

Abstract Abstract
Changes in voltage-gated K+ (Kv) channel function contribute to the pathogenesis of pulmonary hypertension. Yet the mechanisms underlying Kv channel impairments in the pulmonary circulation remain unclear. We tested the hypothesis that reactive oxygen species (ROSs) contribute to the Kv channel dysfunction that develops in resistance-level pulmonary arteries (PRAs) of piglets exposed to chronic in vivo hypoxia. Piglets were raised in either room air (control) or hypoxia for 3 or 10 days. To evaluate Kv channel function, responses to the Kv channel antagonist 4-aminopyridine (4-AP) were measured in cannulated PRAs. To assess the influence of ROSs, PRAs were treated with the ROS-removing agent M40403 (which dismutates superoxide to hydrogen peroxide), plus polyethylene glycol catalase (which converts hydrogen peroxide to water). Responses to 4-AP were diminished in PRAs from both groups of hypoxic piglets. ROS-removing agents had no impact on 4-AP responses in PRAs from piglets exposed to 3 days of hypoxia but significantly increased the response to 4-AP in PRAs from piglets exposed to 10 days of hypoxia. Kv channel function is impaired in PRAs of piglets exposed to 3 or 10 days of in vivo hypoxia. ROSs contribute to Kv channel dysfunction in PRAs from piglets exposed to hypoxia for 10 days but are not involved with the Kv channel dysfunction that develops within 3 days of exposure to hypoxia. Therapies to remove ROSs might improve Kv channel function and thereby ameliorate the progression, but not the onset, of pulmonary hypertension in chronically hypoxic newborn piglets.
Keywords: membrane potential, membrane depolarization, apocynin, NADPH oxidase
Introduction
Potassium (K+) channels play an essential role in regulating vascular tone and the resting membrane potential (Em) of smooth muscle cells.1-3 A number of functionally distinguishable K+ channels, including voltage-gated K+ (Kv) channels, KCNQ channels (a subfamily of Kv channels), adenosine triphosphate (ATP)-sensitive K+ (KATP) channels, two-pore domain K+ channels, and large-conductance Ca2+-activated K+ (BKCa) channels, have been identified in pulmonary artery smooth muscle cells (PASMCs).3-9 Which K+ channel contributes most to resting Em in PASMCs remains controversial. However, current evidence suggests that Kv channels are involved in regulating the resting Em in adult PASMCs.3,4,7,9 Moreover, although changes in other K+ channels may also contribute,5,6,8 there is substantial evidence that changes in Kv channel function and/or expression, along with a concomitant influence on Em, contribute to the elevated pulmonary vascular tone found in adults with pulmonary hypertension.10-12 For example, Kv channel function and expression are altered in PASMCs of adult rats with chronic hypoxia–induced pulmonary hypertension and in PASMCs of adult humans with either primary pulmonary hypertension or anorexigen-induced pulmonary hypertension.13-18
Although the number of studies are few, Kv channel dysfunction and altered PASMC membrane properties have also been reported in newborn-animal models of pulmonary hypertension.19,20 In particular, we previously showed that Kv channel function was impaired concomitant with depolarization of PASMC Em when newborn pigs were exposed to chronic hypoxia for 3 days.19 However, the mechanisms underlying Kv channel impairments in either newborn- or adult-animal models of pulmonary hypertension remain unclear. Reactive oxygen species (ROSs) have been shown to inhibit Kv channel function.21-29 Of interest, we have previously provided evidence that ROSs30 may contribute to the pulmonary hypertension that develops when newborn piglets are exposed to chronic hypoxia. The major purpose of this study was to test the hypothesis that ROSs play a role in the Kv channel dysfunction that develops in resistance-level pulmonary arteries (PRAs) of piglets exposed to in vivo chronic hypoxia. We have previously shown that pulmonary hypertension worsens when hypoxic exposure is extended from 3 to 10 days.31 Therefore, we also performed studies to test the hypothesis that the contribution of ROSs to Kv channel dysfunction would be greater with 10 days than with 3 days of exposure to in vivo chronic hypoxia.
Methods
Animals
Newborn pigs were obtained from the vendor on day of life 2 and raised in a hypoxic, normobaric environment until either day of life 5 (3 days of hypoxia) or day of life 12 (10 days of hypoxia), following our previously described methods.19,30 Normobaric hypoxia was produced by delivering compressed air and N2 to a chamber so that oxygen content was maintained at 10%–11% O2 (PO2: 60–72 Torr). CO2 was maintained at 3–6 Torr by absorption with soda lime. The chamber was opened twice each day for cleaning and to weigh the animals. The animals were fed ad libitum with an artificial sow milk replacer from a feeding device attached to the chamber. Normoxic, age-matched control animals were either 5 or 12 days old when obtained from the vendor and were studied on the day of arrival, i.e., at the same postnatal ages as the hypoxic piglets. We have previously found no differences in vascular responses between piglets raised in a room-air environment and piglets raised on a farm. Hereafter the piglets are referred to as the 3-day exposure group (piglets raised in hypoxia for 3 days and comparable-age control piglets) and the 10-day exposure group (piglets raised in hypoxia for 10 days and comparable-age control piglets).
On the day of study, the piglets were preanesthetized with acepromazine (2 mg/kg intramuscularly [im]) and ketamine (10 mg/kg im) and then anesthetized with pentobarbital (30 mg/kg intravenously [iv]). All animals were given heparin (1,000 IU/kg iv) and then exsanguinated. The thorax was opened, and the lungs were removed and placed in cold (4°C) physiological saline solution (PSS) until use. The PSS had the following composition (in mM): Na+, 141; K+, 4.7; Cl−, 125; Ca2+, 2.5; Mg2+, 0.72; H2PO4−, 1.7; HCO3−, 25; and glucose, 11. All procedures involving animals adhered to the National Institutes of Health guidelines for the use of experimental animals and were approved by the Institutional Animal Care and Use Committee at Vanderbilt University School of Medicine.
Cannulated-artery preparation
Immediately prior to use, segments of 100–300-μm diameter pulmonary arteries were dissected from a lung lobe. The system used to study cannulated arteries has been described in detail previously.32 The exterior of the artery was suffused with PSS from a reservoir at 37°C and aerated with a gas mixture containing 21% O2, 5% CO2, and 74% N2. The arterial lumen was filled from a syringe containing PSS, aerated with the same gas mixture as the reservoir, and connected to the cannula with polyethylene tubing. Gas concentrations and pH in all solutions were monitored with a blood gas analyzer. Inflow pressure was adjusted by changing the height of the reservoir. The artery was observed continuously with a video system containing a color camera (Hitachi VCC151) and a color television monitor. Vessel diameters were measured with a videoscaler (FORA IV).
