

Abstract Abstract
Pulmonary or systemic infections and hypoxemic respiratory failure are among the leading causes of admission to intensive care units, and these conditions frequently exist in sequence or in tandem. Inflammatory responses to infections are reproduced by lipopolysaccharide (LPS) engaging Toll-like receptor 4 (TLR4). Apoptosis is a hallmark of lung injury in sepsis. This study was conducted to determine whether preexposure to LPS or hypoxia modulated the survival of pulmonary artery endothelial cells (PAECs). We also investigated the role TLR4 receptor expression plays in apoptosis due to these conditions. Bovine PAECs were cultured in hypoxic or normoxic environments and treated with LPS. TLR4 antagonist TAK-242 was used to probe the role played by TLR4 receptors in cell survival. Cell apoptosis and survival were measured by caspase 3 activity and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) incorporation. TLR4 expression and tumor necrosis factor α (TNF-α) production were also determined. LPS increased caspase 3 activity in a TAK-242-sensitive manner and decreased MTT incorporation. Apoptosis was decreased in PAECs preconditioned with hypoxia prior to LPS exposure. LPS increased TNF-α production, and hypoxic preconditioning blunted it. Hypoxic preconditioning reduced LPS-induced TLR4 messenger RNA and TLR4 protein. TAK-242 decreased to baseline the LPS-stimulated expression of TLR4 messenger RNA regardless of environmental conditions. In contrast, LPS followed by hypoxia substantially increased apoptosis and cell death. In conclusion, protection from LPS-stimulated PAEC apoptosis by hypoxic preconditioning is attributable in part to reduction in TLR4 expression. If these signaling pathways apply to septic patients, they may account for differing sensitivities of individuals to acute lung injury depending on oxygen tensions in PAECs in vivo.
Keywords: endotoxin; hypoxia; caspase 3; Toll-like receptor 4 (TLR4); 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT); pulmonary artery endothelial cells.
Introduction
Acute lung injury (ALI) is a common indication for admission to the intensive care unit for both children and adults.1 Pulmonary or systemic infections are the leading cause of ALI. Severe sepsis and ALI are associated with high mortality despite early and judicious administration of antibiotic therapy.2 Novel, mechanistically based strategies to prevent ALI are needed to reduce mortality and morbidity in this condition. Extensive studies using animal models and human observations show that pulmonary inflammation often precedes the clinical onset and progression of ALI3 and that these inflammatory responses persist even after infections are controlled. Apoptosis of alveolar epithelial cells and pulmonary vascular endothelial cells is a consistent observation in animal models of ALI.3
Much of the inflammatory response in ALI can be attributed to activation of the innate immune system. Mammalian Toll-like receptors (TLRs) are part of the innate immune system that recognizes specific patterns of microbial components that are conserved among pathogens but that are not found in mammals. The TLR family consists of 10 members (TLR1–TLR10). The endotoxin lipopolysaccharide (LPS), a gram-negative bacterial cell wall product, is recognized by the TLR4 receptor, which is critical for the initiation of a cascade of inflammatory response by mammalian cells.4 TLR4 receptor activation can lead to the induction of proinflammatory genes, such as those encoding tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), and IL-2, via activation of the transcription factor nuclear factor κB (NFκB).4,5 TAK-242 (ethyl-(6R)-[N-(2-chloro-4-fluorophenyl]sulfamoyl]cyclohex-1-hene-1-carboxylate) specifically inhibits TLR4 receptor–mediated signaling by binding to Cys747 in the intracellular domain, leading to suppression of the LPS-mediated inflammatory response.6
The expression of TLR4 on endothelial cells is important for endotoxin-evoked infiltration of neutrophils into the lung tissue, increased lung microvascular permeability, and host survival.7 LPS enhances TLR4 expression in several cell types and injury models.8,9 Nevertheless, there are conflicting reports regarding LPS-mediated effects on TLR4 expression in pulmonary artery endothelial cells (PAECs). For example, intratracheal administration of LPS in mice increases the expression of TLR4 receptors on bronchial epithelial cells and macrophages but not PAECs.9 On the other hand, intratracheal LPS in rats is reported to decrease TLR4 messenger RNA (mRNA) and protein in lung homogenates and lavaged alveolar macrophages.10 Responses of endothelial cells to LPS in these intact animal studies are likely influenced by a number of variables, such as the host immune response and release of cytokines from other cell types in the lungs. Therefore, we examined the specific effect of LPS on TLR4 expression in cultured PAECs.
Studies of LPS and TLR4 in a hypoxic tissue environment, particularly in PAECs, are limited. The effect of hypoxia on LPS-mediated expression of TLR4 in lung cells has not been reported. Because hypoxia and sepsis frequently coexist in critically ill patients, we investigated the effect of hypoxic environment on LPS-mediated PAEC survival and apoptosis and the role played by TLR4 receptors in this process. We hypothesized that incubation of PAECs in hypoxic conditions prior to LPS exposure would diminish the severity of LPS-mediated cell death and decrease TLR4 expression and signaling in PAECs.
Material and methods
Growth and culture of bovine PAECs (BPAECs)
Primary isolates of BPAECs were cultured at 37°C with 5% CO2 in RPMI medium (Gibco, Carlsbad, CA) containing 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Hyclone, Logan, Utah) in a manner reported elsewhere by us.11 When the cells were ∼80% confluent, they were washed twice with phosphate-buffered saline (PBS; Gibco), and the medium was changed to serum-free RPMI medium. Cells were studied in groups as defined below.
Experimental groups
To determine the effect of hypoxia on LPS-induced apoptosis and survival, PAECs were maintained in an atmosphere of 95% N2 and 5% CO2 (hypoxia) or 95% air and 5% CO2 (normoxia). The environmental oxygen during hypoxia (FiO2) was continuously monitored (Pro-Ox 110; Biospherix, Redfield, NY) and did not measure above 2% at any time. Cells were washed and then placed in medium containing LPS at 0.5–0.75 μg/mL (derived from Escherichia coli serotype O55:B5, source strain CDC 1644-70; Sigma, St. Louis, MO) or vehicle and maintained in either environment for 24 hours.
