the intracellular DDAH activity is a more important determinant of cell responses than the extracellular levels of ADMA.
In HPAECs and blood-derived endothelial-like cells, DDAHI was the predominant isoform of DDAH, and manipulation of DDAHI expression did not induce compensatory changes in DDAHII, consistent with observations in mice.12 Endothelial-like cells from patients with IPAH showed reduced expression of DDAHI and Cx43 proteins, increased ADMA production, and significantly increased endothelial permeability, as compared with cells from healthy volunteers. In addition, IPAH cells also showed abnormal capillary network formation in vitro. This abnormal phenotype was improved by overexpression of DDAHI, Cx43, or treatment with rotigaptide.
Endothelial-like cells from IPAH patients exhibited a distinct phenotype in culture that may reflect some of the pathological changes described in the diseased lung itself, including reduction in DDAH expression, increased ADMA levels,5 loss of endothelial barrier function, and abnormal angiogenesis.1,29 Interestingly, functional studies of late-outgrowth progenitor cells from patients with PAH with BMPRII mutations revealed a hyperproliferative phenotype with impaired ability to form vascular networks.29 Blood-derived endothelial-like cells are typically considered to be the progeny of rare circulating progenitor cells that also reside locally in blood vessels, including those in the lung.30 Blood-derived endothelial-like cells may represent an accessible surrogate cell type to study endothelial dysfunction in PAH.
In conclusion, our findings demonstrate the importance of DDAHI/NO/Cx43 pathway in the regulation of pulmonary endothelial function in vitro and provide evidence of the abnormal DDAHI/Cx43 signaling in endothelial-like cells from IPAH patients, improved by treatment with rotigaptide. Dysfunction of DDAH/Cx43 pathway may contribute to vascular remodeling associated with PAH.
Acknowledgments
We thank Prof. David Becker (University College London) for Cx43 expression plasmid, Dr. Lan Zhao (Imperial College London) for help with the lung perfusion study, and the staff of the National Institute for Health Research/Wellcome Trust Imperial Clinical Research Facility, Hammersmith Hospital.
Source of Support: This work was supported by the British Heart Foundation (grant PG/09/045/27570).
Conflict of Interest: None declared.
References
- 1.Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation 2004;109:159–165. [DOI] [PubMed]
- 2.Pullamsetti S, Kiss L, Ghofrani HA, et al. Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. FASEB J 2005;19:1175–1177. [DOI] [PubMed]
- 3.Leiper J, Nandi M, Torondel B, et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med 2007;13:198–203. [DOI] [PubMed]
- 4.Wojciak-Stothard B, Torondel B, Zhao L, Renne T, Leiper JM. Modulation of Rac1 activity by ADMA/DDAH regulates pulmonary endothelial barrier function. Mol Biol Cell 2009;20:33–42. [DOI] [PMC free article] [PubMed]
- 5.Millatt LJ, Whitley GS, Li D, et al. Evidence for dysregulation of dimethylarginine dimethylaminohydrolase I in chronic hypoxia-induced pulmonary hypertension. Circulation 2003;108:1493–1498. [DOI] [PubMed]
- 6.Fiedler LR, Bachetti T, Leiper J, et al. The ADMA/DDAH pathway regulates VEGF-mediated angiogenesis. Arterioscler Thromb Vasc Biol 2009;29:2117–2124. [DOI] [PubMed]
- 7.Jia SJ, Zhou Z, Zhang BK, Hu ZW, Deng HW, Li YJ. Asymmetric dimethylarginine damages connexin43-mediated endothelial gap junction intercellular communication. Biochem Cell Biol 2009;87:867–874. [DOI] [PubMed]
- 8.Reaume AG, de Sousa PA, Kulkarni S, et al. Cardiac malformation in neonatal mice lacking connexin43. Science 1995;267:1831–1834. [DOI] [PubMed]
- 9.Simon AM, McWhorter AR. Vascular abnormalities in mice lacking the endothelial gap junction proteins connexin37 and connexin40. Dev Biol 2002;251:206–220. [DOI] [PubMed]