All arteries were equilibrated for 30–60 minutes at transmural pressures that approximated in vivo pressures previously measured in normoxic and hypoxic piglets:31 control arteries were equilibrated at a transmural pressure of 11 mmHg, and the hypoxic arteries were equilibrated at a transmural pressure of 18 mmHg. Following equilibration and establishment of basal tone, all arteries were tested for viability by contraction to U46619 (10−8 M). The presence of a functional endothelium was determined in control arteries by dilation of the vessel to acetylcholine (ACh, 10−6 M; average dilation: 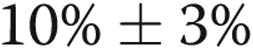 ). We previously found that although hypoxic arteries constricted to ACh, they did dilate to the calcium ionophore A23187 (10−6 M; average dilation:
). We previously found that although hypoxic arteries constricted to ACh, they did dilate to the calcium ionophore A23187 (10−6 M; average dilation: 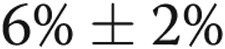 ),32 which also requires the endothelium for its function as a dilator. Therefore, changes in vessel diameter to A23187 were used to check for a functional endothelium in hypoxic arteries.
),32 which also requires the endothelium for its function as a dilator. Therefore, changes in vessel diameter to A23187 were used to check for a functional endothelium in hypoxic arteries.
Smooth muscle cell membrane potential (SMC Em) determination
We previously found that SMC Em is more depolarized (less negative) in PRAs of hypoxic than in those of normoxic control piglets of the 3-day exposure group.19 For this study, we wanted to determine the effect on SMC Em of a longer, 10-day exposure to in vivo hypoxia. With our previously published methods, arteries from some control and hypoxic piglets of the 10-day exposure group were cannulated, pressurized, and equilibrated as described above. SMCs were impaled with microelectrodes filled with 3 M KCl and having impedances between 40 and 90 MΩ. Each microelectrode was connected to a high-impedance biological amplifier (Dagan) that was attached to a digital data acquisition system (PowerLab, ADI Instruments). By means of a micromanipulator, the electrode was advanced into an SMC from the adventitial side of the artery. Criteria for a successful impalement included an abrupt drop in potential to a new steady state value that is maintained for a minimum of 20 seconds and returns to the original baseline value when the microelectrode is removed from the SMC. Multiple successful impalements of at least three vascular SMCs were averaged for a single artery.
We also evaluated the influence of transmural pressure on SMC Em. To do this, we measured SMC Em in some control arteries that were pressurized to the transmural pressure used for hypoxic arteries, 18 mmHg, and in some hypoxic arteries that were pressurized to the transmural pressure used for control arteries, 11 mmHg. To assess the influence of the endothelium, SMC Em was measured in some arteries from control and hypoxic piglets after the endothelium was disrupted with air infusion. Effective functional disruption of the endothelium was verified by loss of relaxation to A23187. To evaluate the influence of Kv channels on SMC Em, in some other control and hypoxic arteries, we measured SMC Em before and 20–30 minutes after adding the Kv channel antagonist 4-aminopyridine (4-AP, 10−3 M) to the reservoir.
We also performed studies to determine whether ROS-removing agents might restore the SMC Em measured in hypoxic arteries to levels similar to those measured in control arteries. SMC Em was measured before and 20–30 minutes after adding to the reservoir either (1) a combination of a cell-permeable superoxide dismutase mimetic, M40403 (3 μg/mL), which dismutates superoxide to hydrogen peroxide (H2O2), and a cell-permeable H2O2-decomposing enzyme, polyethylene glycol-catalase (PEG-CAT, 250 U), or (2) apocynin (APO, 10−6 M) to inhibit NADPH oxidase (NOX), a potential enzymatic source of ROSs.
Cannulated-artery protocols to assess Kv channel function
To evaluate the effect of in vivo chronic hypoxia on Kv channel function, we measured changes in vessel diameter in response to pharmacologic blockade with the Kv channel antagonist 4-AP, 10−5 to 10−3 M, in PRAs from control and hypoxic piglets. The influence of the endothelium on 4-AP responses was assessed in PRAs after disruption of the endothelium with air infusion. Functional disruption of the endothelium was assessed as loss of relaxation to the calcium ionophore A23187. To assess the influence from ROSs on Kv channel function, a combination of M40403 (3 μg/mL) and PEG-CAT (250 U) was added to the reservoir and allowed to circulate for 20 minutes, after which changes in vessel diameter to cumulative doses of 4-AP were measured. To evaluate the influence of NOX, an enzymatic source of ROSs, on 4-AP responses, we measured changes in vessel diameter to 4-AP after adding the NOX inhibitor APO (10−6 M) to the reservoir. To assess whether exogenous ROSs might convert 4-AP responses in control arteries to levels similar to those in hypoxic arteries, we measured responses to 4-AP after adding the ROS generator menadione (10−5 M)33,34 to the reservoir of control arteries.
Cannulated-artery protocols to assess other K+ channel function
The influence of chronic hypoxia on another K+ channel family, KATP channels, was assessed by pharmacologic activation with levcromakalim. For these studies, vessel tone was elevated by 40%–50% with either U46619 or endothelin-1 prior to measurement of changes in vessel diameter in response to levcromakalim, 10−9 to 10−5 M.
Vessel viability was tested at the completion of all cannulated-artery studies by addition of U-46619. In some studies, vessel responses to the vehicle used for solubilization of each agent were evaluated.
Immunoblot analyses of Kv1.2, Kv1.5, and Kv2.1
Pulmonary arteries (20–500-μm diameter) were dissected from lungs of control and hypoxic piglets, frozen in liquid nitrogen, and stored at −80°C until use for immunoblot analysis.
For each protein, we performed preliminary studies with different amounts of total protein in order to determine the dynamic range of the immunoblot analysis. An amount of protein within the dynamic range of the immunoblot analysis for that protein (for Kv1.2 and Kv1.5 analyses we used 20-μg protein samples, and for Kv2.1 analysis we used 50-μg protein samples) was then used to compare protein abundance between homogenates of small pulmonary arteries from control and hypoxic piglets as follows.