After 24 hours, medium was replaced for the second incubation period (with LPS or vehicle as indicated in figure legends), and cells were returned to normoxic or hypoxic environments for a second period before assays were performed. The duration of the second period varied on the basis of the end point of interest (e.g., shorter for mRNA than protein) and is defined in “Results” or in figure legends. Two protocols of hypoxia alone were tested: the first included two periods of 24 hours each of hypoxia separated by 5–10 minutes of normoxia to exchange medium to match the treatment protocols. The second consisted of 48 hours of hypoxia without medium exchange. The specific hypoxia protocol is indicated in the text and figure legends. These exposure protocols were chosen to optimize caspase 3 activation while limiting cell detachment and death on the basis of pilot experiments.
To determine whether the LPS effects were mediated by activation of TLR4 receptors, PAECs were preincubated with a specific TLR4 inhibitor, TAK-242 (1.0 μM; tlr-cli95; InvivoGen, San Diego, CA), before treatment with LPS. TLR4 signaling is inhibited by TAK-242 binding to a specific amino acid, Cys747, in the intracellular domain of TLR4. Signaling through TLR2, TLR35, TLR9, and TLR adapter molecules or MD-2 (lymphocyte antigen 96) is not triggered by TAK-242.12,13
At the end of the second treatment period, PAECs were harvested for survival assays or for determination of TLR4 expression. For some experiments, the cell-free medium was saved for detection of TNF-α. For cell survival experiments, adherent cells were washed with PBS, and the cells were scraped and collected in 65 μL of lysis buffer (R&D Systems, Minneapolis, MN). The cell lysate was centrifuged at 14,000 g for 10 minutes at 4°C, and the supernatant was collected and frozen at −80°C for determination of caspase 3 activity. Protein concentrations were estimated using the BioRad Protein Dye reagent (BioRad, Hercules, CA).
Caspase 3 activity
Caspase 3 activity was determined as an apoptotic index because it represents the final common pathway for activation of apoptosis through extrinsic and intrinsic mechanisms, thus measuring any caspase-associated programmed cell death.14 The Caspase 3 Colorimetric Assay (R&D Systems) was used to determine the amount of caspase 3 activity in each sample of cell lysates, as reported elsewhere by us.15 This assay uses Asp-Glu-Val-Asp-pNA as a substrate for caspase 3, with spectrophotometric detection of purple an indicator of caspase 3 activity. Caspase 3 activity was normalized for protein concentration and reported as a percentage of caspase 3 activity in the normoxia vehicle control.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay
Survival of cells was determined by MTT assay as described in the manufacturer’s protocol (V-13154; Molecular Probes, Eugene, OR).16 Briefly, washed cells were incubated for 3 hours in phenol red-free medium containing 0.5% of the yellow mitochondrial dye MTT+. The amount of blue formazan dye generated from MTT+ is proportional to the number of live cells. The readings for the test groups of cells were expressed as percentages of normoxia vehicle control.
TNF-α production by BPAECs
A bovine TNF-α enzyme-linked immunosorbent assay kit (DY2279; R&D Systems) was used to determine TNF-α levels in the cell culture medium according to the instructions provided by the manufacturer. Briefly, 96-well plates (DY990; R&D Systems) coated with capture antibody and detection antibodies were used with a streptavidin–horseradish peroxidase visualization system. TNF-α levels in cell culture medium were estimated using the standard curve and expressed as picograms per milliliter. This assay was chosen as functional index of binding to TLR-4 receptors in our cells because it is a well-recognized consequence of receptor engagement in endothelial as well as other cell types.5
TLR4 mRNA expression
Expression of TLR4 mRNA was determined by real-time polymerase chain reaction (PCR). Total RNA was extracted by means of Trizol (Invitrogen, Carlsbad, CA), and 1.0 μg was reverse transcribed for first-strand complementary DNA (cDNA) synthesis using Oligo(dT) primers according to the manufacturer’s protocol (Invitrogen). This first-strand cDNA was then used for real-time PCR. Bovine TLR4 (forward, ACTGCAGCTTCAACCGTATC; reverse, TAAAGGCTCTGCACACATCA) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; forward, TCAAGAAGGTGGTGAAGCAG; reverse, TGTCGTACCAGGAAATGAGC) primers were constructed using sequences from Ibeagha-Awemu et al.17 Real-time PCR was performed with the iQ5 Multicolor Real-Time PCR Detection System using SYBR Green Supermix (BioRad). A two-step amplification and thermal denaturation protocol was used for real-time PCR. iQ5 Optical System software (ver. 2.1; BioRad) was used to calculate the relative amounts of genes of interest. The comparative Ct method (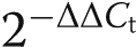 ) was used for quantification. This method entails normalization of the samples with the housekeeping gene (GAPDH) followed by comparison of the Ct values in the samples of interest to those of untreated controls. The results are expressed as fold change compared with control.
) was used for quantification. This method entails normalization of the samples with the housekeeping gene (GAPDH) followed by comparison of the Ct values in the samples of interest to those of untreated controls. The results are expressed as fold change compared with control.
TLR4 protein expression determined by Western blot analysis
Western blots were performed on 50 μg of cell lysate protein. Membranes were probed with anti-TLR4 (AF1478; R&D Systems) or anti-β-actin (A2228; Sigma) antibodies. The density of bands in an image of the Western blot was determined (ImageJ software; National Institutes of Health), and the results were expressed as the ratio of TLR4 to β-actin.
Statistical analysis
For each assay, 3–13 separate PAEC isolates (n values) were studied. The number of replicates for each group appears in figure legends, figures, or text. Comparisons between groups for all experiments were performed using one-way analysis of variance unless only one treatment was studied, in which case t tests were performed. When between-group differences were found, a Newman-Keuls post hoc test was employed for pairwise multiple comparisons. Between-group and pairwise P values are provided with symbols to indicate those that are significant. All grouped data are presented as mean ± standard error of the mean.
Results
LPS increases apoptosis and decreases cell survival in PAECs
Treatment with LPS for 48 hours increased caspase 3 activity (Fig. 1, left) and decreased MTT incorporation (Fig. 1, right) in PAECs relative to vehicle controls.
Figure 1.

Caspase 3 activity (left) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; right) were determined in pulmonary artery endothelial cells (PAECs) incubated in normoxia and treated with vehicle or lipopolysaccharide (LPS) for 48 hours to determine the time and concentration of maximum response. The number of experiments appears in the bars. The vehicle for LPS was physiological saline. Caspase 3 activity was increased and MTT incorporation was decreased by exposure for 48 hours to 0.5 μg/mL LPS (t tests), consistent with apoptosis and necrosis injury of these cells. LPS-induced increments in these end points were lower in cells treated for greater or lesser time periods or with differing concentrations of LPS.