- 10.Yoder MC, Mead LE, Prater D, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 2007;109:1801–1809. [DOI] [PMC free article] [PubMed]
- 11.Wojciak-Stothard B, Torondel B, Tsang LYF, et al. The ADMA/DDAH pathway is a critical regulator of endothelial cell motility. J Cell Sci 2007;120:929–942. [DOI] [PubMed]
- 12.Hu X, Atzler D, Xu X, et al. Dimethylarginine dimethylaminohydrolase-1 is the critical enzyme for degrading the cardiovascular risk factor asymmetrical dimethylarginine. Arterioscler Thromb Vasc Biol 2011;31:1540–1546. [DOI] [PMC free article] [PubMed]
- 13.Tilghman RW, Hoover RL. E-selectin and icam-1 are incorporated into detergent-insoluble membrane domains following clustering in endothelial cells. FEBS Lett 2002;525:83–87. [DOI] [PubMed]
- 14.Julià E, Edo MC, Horga A, Montalban X, Comabella M. Differential susceptibility to apoptosis of CD4+T cells expressing CCR5 and CXCR3 in patients with MS. Clin Immunol 2009;133:364–374. [DOI] [PubMed]
- 15.Pilz RB, Suhasini M, Idriss S, Meinkoth JL, Boss GR. Nitric oxide and cGMP analogs activate transcription from AP-1-responsive promoters in mammalian cells. FASEB J 1995;9:552–558. [DOI] [PubMed]
- 16.Mitchell JA, Lye SJ. Differential activation of the connexin 43 promoter by dimers of activator protein-1 transcription factors in myometrial cells. Endocrinology 2005;146:2048–2054. [DOI] [PubMed]
- 17.Wang HH, Kung CI, Tseng YY, et al. Activation of endothelial cells to pathological status by down-regulation of connexin43. Cardiovasc Res 2008;79:509–518. [DOI] [PubMed]
- 18.Giepmans BN. Gap junctions and connexin-interacting proteins. Cardiovasc Res 2004;62:233–245. [DOI] [PubMed]
- 19.Walker DL, Vacha SJ, Kirby ML, Lo CW. Connexin43 deficiency causes dysregulation of coronary vasculogenesis. Dev Biol 2005;284:479–498. [DOI] [PubMed]
- 20.Gairhe S, Bauer NN, Gebb SA, McMurtry IF. Myoendothelial gap junctional signaling induces differentiation of pulmonary arterial smooth muscle cells. Am J Physiol 2011;301:L527–L535. [DOI] [PubMed]
- 21.Gairhe S, Bauer NN, Gebb SA, McMurtry IF. Serotonin passes through myoendothelial gap junctions to promote pulmonary arterial smooth muscle cell differentiation. Am J Physiol 2012;303:L767–L777. [DOI] [PubMed]
- 22.O’Quinn MP, Palatinus JA, Harris BS, Hewett KW, Gourdie RG. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ Res 2011;108:704–715. [DOI] [PMC free article] [PubMed]
- 23.Lang NN, Myles RC, Burton FL, et al. The vascular effects of rotigaptide in vivo in man. Biochem Pharmacol 2008;76:1194–1200. [DOI] [PubMed]
- 24.Axelsen LN, Haugan K, Stahlhut M, et al. Increasing gap junctional coupling: a tool for dissecting the role of gap junctions. J Membr Biol 2007;216:23–35. [DOI] [PubMed]
- 25.Pedersen CM, Lang N, Venkatasubramanian S, et al. Augmentation of connexin-43 conductance reduces ischemia-reperfusion injury and myocardial infarct size. Circulation 2012;126:A11360.
- 26.Axelsen LN, Stahlhut M, Mohammed S, et al. Identification of ischemia-regulated phosphorylation sites in connexin43: a possible target for the antiarrhythmic peptide analogue rotigaptide (zp123). J Mol Cell Cardiol 2006;40:790–798. [DOI] [PubMed]
- 27.Jeyaraman MM, Srisakuldee W, Nickel BE, Kardami E. Connexin43 phosphorylation and cytoprotection in the heart. Biochim Biophys Acta 2012;1818:2009–2013. [DOI] [PubMed]
- 28.Parthasarathi K, Ichimura H, Monma E, Lindert J, Quadri S, Issekutz A, Bhattacharya J. Connexin 43 mediates spread of Ca2+-dependent proinflammatory responses in lung capillaries. J Clin Invest 2006;116:2193–2200. [DOI] [PMC free article] [PubMed]
- 29.Toshner M, Voswinckel R, Southwood M, et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med 2009;180:780–787. [DOI] [PMC free article] [PubMed]
- 30.Duong HT, Comhair SA, Aldred MA, et al. Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulm Circ 2011;1:475–486. [DOI] [PMC free article] [PubMed]