Frozen samples of small pulmonary arteries (20–500-μm diameter) from control and hypoxic piglets were crushed under liquid N2 in a prechilled mortar and pestle into a fine powder, transferred to a tube containing homogenate buffer, and then sonicated with three 15-second pulses; care was taken not to foam the sample. The supernatant was centrifuged, and the protein concentration was determined by protein assay (Bradford). With our previously described methods, supernatants were applied to tris-glycine precast 8% polyacrylamide gels (Novex) so that equal amounts of protein were loaded. Electrophoresis was carried out, and the proteins were transferred from the gel to a nitrocellulose membrane (Novex). The membrane was incubated for 1 hour at room temperature in phosophate-buffered saline (PBS) containing 10% nonfat dried milk and 0.1% Tween-20 to block nonspecific protein binding. To detect the protein of interest, the nitrocellulose membrane was incubated overnight at 4°C with the primary antibody (Kv1.2, 1∶500 from Upstate; Kv1.5 and Kv2.1, 1∶500 from Alomone) diluted in PBS containing 0.1% Tween-20 and 1% nonfat dried milk (carrier buffer), followed by incubation for 1 hour at room temperature with a horseradish peroxidise-conjugated secondary antibody (Zymed) diluted in the carrier buffer. To visualize the antibody, the membranes were developed using enhanced chemiluminescence reagents (ECL, Amersham), and the chemiluminescent signal was captured on x-ray film (ECL Hyperfilm, Kodak). Similar procedures were followed to reprobe the membranes for β smooth muscle actin (Sigma). The bands for each protein were quantified via densitometry.
Statistics
Data are presented as means ± SEM. An unpaired t test or ANOVA with post hoc multiple-comparison test was used to compare data between groups as appropriate; 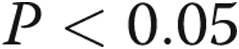 was considered significant.
was considered significant.
Materials
Concentrations for each drug listed in cannulated-artery protocols are expressed as final molar concentrations in the vessel bath. The 4-AP, PEG-CAT, and menadione were obtained from Sigma Chemical and were solubilized, respectively, in water and pH titrated to 7.4 with 12 N HCl, distilled water, and ethanol (EtOH). The M40403 was a generous gift from Activbiotics (Lexington, MA) and was solubilized in 26-mM NaHCO3 buffer. The apocynin was from EMD Biosciences and was solubilized in dimethyl sulfoxide (DMSO). The A23187 and U46619 were from Biomol and were solubilized, respectively, in DMSO and EtOH. The levcromakalim was from Tocris and was solubilized in DMSO.
Results
PASMCs are depolarized after 10 days of exposure to in vivo hypoxia
As shown in Table 1, regardless of transmural pressure, PASMC Em measurements in PRAs from piglets exposed to 10 days of hypoxia were less negative (depolarized) than those in PRAs from piglets exposed to 10 days of normoxia. These differences in PASMC Em between PRAs isolated after 10 days of normoxic versus hypoxic exposure (Table 1) mirrored the differences in PASMC Em measured in PRAs from piglets exposed to 3 days of normoxia (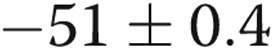 mV) versus 3 days of hypoxia (
mV) versus 3 days of hypoxia (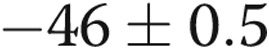 mV) in our previous study.19 Moreover, findings of PASMC Em in arteries from piglets exposed to 10 days of either normoxia or hypoxia were similar for air-infused and intact vessels (for normoxic, control arteries, PASMC Em was
mV) in our previous study.19 Moreover, findings of PASMC Em in arteries from piglets exposed to 10 days of either normoxia or hypoxia were similar for air-infused and intact vessels (for normoxic, control arteries, PASMC Em was 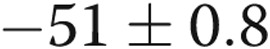 and
and 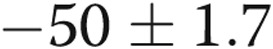 mV for intact [
mV for intact [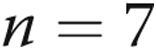 ] and air-infused [
] and air-infused [ ] arteries, respectively; for hypoxic arteries, PASMC Em was
] arteries, respectively; for hypoxic arteries, PASMC Em was 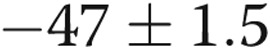 and
and 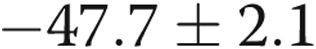 mV for intact [
mV for intact [ ] and air-infused [
] and air-infused [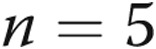 ] arteries, respectively). Thus, neither the presence of the endothelium nor differing transmural pressures explain differences in PASMC Em between control and hypoxic arteries. Furthermore, PASMC Em measurements in hypoxic PRAs were not significantly changed by treatment with ROS-removing agents: PASMC Em was
] arteries, respectively). Thus, neither the presence of the endothelium nor differing transmural pressures explain differences in PASMC Em between control and hypoxic arteries. Furthermore, PASMC Em measurements in hypoxic PRAs were not significantly changed by treatment with ROS-removing agents: PASMC Em was 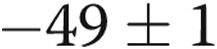 ,
, 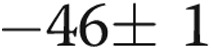 , and
, and 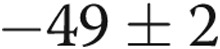 mV, respectively, for untreated arteries (
mV, respectively, for untreated arteries (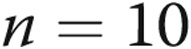 ), M40403+PEG-CAT-treated arteries (
), M40403+PEG-CAT-treated arteries (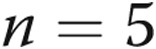 ), and apocynin-treated arteries (
), and apocynin-treated arteries (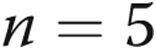 ).
).
Table 1.
Membrane potential (Em) determinations in 100–300-μm-diameter pulmonary arteries of normoxic and hypoxic piglets from the 10-day exposure group measured at transmural pressures of 11 and 18 mmHg
| Group | Transmural pressure, mmHg | n | Mean Em ± SEM, mV |
|---|---|---|---|
| 10-day normoxic | 11 | 23 | −55 ± 1 |
| 10-day normoxic | 18 | 5 | −58 ± 1.5 |
| 10-day hypoxic | 11 | 6 | −51 ± 0.8a |
| 10-day hypoxic | 18 | 18 | −49 ± 1a |
Significantly different (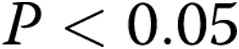 ) from normoxic piglets at both 11 and 18 mmHg.
) from normoxic piglets at both 11 and 18 mmHg.
Kv channels are dysfunctional after 10 days of exposure to in vivo hypoxia
Pharmacological blockade of Kv channels with 4-AP affected the Em of control arteries more than that of the hypoxic arteries. Specifically, as shown in Figure1A, the Em of the control arteries depolarized (became more positive) by 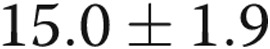 mV (from
mV (from 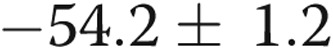 to
to 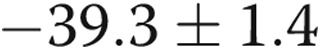 mV;
mV; 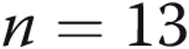 ) with 4-AP (10−3 M), whereas the Em of the hypoxic arteries depolarized only
) with 4-AP (10−3 M), whereas the Em of the hypoxic arteries depolarized only 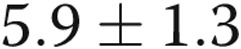 mV (from
mV (from 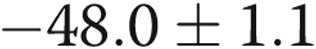 to
to 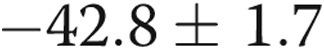 mV,
mV, 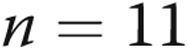 ) with 4-AP (10−3 M). These findings support Kv channel dysfunction in the PRAs of piglets exposed to 10 days of hypoxia.