Preconditioning decreases LPS-evoked stimulated caspase 3 activity in PAECs
Next we investigated the effects of environmental FiO2 on LPS-stimulated apoptosis. Treatment of PAECs with LPS in normoxia for 24 or 48 hours increased caspase 3 activity to values more than four times that of cells treated with vehicle (Fig. 2; 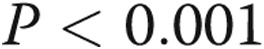 relative to normoxia vehicle). Hypoxia alone for 48 hours led to a twofold increase in caspase 3 activity compared with that of normoxic vehicle control. Preconditioning with hypoxia for 24 hours decreased LPS-induced increments in caspase 3 activity compared with those of PAECs kept in normoxia for 24 hours and then treated with LPS (
relative to normoxia vehicle). Hypoxia alone for 48 hours led to a twofold increase in caspase 3 activity compared with that of normoxic vehicle control. Preconditioning with hypoxia for 24 hours decreased LPS-induced increments in caspase 3 activity compared with those of PAECs kept in normoxia for 24 hours and then treated with LPS (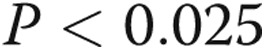 ).
).
Figure 2.
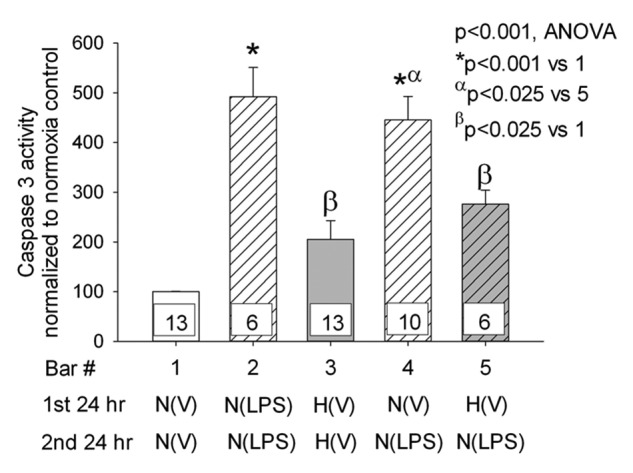
Caspase 3 activity was determined in pulmonary artery endothelial cells (PAECs) incubated in hypoxia or normoxia and treated with vehicle or lipopolysaccharide (LPS). Five groups of PAECs were studied: (1) vehicle for 48 hours in normoxia, (2) LPS for 48 hours in normoxia, (3) vehicle alone for 48 hours in hypoxia, (4) maintenance in normoxia for 24 hours followed by LPS for 24 hours in normoxia, and (5) hypoxia for 24 hours followed by LPS for 24 hours in normoxia. P values for between-group (analysis of variance [ANOVA]) and pairwise multiple comparisons appear in the graph. The number of samples in each group are provided within the bars of the figure. For all figures, the letters below the X-axis indicate the following: N = PAECs incubated in normoxia, H = PAECs incubated in hypoxia, V = cells treated with vehicle, LPS = cells treated with LPS, N(V) = cells incubated in normoxia treated with vehicle, N(LPS) = cells incubated in normoxia treated with LPS, and H(V) = cells incubated in hypoxia treated with vehicle. ANOVA for five groups revealed between-group differences of 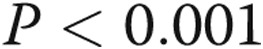 . Caspase 3 activity was increased with all treatments compared with untreated normoxia controls and was increased in PAECs kept in normoxia for 24 hours followed by LPS treatment for 24 or 48 hours (*
. Caspase 3 activity was increased with all treatments compared with untreated normoxia controls and was increased in PAECs kept in normoxia for 24 hours followed by LPS treatment for 24 or 48 hours (*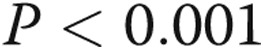 , bars 1 vs. 4 and bars 1 vs. 2). LPS also increased caspase 3 activity in cells kept in hypoxic conditions prior to treatment with LPS in normoxia (
, bars 1 vs. 4 and bars 1 vs. 2). LPS also increased caspase 3 activity in cells kept in hypoxic conditions prior to treatment with LPS in normoxia (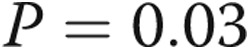 , bars 1 vs. 5). However, the increase was lower than that observed with cells incubated in normoxia prior to treatment with LPS (α
, bars 1 vs. 5). However, the increase was lower than that observed with cells incubated in normoxia prior to treatment with LPS (α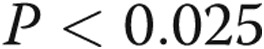 , bars 4 vs. 5). Caspase 3 activity was assessed in four additional paired samples not shown in this graph. The first was maintained in normoxia for 24 hours, and the second was maintained in hypoxia for 24 hours followed by 10 minutes of reoxygenation for exchange of medium followed by a second 24-hour period of hypoxia. The hypoxia-induced increment in caspase 3 activity in this group of cells (caspase 3 activity was 212% ± 68% of normoxia control) was not different from that of cells maintained in hypoxia for 48 hours without reoxygenation for medium change (
, bars 4 vs. 5). Caspase 3 activity was assessed in four additional paired samples not shown in this graph. The first was maintained in normoxia for 24 hours, and the second was maintained in hypoxia for 24 hours followed by 10 minutes of reoxygenation for exchange of medium followed by a second 24-hour period of hypoxia. The hypoxia-induced increment in caspase 3 activity in this group of cells (caspase 3 activity was 212% ± 68% of normoxia control) was not different from that of cells maintained in hypoxia for 48 hours without reoxygenation for medium change (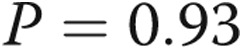 ). Data in the graph represent data from cells that we maintained for 48 hours in hypoxia without medium exchange or reoxygenation.
). Data in the graph represent data from cells that we maintained for 48 hours in hypoxia without medium exchange or reoxygenation.
LPS mediates PAEC apoptosis through TLR4 receptor binding
To confirm that LPS-mediated apoptosis depends on binding to TLR4 receptors in our PAECs, we pretreated cells with TAK-242 or vehicle and then measured LPS-stimulated caspase 3 activity. TAK-242, a specific inhibitor of the signaling mechanism activated by LPS through TLR4 engagement, abolished LPS-mediated PAEC apoptosis (Fig. 3; 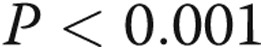 ).
).
Figure 3.