) with 4-AP (10−3 M). These findings support Kv channel dysfunction in the PRAs of piglets exposed to 10 days of hypoxia.
Figure 1.
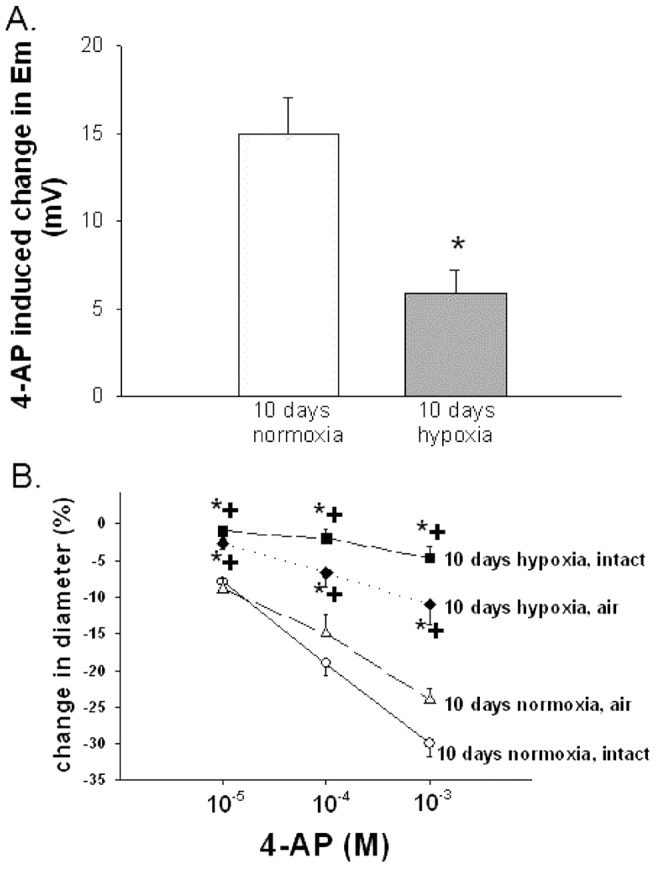
A, 4-aminopyridine (4-AP; 10−3 M)-induced change in membrane potential (Em) for arteries from normoxic ( ) and hypoxic (
) and hypoxic ( ) piglets from the 10-day exposure group. An asterisk indicates significant difference from normoxia (
) piglets from the 10-day exposure group. An asterisk indicates significant difference from normoxia ( ). B, 4-AP-induced changes in diameter for endothelium-intact and air-infused arteries from normoxic and hypoxic piglets from the 10-day exposure group (for normoxic arteries,
). B, 4-AP-induced changes in diameter for endothelium-intact and air-infused arteries from normoxic and hypoxic piglets from the 10-day exposure group (for normoxic arteries, 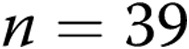 intact arteries and
intact arteries and 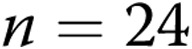 air-infused arteries; for hypoxic arteries,
air-infused arteries; for hypoxic arteries, 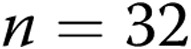 intact arteries and
intact arteries and 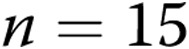 air-infused arteries). An asterisk indicates significant difference from normoxic intact arteries, and a plus sign (B) indicates significant difference from normoxic air-infused arteries (
air-infused arteries). An asterisk indicates significant difference from normoxic intact arteries, and a plus sign (B) indicates significant difference from normoxic air-infused arteries (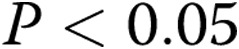 ). In both panels, values are mean ± SEM.
). In both panels, values are mean ± SEM.
Also supportive of Kv channel dysfunction, PRAs from piglets exposed to 10 days of hypoxia constricted less with 4-AP than did PRAs from comparable-age normoxic, control piglets (Fig. 1B). Of note, 4-AP responses were also lower in air-embolized PRAs from hypoxic arteries than in those from normoxic arteries (Fig. 1B), indicative that PASMCs and not endothelial cells are the cellular source of Kv dysfunction.
ROSs contribute to Kv dysfunction after 10 days of exposure to in vivo hypoxia
Treatments to remove ROSs and to inhibit NOX, a potential enzymatic source of ROSs, increased the constrictor response to 4-AP in PRAs from piglets of the 10-day hypoxic exposure group (Fig. 2). In contrast, treatments to remove ROSs and to inhibit NOX had inhibitory effects on the constrictor response to 4-AP in PRAs from comparable-age normoxic, control piglets (Fig. 2). Moreover, addition of the ROS generator menadione reduced constriction to 4-AP in normoxic, control arteries from the 10-day exposure group (Fig. 3). Thus, exogenous ROSs impair Kv channel function in control PRAs, and ROS-removing agents improve Kv channel function in PRAs following exposure to hypoxia for 10 days.
Figure 2.