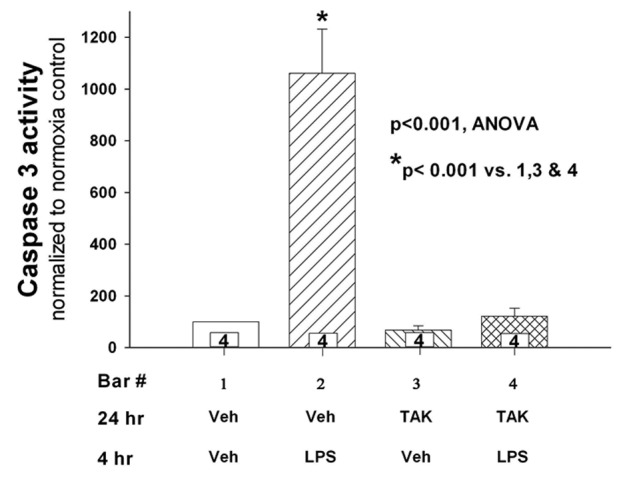
Caspase 3 activity was determined in endothelial cells incubated for 24 hours with TAK-242 or vehicle followed by treatment with lipopolysaccharide (LPS) or vehicle for an additional 4 hours ( for each group). The vehicle for TAK-242 was dimethyl sulfoxide diluted 1∶1,000 with phosphate-buffered saline. LPS-mediated apoptosis was reduced to baseline in cells treated with TAK-242 (
for each group). The vehicle for TAK-242 was dimethyl sulfoxide diluted 1∶1,000 with phosphate-buffered saline. LPS-mediated apoptosis was reduced to baseline in cells treated with TAK-242 (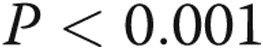 ). TAK-242 treatment by itself did not affect apoptosis relative to vehicle control (bar 3). n values appear in the bars. ANOVA: analysis of variance.
). TAK-242 treatment by itself did not affect apoptosis relative to vehicle control (bar 3). n values appear in the bars. ANOVA: analysis of variance.
Effects of hypoxic preconditioning on LPS-induced TNF-α production
We measured LPS-stimulated TNF-α production as a biological indicator of signaling initiated by TLR4 binding and as an index of proinflammatory response and injury. LPS increased the expression of TNF-α in endothelial cells grown in normoxic conditions (Fig. 4). Hypoxic treatment of PAECs alone did not affect TNF-α levels. Incubation of cells for 24 hours in hypoxia prior to treatment with LPS resulted in reduction of the LPS-stimulated TNF-α levels to levels not different from those of cells treated with hypoxia and vehicle.
Figure 4.
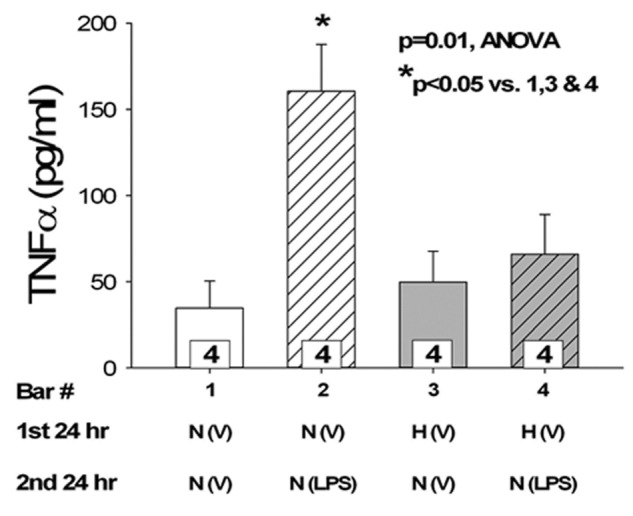
Tumor necrosis factor α (TNF-α) induction by lipopolysaccharide (LPS) is blunted by hypoxia preconditioning. Pulmonary artery endothelial cells (PAECs) were cultured in conditions of normoxia or hypoxia for 24 hours followed by normoxia with vehicle or LPS for an additional 24 hours (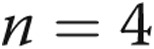 for all groups). LPS increases the expression of TNF-α in endothelial cells kept in normoxic conditions (
for all groups). LPS increases the expression of TNF-α in endothelial cells kept in normoxic conditions (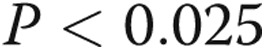 ). Pretreatment with hypoxia for 24 hours prior to LPS leads to reduced LPS-mediated TNF-α levels (
). Pretreatment with hypoxia for 24 hours prior to LPS leads to reduced LPS-mediated TNF-α levels (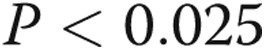 ). ANOVA: analysis of variance.
). ANOVA: analysis of variance.
Effects of hypoxic preconditioning and TAK-242 on LPS-mediated TLR4 expression
TLR4 mRNA was measured in PAECs preconditioned with normoxic or hypoxic pretreatment for 24 hours followed by 4 hours of vehicle or LPS treatment (Fig. 5). The effect of the TLR4 inhibitor TAK-242 on TLR4 expression was also determined. LPS increased TLR4 mRNA by more than eightfold compared with the vehicle-treated cells kept in normoxia (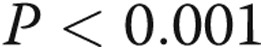 ). Preexposure to hypoxia attenuated the LPS-mediated increase in TLR4 expression to approximately half that observed in LPS-treated cells kept in normoxia (
). Preexposure to hypoxia attenuated the LPS-mediated increase in TLR4 expression to approximately half that observed in LPS-treated cells kept in normoxia (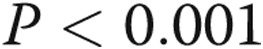 ). TAK-242 decreased the LPS-stimulated expression of TLR4 in PAECs incubated in both normoxic and hypoxic conditions. Eight hours after preconditioning with normoxia or hypoxia, TLR4 protein was lower in LPS-stimulated PAECs kept in hypoxic compared with normoxic conditions (Fig. 6;
). TAK-242 decreased the LPS-stimulated expression of TLR4 in PAECs incubated in both normoxic and hypoxic conditions. Eight hours after preconditioning with normoxia or hypoxia, TLR4 protein was lower in LPS-stimulated PAECs kept in hypoxic compared with normoxic conditions (Fig. 6; 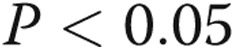 ).
).
Figure 5.
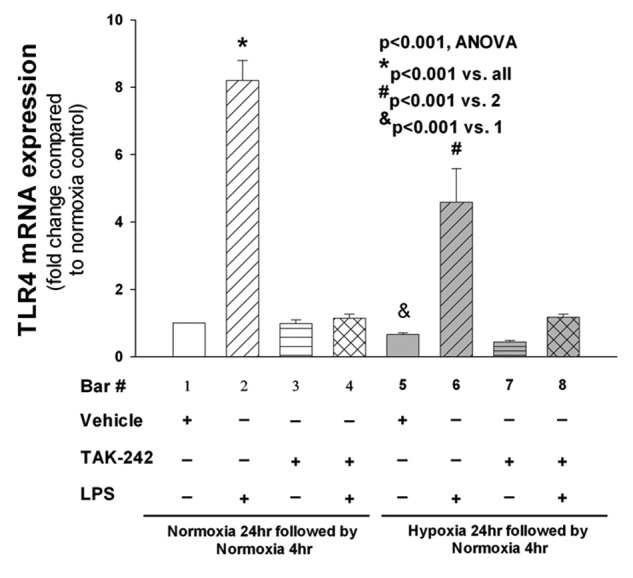
Toll-like receptor 4 (TLR4) expression in pulmonary artery endothelial cells (PAECs) preconditioned in hypoxia and treated with lipopolysaccharide (LPS) and TAK-242. Preliminary experiments showed that LPS-stimulated expression of TLR4 messenger RNA (mRNA) peaked 4 hours after exposure followed by a decrease to basal levels at 24 hours (not shown). Accordingly, PAECs were incubated in conditions of hypoxia or normoxia for 24 hours followed by treatment with LPS or vehicle for an additional 4 hours, and then TLR4 mRNA was measured (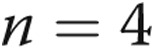 for all groups). LPS increases the expression of TLR4 severalfold compared with untreated cells (
for all groups). LPS increases the expression of TLR4 severalfold compared with untreated cells (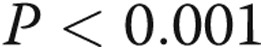 , bars 1 vs. 2). A t test of TLR4 mRNA from cells treated with hypoxia alone compared with normoxia (bars 1 vs. 5) revealed a hypoxia-induced decrease in TLR4 (
, bars 1 vs. 2). A t test of TLR4 mRNA from cells treated with hypoxia alone compared with normoxia (bars 1 vs. 5) revealed a hypoxia-induced decrease in TLR4 (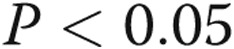 ). Preconditioning of the cells in hypoxia prior to treatment with LPS leads to a reduction in TLR4 expression (
). Preconditioning of the cells in hypoxia prior to treatment with LPS leads to a reduction in TLR4 expression (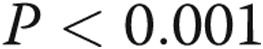 , bars 2 vs. 6). The effect of TAK-242 on LPS-stimulated TLR4 mRNA expression was determined by incubating PAECs in hypoxic or normoxic conditions for 24 hours with TAK-242 or vehicle followed by treatment with LPS or vehicle for an additional 4 hours (
, bars 2 vs. 6). The effect of TAK-242 on LPS-stimulated TLR4 mRNA expression was determined by incubating PAECs in hypoxic or normoxic conditions for 24 hours with TAK-242 or vehicle followed by treatment with LPS or vehicle for an additional 4 hours (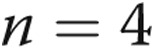 separate isolates of PAECs). TAK-242 decreased the expression of LPS-stimulated TLR4 cells incubated in both normoxic and hypoxic conditions (
separate isolates of PAECs). TAK-242 decreased the expression of LPS-stimulated TLR4 cells incubated in both normoxic and hypoxic conditions (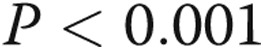 , analysis of variance [ANOVA]; other pairwise comparisons are as per the graph).
, analysis of variance [ANOVA]; other pairwise comparisons are as per the graph).
Figure 6.
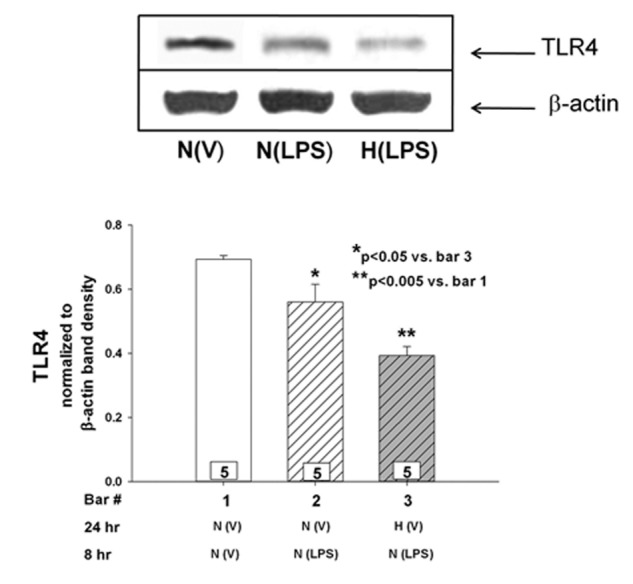
Effect of lipopolysaccharide (LPS) on Toll-like receptor 4 (TLR4) protein in pulmonary artery endothelial cells (PAECs). Cells were treated with LPS or vehicle for 8 hours. In data not shown, hypoxia alone had no effect on TLR4 protein relative to PAECs in normoxia. Eight hours after LPS treatment, TLR4 protein in PAECs preconditioned with hypoxia was less than that observed in the cells kept in normoxic environment. n values appear in the bars, and P values with symbols appear within the graphs.
Hypoxia after LPS worsens injury
To determine whether the order of exposure to hypoxia or LPS affected injury to PAECs, caspase 3 activity and MTT were assayed in cells treated first with LPS in normoxia for 24 hours followed by an additional 24 hours in either normoxia or hypoxia. In these experiments, hypoxia worsened apoptosis, as indicated by higher caspase 3 activity and lower MTT relative to values for cells maintained in normoxia after LPS (Fig. 7).
Figure 7.
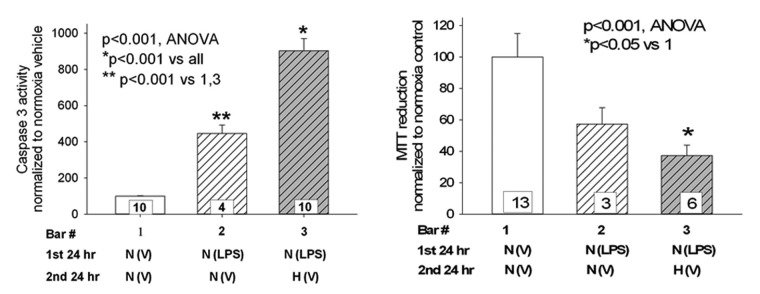
Hypoxia after lipopolysaccharide (LPS) exacerbates injury. Pulmonary artery endothelial cells (PAECs) were treated with 0.5 μg/mL LPS for 24 hours in a normoxic environment, after which time the medium was exchanged for LPS-free medium. Cells were then maintained another 24 hours in either normoxia or hypoxia, after which time caspase 3 activity (left) or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; right) was measured. Relative to cells maintained in normoxia after LPS treatment, caspase 3 activity was increased and MTT was decreased in PAECs kept in a hypoxic environment. n values appear in the bars, and P values with symbols appear within the graphs.