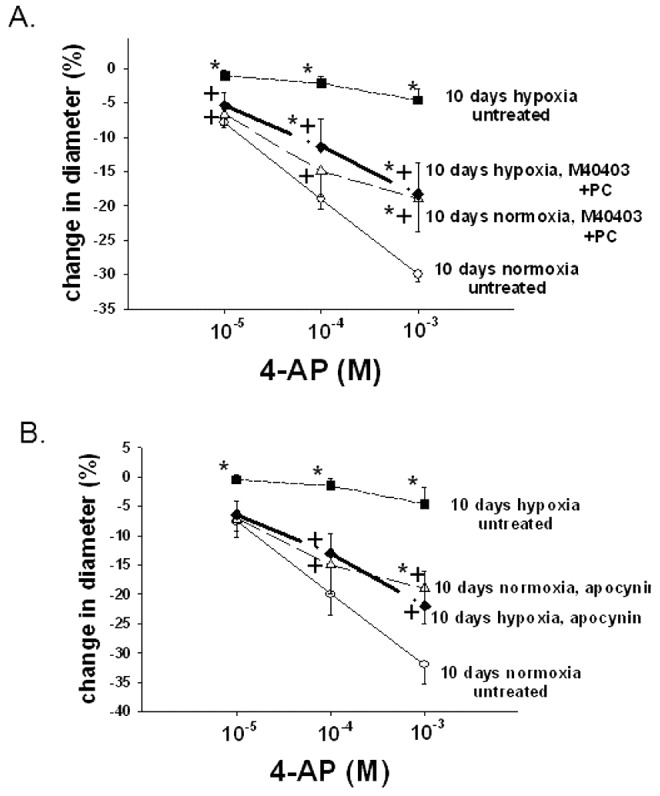
A, Effect of treatment with the superoxide dismutase mimetic M40403 and the H2O2-decomposing agent polyethylene glycol catalase (PEG-CAT) on responses to 4-aminopyridine (4-AP) in arteries from normoxic and hypoxic piglets of the 10-day exposure group (for normoxic arteries: 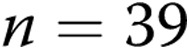 untreated arteries and
untreated arteries and 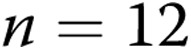 M40403+PEG-CAT-treated arteries; for hypoxic arteries:
M40403+PEG-CAT-treated arteries; for hypoxic arteries: 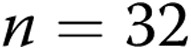 untreated arteries and
untreated arteries and 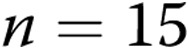 M40403+PEG-CAT-treated arteries). B, Effect of treatment with the NADPH oxidase inhibitor apocynin on responses to 4-AP in arteries from normoxic and hypoxic piglets of the 10-day exposure group (for normoxic arteries:
M40403+PEG-CAT-treated arteries). B, Effect of treatment with the NADPH oxidase inhibitor apocynin on responses to 4-AP in arteries from normoxic and hypoxic piglets of the 10-day exposure group (for normoxic arteries: 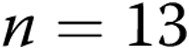 untreated arteries and
untreated arteries and 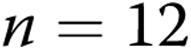 apocynin-treated arteries; for hypoxic arteries:
apocynin-treated arteries; for hypoxic arteries: 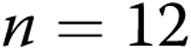 for untreated arteries and
for untreated arteries and 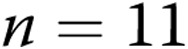 apocynin treated arteries). In both panels, values are mean ± SEM, an asterisk indicates a significant difference from untreated normoxic arteries, and a plus sign indicates a significant difference from untreated hypoxic arteries (
apocynin treated arteries). In both panels, values are mean ± SEM, an asterisk indicates a significant difference from untreated normoxic arteries, and a plus sign indicates a significant difference from untreated hypoxic arteries (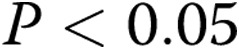 ).
).
Figure 3.
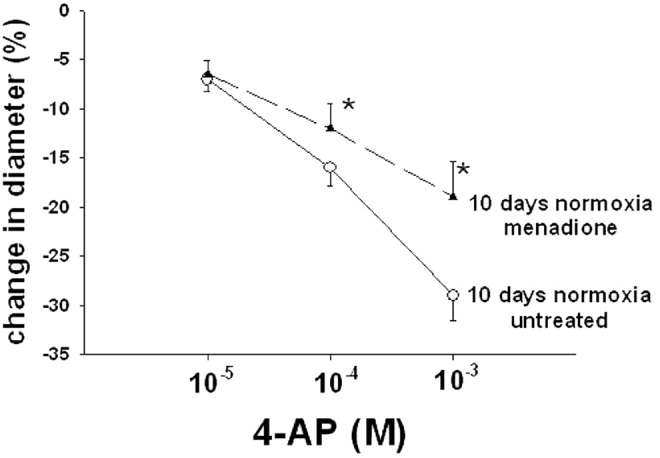
Effect of treatment with the ROS generator menadione on responses to 4-aminopyridine (4-AP) in arteries from normoxic piglets of the 10-day exposure group (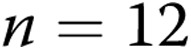 untreated arteries and
untreated arteries and 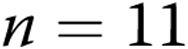 menadione-treated arteries). All values are mean ± SEM. An asterisk indicates a significant difference from untreated arteries (
menadione-treated arteries). All values are mean ± SEM. An asterisk indicates a significant difference from untreated arteries (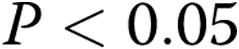 ).
).
ROSs do not contribute to Kv dysfunction after 3 days of exposure to in vivo hypoxia
We previously showed that functional activity of Kv channels was lower in PRAs of piglets exposed to 3 days of hypoxia than in those of comparable-age normoxic piglets.19 Based on our findings with PRAs from the 10-day group, we wanted to determine whether ROSs might contribute to Kv dysfunction in PRAs from piglets exposed to a shorter, 3-day duration of in vivo hypoxia. Unlike the longer duration of hypoxia (Fig. 2), treatments to remove ROSs or to inhibit NOX had no impact on constrictor responses to 4-AP in pulmonary arteries of piglets exposed to hypoxia for only 3 days (Fig. 4). Findings for control PRAs (Fig. 4) were similar to those from the older age group of piglets (Fig. 2).
Figure 4.
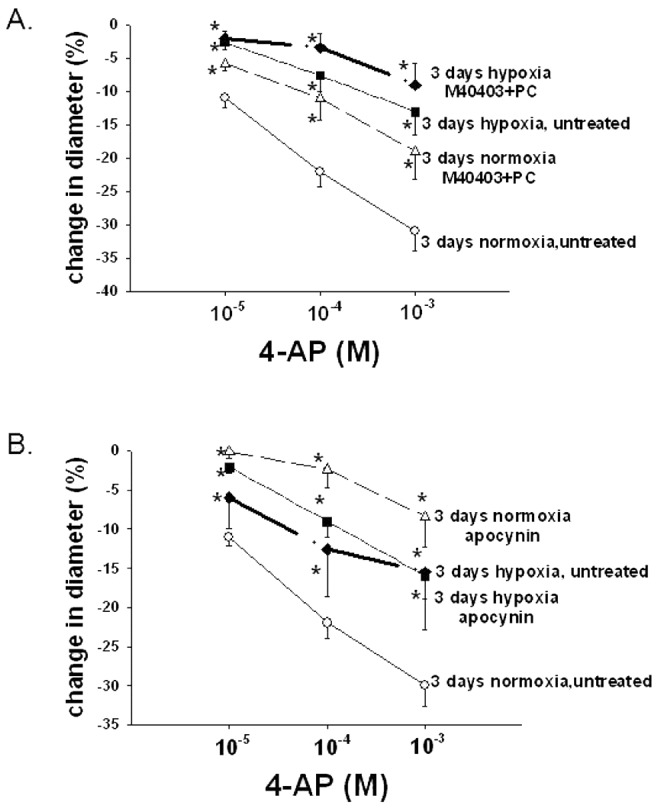
A, Effect of treatment with the superoxide dismutase mimetic M40403 and the H2O2-decomposing agent polyethylene glycol catalase (PEG-CAT) on responses to 4-aminopyridine (4-AP) in arteries from normoxic and hypoxic piglets of the 3-day exposure group (for normoxic arteries: 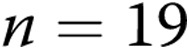 untreated arteries and
untreated arteries and 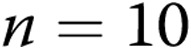 M40403+PEG-CAT-treated arteries; for hypoxic arteries:
M40403+PEG-CAT-treated arteries; for hypoxic arteries: 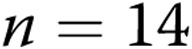 untreated arteries and
untreated arteries and 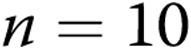 M40403+PEG-CAT-treated arteries). B, Effect of treatment with the NADPH oxidase inhibitor apocynin on responses to 4-AP in arteries from normoxic and hypoxic piglets of the 3-day exposure group (for normoxic arteries:
M40403+PEG-CAT-treated arteries). B, Effect of treatment with the NADPH oxidase inhibitor apocynin on responses to 4-AP in arteries from normoxic and hypoxic piglets of the 3-day exposure group (for normoxic arteries: 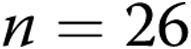 untreated arteries and
untreated arteries and 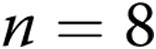 apocynin-treated arteries; for hypoxic arteries:
apocynin-treated arteries; for hypoxic arteries: 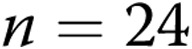 for untreated arteries and
for untreated arteries and 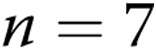 apocynin-treated arteries). In both panels, all values are mean ± SEM, and an asterisk indicates a significant difference from untreated normoxic arteries (
apocynin-treated arteries). In both panels, all values are mean ± SEM, and an asterisk indicates a significant difference from untreated normoxic arteries (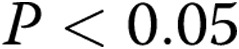 ).