Discussion
LPS, the laboratory mimic of sepsis, is well recognized to trigger apoptosis in endothelial cells. For example, LPS evokes apoptosis in porcine aortic endothelial cells in a concentration- and time-dependent manner.18 Thambiayya et al.19 demonstrated that transcellular movement of extracellular zinc and nitric oxide contribute to LPS-induced apoptosis in cultured sheep PAECs. However, PAECs are structurally and functionally different from systemic endothelial cells, particularly with respect to native oxygen tensions and their innate immune responses. Most relevant to the present study, Sampath et al.20 reported LPS-induced apoptosis and reactive oxygen species (ROS) production in fetal ovine PAECs, which was ameliorated by preexposure to hypoxia. Our study shows for the first time that hypoxic preconditioning protects PAECs from LPS-stimulated TNF-α production and increases in caspase 3 activity. We also observed large LPS-induced increases in TLR4 mRNA transcripts, and these increases were blunted by hypoxic preconditioning. These data are consistent with LPS-induced upregulation of TLR4 expression in other cell types, but they are the first to describe this upregulation in PAECs, to our knowledge. We observed LPS-associated decreases TLR4 protein in the setting of increased TLR4 transcripts in PAECs, a phenomenon previously reported in phagocytes but not in endothelial cells. Finally, these data are the first to identify enhanced sensitivity to apoptosis in BPAECs treated first with LPS followed by hypoxia.
The effect of LPS on TLR4 expression has been studied in a variety of different models and cell types with disparate results. Intratracheal LPS in mice leads to increases in both mRNA and protein levels of TLR4 in bronchial epithelial cells and alveolar macrophages but not in endothelial cells.9 Intratracheal LPS in rats is reported to result in both decreases10 in TLR4 mRNA and protein in lung homogenates and increases21 in TLR4 mRNA in lung homogenates, accompanied by increased expression in endothelium determined by immunohistochemistry. Cultured rat lung pericytes exhibit upregulated TLR4 in response to LPS.8 We find no previous reports of the effects of LPS on TLR4 expression in PAECs. In human circulating mononuclear phagocytes, LPS increases TLR4 mRNA but decreases the surface expression of these receptors, similar to our observations in cultured PAECs.22 The authors hypothesize that the increase in TLR4 mRNA by LPS counters the reduction of surface expression. LPS binding to TLR4 surface receptors initiates internalization and destruction of these proteins. The rate of loss of these receptors exceeds the rate for replenishment; therefore, there is a net reduction in the steady-state surface expression of these receptors.22 Our findings show that LPS stimulated the TLR4 transcript in PAECs and that this increase was blunted in cells pretreated with hypoxia. Similarly, TLR4 protein density was higher in LPS-treated PAECs kept in normoxic relative to hypoxic conditions (Fig. 7). Functional implications of changes in TLR4 expression should be more closely linked to protein density rather than transcript numbers. Thus, our observations are consistent with the data in phagocytes,22 although though new for PAECs. They support the hypothesis that (i) hypoxic preconditioning protects from LPS partially through decreased expression of TLR4 and (ii) LPS stimulation of TLR4 transcripts could be instrumental in the maintenance receptor density in PAECs. Nevertheless, direct investigations of the role played by LPS in internalization or destruction of TLR4 receptors and the time frame over which this occurs in PAECs remain to be undertaken.
Seki et al.23 and Sha et al.24 have reported that inhibition of TLR4 with TAK-242 protects mice from LPS-induced increases in pulmonary polymorphonuclear leukocyte infiltration, increased microvascular permeability, and cytokine release into the bronchoalveolar lavage and that it increases survival. Our studies show that TAK-242 not only decreases PAEC apoptosis but also decreases LPS-stimulated TLR4 mRNA. These data add further support to the hypothesis that increments of TLR4 mRNA may be necessary for the full activation of the inflammatory pathway by LPS in PAECs.
We found increases in TNF-α in PAECs treated with LPS, verifying an inflammatory response initiated by binding to TLR4 receptors in our BPAECs, as has been reported in a number of cell types.25,26 Activation of TLR4 induces inflammatory responses by initiating multiple intracellular signaling events, including the activation of NFκB, which initiates synthesis and release of many proinflammatory mediators and adhesion molecules, such as IL-1, IL-6, IL-8, TNF-α, and intercellular adhesion molecule 1 (ICAM-1). These signaling pathways have been verified in PAECs.25 Furthermore, TNF-α causes apoptosis in PAECs, liver endothelial cells, and bovine aorta endothelial cells in culture.27-29 Since TLR4 receptors are preferentially utilized by LPS for the induction of the inflammatory pathway,5,30 a reduction in these receptors could lead to a reduction in the LPS-stimulated activation of the inflammatory pathway. Some of these inflammatory pathways may be related to the increased sensitivity of BPAECs to hypoxia-induced apoptosis after treatment with LPS, but our data do not permit us to address this question.
We are keenly interested in the mechanisms through which hypoxic exposure may be protective from subsequent exposure to LPS. Perhaps the best-recognized signaling pathway by hypoxia and anoxia is hypoxia inducible factor 1α (HIF-1α). Hypoxia triggers decreased degradation, nuclear translocation, and binding of HIF-1α to hypoxia-response elements. Induction of HIF-1α protein and its transcriptional activation by hypoxia and oxidative stress are also regulated by signaling pathways, including PI3K/AKT/FRAP30–32, p38, and extracellular signal-regulated protein kinase.31-33 Redox-sensitive factors Ref-1 and thioredoxin and the Rho family small GTPase Rac1 have been shown to play a role.32-34 Enhanced nitric oxide release and activation of Bcl-2 are other prosurvival pathways activated by hypoxia.35 Protection from oxidative stress–induced apoptosis in cortical neuronal cultures by iron chelators correlates to enhanced DNA binding of HIF-1α and ATF-1/CREB transcription factors and to increased expression of glycolytic enzymes.34 HIF-1α can mediate crosstalk between hypoxia and glucose metabolism via glucose-response elements.36 Our experiments show that hypoxia in PAECs blocks LPS-induced increments in TNF-α, suggesting that one pathway of protection from apoptosis by hypoxic preconditioning may be through attenuated synthesis and release of this cytokine involving one or more of the above-described signaling cascades.