).
Selective Kv channel proteins are downregulated after 10 days of exposure to in vivo hypoxia
ROS removal did not completely restore to control levels the Kv channel function in PRAs of piglets exposed to 10 days of hypoxia (Fig. 2A). We therefore investigated whether expression of Kv channels was reduced during the 10-day exposure to hypoxia. Immunoblot analyses for Kv1.2, Kv1.5, and Kv2.1 α subunits in small pulmonary artery homogenates from control and hypoxic piglets of the 10-day exposure group are shown in Figure5. As determined by densitometry, Kv1.2 expression was decreased in pulmonary arteries from hypoxic piglets (Fig. 5A), while expression levels of Kv1.5 and Kv2.1 were similar for hypoxic and control pulmonary arteries (Figs. 5B and 5C, respectively).
Figure 5.

A, Immunoblot results and corresponding densitometry for Kv1.2 α subunit protein relative to β actin in pulmonary arteries from normoxic and hypoxic piglets of the 10-day exposure group. The plus sign denotes positive control (rat brain). An asterisk indicates a significant different from normoxic piglets (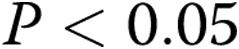 ). B, Immunoblot results and corresponding densitometry for Kv1.5 α subunit protein relative to β actin in pulmonary arteries from normoxic and hypoxic piglets of the 10-day exposure group. C, Immunoblot results and corresponding densitometry for Kv2.1 α subunit protein relative to β actin in pulmonary arteries from normoxic and hypoxic piglets of the 10-day exposure group. The plus sign denotes positive control (rat brain). In all panels, all values are mean ± SEM.
). B, Immunoblot results and corresponding densitometry for Kv1.5 α subunit protein relative to β actin in pulmonary arteries from normoxic and hypoxic piglets of the 10-day exposure group. C, Immunoblot results and corresponding densitometry for Kv2.1 α subunit protein relative to β actin in pulmonary arteries from normoxic and hypoxic piglets of the 10-day exposure group. The plus sign denotes positive control (rat brain). In all panels, all values are mean ± SEM.
KATP channel function is preserved following 3 or 10 days of exposure to in vivo hypoxia
We explored the possibility that in addition to alterations in Kv channel expression and function, exposure to in vivo hypoxia might impair the function of other K+ channels. As shown in Figure6, diameters of both control and hypoxic arteries from both 3- and 10-day exposure groups responded similarly to the KATP channel agonist levcromakalim.
Figure 6.
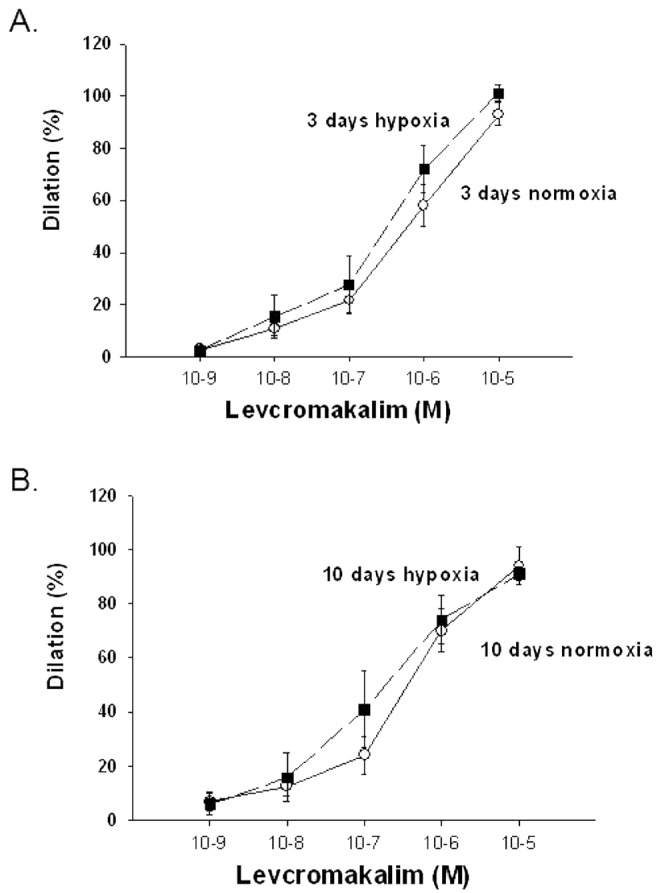
A, Levcromakalim-induced changes in diameter in arteries with elevated tone from normoxic (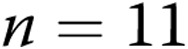 ) and hypoxic (
) and hypoxic (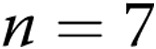 ) piglets of the 3-day exposure group. B, Levochromakalin-induced changes in diameter in arteries with elevated tone from normoxic (
) piglets of the 3-day exposure group. B, Levochromakalin-induced changes in diameter in arteries with elevated tone from normoxic (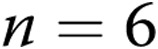 ) and hypoxic (
) and hypoxic (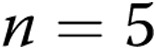 ) piglets of the 10-day exposure group. In both panels, all values are mean ± SEM.
) piglets of the 10-day exposure group. In both panels, all values are mean ± SEM.
Discussion
In this study, we show that PASMCs are depolarized and that 4-AP-sensitive K+ channel function is diminished in PRAs of newborn piglets exposed to in vivo hypoxia for 10 days. We previously reported similar abnormalities in PASMC membrane properties in PRAs from piglets exposed to in vivo hypoxia for 3 days.19 Thus, our new findings support and extend the link between impairments in Kv channel function and the pathogenesis of chronic hypoxia–induced pulmonary hypertension in a newborn-animal model. Important new findings in this study are that ROSs contribute to the K+ channel dysfunction that occurs after the longer 10-day exposure to in vivo hypoxia, whereas ROSs do not play a prominent role in the K+ channel dysfunction that develops within 3 days of exposure to in vivo hypoxia. Taken together, these findings indicate that ROS-removing agents may improve 4-AP- dependent K+ channel function in the pulmonary circulation at more progressive, but not earlier, stages of pulmonary hypertension.
We are not the first to provide evidence that ROSs contribute to K+ channel dysfunction.20,25,28,35,36 More than 10 years ago, ROSs were shown to contribute to K+ channel dysfunction in the systemic circulation.25,35,36 More recently, two studies provided evidence that ROSs contribute to K+ channel dysfunction in the pulmonary circulation.20,28 Specifically, in a fetal ductal-ligation lamb model of persistent pulmonary hypertension of the newborn (PPHN), the O2 scavenger tiron was shown to improve both Kv currents and 4-AP-dependent K+ channel responses in pulmonary arteries from PPHN lambs.20 In a series of studies evaluating the effect of in vitro hypoxia, investigators found that PASMCs cultured for 72 hours under hypoxic conditions exhibited reductions in Kv channel currents that were restored by treatments to remove NOX-derived ROSs.28 We add to the literature and are the first to show that the ability of ROS-removing agents to improve 4-AP-dependent K+ channel function in the pulmonary circulation is dependent on the length of in vivo exposure to chronic hypoxia and the stage of pulmonary hypertension.
The mechanisms by which ROSs impair K+ channel function are not yet clear. One possibility is that ROSs directly interact with and modify Kv channel-gating properties via the oxidation of amino acid residues within the channel.23,24,37 Another possibility is that ROSs interact with nitric oxide (NO) to form peroxynitrite, which, in turn, causes nitration and functional impairment of Kv channel proteins.24 In addition to promoting nitration, NO has been shown to inhibit Kv channel current by S-nitrosylation.38 It should also be noted that Kv channel function is inhibited by elevated cytosolic calcium levels.39,40 Hence, the ability to trigger intracellular calcium release could be another mechanism by which ROSs contribute to impaired Kv channel function.41,42 Relevant to this in our study ROS-removing agents did not restore the PASMC Em of hypoxic arteries to control levels. Thus, our findings are consistent with the possibility that ROSs, rather than being an influence on PASMC Em, alter Kv-mediated vascular contraction via changes in Ca2+ sensitization.
Phosphorylation of channel molecules also regulates membrane channel activity.43-45 ROSs could indirectly affect Kv channel protein function by mediating changes in phosphatases and kinases that in turn modulate phosphorylation of channel proteins.11,26,46 For example, activation of protein kinase C (PKC) by H2O2 has been shown to underlie Kv channel inhibition.47,48 Along this line, PKC has been shown to be activated in the pulmonary vasculature of chronically hypoxic newborn piglets.49 It is possible that during progressive exposure to chronic hypoxia, specific PKC isozymes are activated by ROSs and contribute to Kv channel dysfunction.
The sources of ROSs that impair Kv channel function merit discussion. Our findings with the pharmacologic NOX inhibitor apocynin suggest that NOX-family enzymes could be one source of ROSs that contributes to inhibition of Kv channel function. Further supportive of a role for NOX-derived ROSs, another group of investigators found that both apocynin and NOX4 silencing RNA reversed the decrease in Kv current that occurred in PASMCs cultured under hypoxic conditions for 72 hours.28 The mitochondrial electron transport chain is another possible source of ROSs.48 Indeed, mitochondria-derived ROSs have been shown to activate NOX and create a positive-feedback mechanism that augments intracellular ROS generation.48 A role for mitochondria-derived ROSs in modulating K+ channels is supported by studies showing that knockdown of mitochondrial superoxide dismutase downregulates Kv1.5 channel protein expression in PASMCs from normal rats.50
It is of interest that we found that when exposed to an ROS-generating agent, PRAs from normoxic piglets exhibited evidence of K+ channel dysfunction. Yet our findings also suggest that endogenous ROSs are important for 4-AP-sensitive Kv channel function in normoxic PRAs. The mechanism underlying this apparent differential modulation of Kv channels by endogenous and exogenous ROSs is not known. However, ROSs have been shown to both activate21,51,52 and inhibit22,46 Kv channels. Moreover, although they contribute to disease processes, ROSs also serve as physiologically significant signaling molecules under normal, nondiseased conditions.53,54 The precise mechanisms underlying the change in role of ROSs from facilitating normal intracellular signaling to mediating detrimental effects are not yet understood.
Another finding in this study is that ROS removal did not completely restore Kv channel function in PRAs from the hypoxic piglets. Therefore, other mechanisms of Kv impairment must also be involved. Both our findings and those of others support a contribution from reduced Kv channel expression.13,14,16,17,19 Which Kv channels are reduced by hypoxia appears to differ, depending on the species studied as well as the duration and type (in vitro vs. in vivo) of hypoxic exposure.13,14,16,17,19 We evaluated protein expression of Kv1.2, Kv1.5, and Kv2.1 because homo- or heterotetramers composed of these ion channels have been identified as being oxygen sensitive and having a role in physiological responses of PASMC to hypoxia.55 The potential lack of specificity of available antibodies must be considered in interpretation of Kv channel protein expression findings. In addition, although other channels may also be oxygen sensitive, lack of available antibodies for swine precluded our ability to evaluate all of them. Others have shown that transcriptional regulation of K+ channel expression in chronic hypoxia in adult animals and in adult humans with pulmonary arterial hypertension involves hypoxia-inducible factor (HIF).56,57 In fact, the reduction in Kv channel expression in PASMCs of chronically hypoxic rats and mice is the result of HIF-1-dependent induction of endothelin-1.58 Some K+ channels appear to be regulated by translational control rather than by changes in transcription or degradation changes.59
Other non-ROS-mediated mechanisms of K+ channel dysfunction include abnormalities in Kv channel trafficking/cell surface expression60-62 and modulation of Kv channel activity63-65 by modification of disulfide bridges in the channel protein.66 Cellular localization of mitochondria, as well as mitochondria-dependent changes in intracellular Mg2+ and ATP concentrations, has also been shown to modulate Kv channel function.67,68 The role of these mechanisms in the K+ channel dysfunction found in our neonatal model of chronic hypoxia–induced pulmonary hypertension has yet to be explored.