Some studies suggest that hypoxia-induced modulation of TLR4 expression may be critical to the LPS-mediated inflammatory pathways in nonpulmonary cells.26,37 Hara et al.26 reported that hypoxia reduces TLR4 mRNA in immortalized and cultured corneal epithelial cells, leading to a reduction in NFκB activation with concomitant reduction in IL-6 and IL-8. Ock et al.37 reported that hypoxia results in upregulation of TLR4 expression and enhanced myeloid differentiation factor 88–independent interferon regulatory factor 3 pathway but, interestingly, a decrease in LPS-induced NFκB in microglial cells. Leeper-Woodford and Detmer38 observed that acute hypoxia enhances LPS-stimulated cytokine production by greater activation of NFκB in alveolar macrophages. Sampath et al.,20 on the other hand, reported that preexposure to hypoxia (FiO2 of 0.05 for 24–48 hours) decreases LPS-induced apoptosis and ROS production in fetal ovine PAECs. The study of Sampath et al. did not include investigations of TLR4 receptor expression. In data not shown, we measured LPS-induced increments in dihydroethidium (a fluorescent marker of ROS, particularly superoxide) in BPAECs maintained in normoxia. We found modest time-dependent increments in LPS-induced dihydroethidium fluorescence peaking at 6 hours after exposure, confirming a qualitatively similar response in bovine as in ovine PAECs. Ishida et al.39 demonstrated that hypoxia decreased TLR4 protein and mRNA expression in human PAECs in a manner that depends on mitochondrial ROS. Furthermore, LPS-induced increments in ICAM expression were decreased by hypoxia, suggesting to them that hypoxia-induced downregulation of TLR4 alters cellular responsiveness to endotoxin. Cell survival or apoptosis were not evaluated as an end point in the study of Ishida et al. The role played by mitochondrial ROS in modifying TLR4 expression in hypoxia preconditioning or the mechanisms underlying worsened injury in PAECs exposed to hypoxia after LPS are very interesting questions for future studies.
Both sepsis and hypoxia are common conditions in intensive care units and in fact often coexist in the same patient, either simultaneously or in series. Little information has been available regarding the interaction of these conditions or the mechanism through which one may modulate the effect of the other. Our observations that LPS- and TLR4-mediated induction of the inflammatory pathway and apoptosis can be modulated by preexposure to hypoxia suggest that a hypoxic environment (e.g., in an area of atelectasis) may limit escalation of an inflammatory response in an injured lung. Because apoptosis is enhanced and survival decreased in PAECs first stressed with LPS and then exposed to hypoxia, individuals with subclinical sepsis suffering subsequent insults causing hypoxia may be at risk of particularly severe lung injury. We have previously reported that TLR4 expression is increased in ischemic lung injury in vivo.40 However, the role played by lung preexposure to hypoxia or LPS in vivo remains to be tested.
Acknowledgments
We appreciate the expert technical assistance of Stephanie Gruenloh and Ying Gao in cell culturing and assays.
Source of Support: National Heart, Lung, and Blood Institute: HL-68627, HL-49294, HL-116530, and BX001681 to ERJ; UL1RR031973 to GGK and ERJ; and 8UL1TR000055 to SA.
Conflict of Interest: None declared.
References
- 1.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med 2005;353(16):1685–1693. [DOI] [PubMed]
- 2.Irish Critical Care Trials Group. Acute lung injury and the acute respiratory distress syndrome in Ireland: a prospective audit of epidemiology and management. Crit Care 2008;12(1):R30. [DOI] [PMC free article] [PubMed]
- 3.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2008;295(3):L379–L399. [DOI] [PMC free article] [PubMed]
- 4.Takeda K, Akira S. TLR signaling pathways. Semin Immunol 2004;16(1):3–9. [DOI] [PubMed]
- 5.Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest 2006;86(1):9–22. [DOI] [PubMed]
- 6.Wittebole X, Castanares-Zapatero D, Laterre PF. Toll-like receptor 4 modulation as a strategy to treat sepsis. Mediators Inflamm 2010;2010:568396. [DOI] [PMC free article] [PubMed]
- 7.Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, et al. Endothelium-derived Toll-like receptor–4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest 2003;111(7):1011–1020. [DOI] [PMC free article] [PubMed]
- 8.Edelman DA, Jiang Y, Tyburski J, Wilson RF, Steffes C. Toll-like receptor–4 message is up-regulated in lipopolysaccharide-exposed rat lung pericytes. J Surg Res 2006;134(1):22–27. [DOI] [PubMed]
- 9.Saito T, Yamamoto T, Kazawa T, Gejyo H, Naito M. Expression of Toll-like receptor 2 and 4 in lipopolysaccharide-induced lung injury in mouse. Cell Tissue Res 2005;321(1):75–88. [DOI] [PubMed]
- 10.Fan J, Kapus A, Marsden PA, Li YH, Oreopoulos G, Marshall JC, et al. Regulation of Toll-like receptor 4 expression in the lung following hemorrhagic shock and lipopolysaccharide. J Immunol 2002;168(10):5252–5259. [DOI] [PubMed]
- 11.Medhora M, Chen Y, Gruenloh S, Harland D, Bodiga S, Zielonka J, et al. 20-HETE increases superoxide production and activates NAPDH oxidase in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 2008;294(5):L902–L911. [DOI] [PMC free article] [PubMed]
- 12.Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y, Ii M. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol 2009;157(7):1250–1262. [DOI] [PMC free article] [PubMed]
- 13.Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol 2008;584(1):40–48. [DOI] [PubMed]
- 14.Favaloro B, Allocati N, Graziano V, Di Ilio C, De Laurenzi V. Role of apoptosis in disease. Aging (Albany NY) 2012;4(5):330–349. [DOI] [PMC free article] [PubMed]
- 15.Dhanasekaran A, Bodiga S, Gruenloh S, Gao Y, Dunn L, Falck JR, et al. 20-HETE increases survival and decreases apoptosis in pulmonary arteries and pulmonary artery endothelial cells. Am J Physiol Heart Circ Physiol 2009;296(3):H777–H786. [DOI] [PMC free article] [PubMed]
- 16.Dhanasekaran A, Gruenloh SK, Buonaccorsi JN, Zhang R, Gross GJ, Falck JR, et al. Multiple antiapoptotic targets of the PI3K/Akt survival pathway are activated by epoxyeicosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. Am J Physiol Heart Circ Physiol 2008;294(2):H724–H735. [DOI] [PMC free article] [PubMed]
- 17.Ibeagha-Awemu EM, Lee JW, Ibeagha AE, Bannerman DD, Paape MJ, Zhao X. Bacterial lipopolysaccharide induces increased expression of toll-like receptor (TLR) 4 and downstream TLR signaling molecules in bovine mammary epithelial cells. Vet Res 2008;39(2):11. [DOI] [PubMed]
- 18.Bernardini C, Zannoni A, Turba ME, Fantinati P, Tamanini C, Bacci ML, Forni M. Heat shock protein 70, heat shock protein 32, and vascular endothelial growth factor production and their effects on lipopolysaccharide-induced apoptosis in porcine aortic endothelial cells. Cell Stress Chaperones 2005;10(4):340–348. [DOI] [PMC free article] [PubMed]
- 19.Thambiayya K, Wasserloos KJ, Huang Z, Kagan VE, St. Croix CM, Pitt BR. PS-induced decrease in intracellular labile zinc, [Zn]i, contributes to apoptosis in cultured sheep pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 2011;300(4):L624–L632. [DOI] [PMC free article] [PubMed]
- 20.Sampath V, Radish AC, Eis AL, Broniowska K, Hogg N, Konduri GG. Attenuation of lipopolysaccharide-induced oxidative stress and apoptosis in fetal pulmonary artery endothelial cells by hypoxia. Free Radic Biol Med 2009;46(5):663–671. [DOI] [PMC free article] [PubMed]
- 21.Janardhan KS, McIsaac M, Fowlie J, Shrivastav A, Caldwell S, Sharma RK, et al. Toll like receptor–4 expression in lipopolysaccharide induced lung inflammation. Histol Histopathol 2006;21(7):687–696. [DOI] [PubMed]
- 22.Bosisio D, Polentarutti N, Sironi M, Bernasconi S, Miyake K, Webb GR, et al. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-γ: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood 2002;99(9):3427–3431. [DOI] [PubMed]
- 23.Seki H, Tasaka S, Fukunaga K, Shiraishi Y, Moriyama K, Miyamoto K, et al. Effect of Toll-like receptor 4 inhibitor on LPS-induced lung injury. Inflamm Res 2010;59(10):837–845. [DOI] [PubMed]
- 24.Sha T, Sunamoto M, Kitazaki T, Sato J, Ii M, Iizawa Y. Therapeutic effects of TAK-242, a novel selective Toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur J Pharmacol 2007;571(2–3):231–239. [DOI] [PubMed]
- 25.Cendan JC, Moldawer LL, Souba WW, Copeland EM III, Lind DS. Endotoxin-induced nitric oxide production in pulmonary artery endothelial cells is regulated by cytokines. Arch Surg 1994;129(12):1296–1300. [DOI] [PubMed]
- 26.Hara Y, Shiraishi A, Ohashi Y. Hypoxia-altered signaling pathways of toll-like receptor 4 (TLR4) in human corneal epithelial cells. Mol Vis 2009;15:2515–2520. [PMC free article] [PubMed]
- 27.Marino E, Cardier JE. Differential effect of IL-18 on endothelial cell apoptosis mediated by TNF-α and Fas (CD95). Cytokine 2003;22(5):142–148. [DOI] [PubMed]
- 28.Robaye B, Mosselmans R, Fiers W, Dumont JE, Galand P. Tumor necrosis factor induces apoptosis (programmed cell death) in normal endothelial cells in vitro. Am J Pathol 1991;138(2):447–453. [PMC free article] [PubMed]
- 29.Wendt CH, Polunovsky VA, Peterson MS, Bitterman PB, Ingbar DH. Alveolar epithelial cells regulate the induction of endothelial cell apoptosis. Am J Physiol 1994;267(4 pt 1):C893–C900. [DOI] [PubMed]
- 30.Casanova JL, Abel L, Quintana-Murci L. Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu Rev Immunol 2011;29:447–491. [DOI] [PubMed]
- 31.Yu EZ, Li YY, Liu XH, Kagan E, McCarron RM. Antiapoptotic action of hypoxia-inducible factor–1α in human endothelial cells. Lab Invest 2004;84(5):553–561. [DOI] [PubMed]
- 32.Welsh SJ, Williams RR, Birmingham A, et al. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1α and vascular endothelial growth factor formation. Mol Cancer Ther 2003;2:235–243. [PubMed]
- 33.Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol 2002;64:993–998. [DOI] [PubMed]
- 34.Wang N, Stemerman MB. Ref-1 and transcriptional control of endothelial apoptosis. Circ Res 2001;88:1223–1225. [DOI] [PubMed]
- 35.Marzo F, Lavorgna A, Coluzzi G, Santucci E, Tarantino F, Rio T, Conti E, Autore C, Agati L, Andreotti F. Erythropoietin in heart and vessels: focus on transcription and signalling pathways. J Thromb Thrombolysis 2008;26(3):183–187. [DOI] [PubMed]
- 36.Kietzmann T, Krones-Herzig A, Jungermann K. Signaling cross-talk between hypoxia and glucose via hypoxia-inducible factor 1 and glucose response elements. Biochem Pharmacol 2002;64:903–911. [DOI] [PubMed]
- 37.Ock J, Jeong J, Choi WS, Lee WH, Kim SH, Kim IK, et al. Regulation of Toll-like receptor 4 expression and its signaling by hypoxia in cultured microglia. J Neurosci Res 2007;85(9):1989–1995. [DOI] [PubMed]
- 38.Leeper-Woodford SK, Detmer K. Acute hypoxia increases alveolar macrophage tumor necrosis factor activity and alters NF-κB expression. Am J Physiol 1999;276(6 pt 1):L909–L916. [DOI] [PubMed]
- 39.Ishida I, Kubo H, Suzuki S, Suzuki T, Akashi S, Inoue K, Maeda S, Kikuchi J, Sasaki H, Kondo T. Hypoxia diminishes toll-like-receptor 4 expression through reactive oxygen species generated by mitochondria in endothelial cells. J Immunol 2002;169(4):2069–2075. [DOI] [PubMed]
- 40.Ali I, Gruenloh S, Gao Y, Clough A, Falck JR, Medhora M, Jacobs ER. Protection by 20-5,14-HEDGE against surgically induced ischemia reperfusion lung injury in rats. Ann Thorac Surg 2012;93(1):282–288. [DOI] [PMC free article] [PubMed]