There are some important therapeutic implications from our findings. Restoration of Kv channels has been shown to ameliorate pulmonary hypertension.13 Treatments that remove ROSs have also been shown to prevent or cause regression of pulmonary hypertension in adult-animal models.50 Our findings raise the possibility that ROS-removing agents might improve Kv channel function and thereby prevent the progression, but perhaps not the initial stages, of chronic hypoxia–induced pulmonary hypertension in newborns. Indeed, supportive of a role for ROS removal as a treatment strategy during progression of hypoxia-induced pulmonary hypertension, we recently found that NOX1 expression and superoxide production are greater in PRAs of piglets exposed to 10 days of hypoxia than in those of piglets exposed to 3 days.69 Of note, our findings that ROSs are important in Kv channel function in normoxic vessels indicates that extreme caution should be used when considering ROS-removing therapies. Moreover, it is important to consider that the effectiveness of dilator therapies can be affected by alterations in downstream mechanisms, including activation of specific K+ channels that mediate their response. Thus, our finding of preserved KATP channel function in the face of impaired Kv function has important implications for designing therapies to lower pulmonary vascular resistance in infants with pulmonary hypertension associated with chronic hypoxia.
In summary, the findings in this study continue to support the growing body of evidence that changes in Kv channel function and concomitant alterations in PASMC Em contribute to the pathogenesis of progressive pulmonary hypertension, particularly in conditions associated with chronic hypoxia.10-12,19,20 Our work has potential clinical relevance. Rather than a preventive, prophylactic approach, treatments for progressive, neonatal pulmonary hypertension are often initiated late in the disease process, typically after a diagnosis of right heart dysfunction and cor pulmonale is made. With this in mind, the therapeutic implications of our findings about the effect of ROS removal on Kv channel function merit further study in in vivo models of progressive neonatal pulmonary hypertension. Our findings suggest that once the diagnosis is made, ROS-removing agents could be used to improve Kv function and ameliorate the progressive development of pulmonary hypertension in infants suffering from conditions associated with chronic or intermittent periods of hypoxia. This possibility warrants future investigation, as it could provide a much-needed, novel approach to improving outcomes for an understudied patient population in desperate need of effective therapies.
Source of Support: This work was supported by RO1 HL68572 (CDF) and RO1 HL97566 (CDF).
Conflict of Interest: None declared.
References
- 1.Firth AL, Remillard CV, Platoshyn O, Fantozzi I, Ko EA, Yuan JX-J. Functional ion channels in human pulmonary arterial cells: voltage-dependent cation channels. Pulm Circ 2011;1:48–71. [DOI] [PMC free article] [PubMed]
- 2.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol 1995;268:C799–C822. [DOI] [PubMed]
- 3.Yuan X-J. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res 1995;77:370–378. [DOI] [PubMed]
- 4.Archer SL, Souli E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 1998;101:2319–2330. [DOI] [PMC free article] [PubMed]
- 5.Gurney AM, Osipenko ON, MacMillan D, McFarlane KM, Tate RJ, Kempsill FEJ. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ Res 2003;93:957–964. [DOI] [PubMed]
- 6.Morecroft I, Murray A, Nilsen M, Gurney AM, MacLean MR. Treatment with the Kv7 potassium channel activator flupirtine is beneficial in two independent mouse models of pulmonary hypertension. Br J Pharmacol 2009;157:1241–1249. [DOI] [PMC free article] [PubMed]
- 7.Platoshyn O, Remillard CV, Fantozzi I, Mandegar M, Sison TT, Zhang S, Burg E, Yuan JX-J. Diversity of voltage-dependent K+ channels in human pulmonary artery smooth muscle cells. Am J Physiol 2004;287:L226–L238. [DOI] [PubMed]
- 8.Tang B, Li Y, Nagaraj C, Morty RE, Gabor S, Stacher E, Voswinckel R, et al. Endothelin-1 inhibits background two-pore domain channel TASK-1 in primary human pulmonary artery smooth muscle cells. Am J Resp Cell Mol Biol 2009;41:476–483. [DOI] [PubMed]
- 9.Yuan X-J, Wang J, Juhaszova M, Golovina VA, Rubin LJ. Molecular basis and function of voltage-gated K+ channels in pulmonary arterial smooth muscle cells. Am J Physiol 1998;274:L621–L635. [DOI] [PubMed]
- 10.Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary hypertension for clinicians: new concepts and experimental therapies. Circulation 2010;121:2045–2066. [DOI] [PMC free article] [PubMed]
- 11.Ko EA, Park WS, Firth AL, Kim N, Yuan JX-J, Han J. Pathophysiology of voltage-gated K+ channels in vascular smooth muscle cells: modulation by protein kinases. Prog Biophys Mol Biol 2010;103:95–101. [DOI] [PubMed]
- 12.Michelakis ED, Weir EK. The pathobiology of pulmonary hypertension: smooth muscle cells and ion channels. Clin Chest Med 2001;22:419–432. [DOI] [PubMed]
- 13.Pozeg ZI, Michelakis ED, McMurtry MS, Thébaud B, Wu X-C, Dyck JRB, Hashimoto K, et al. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 2003;107:2037–2044. [DOI] [PubMed]
- 14.Reeve HL, Michelakis E, Nelson DP, Weir EK, Archer SL. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. J Appl Physiol 2001;90:2249–2256. [DOI] [PubMed]
- 15.Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, Nicholson A, et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary hypertension. Am J Physiol 2007;292:C1837–C1853. [DOI] [PubMed]
- 16.Wang J, Juhaszova M, Rubin LL, Yuan X-J. Hypoxia inhibits gene expression of voltage-gated K+ channel a subunits in pulmonary artery smooth muscle cells. J Clin Investig 1997;100:2347–2353. [DOI] [PMC free article] [PubMed]
- 17.Wang J, Weigang L, Wang W, Sylvester JT, Shimoda LA. Chronic hypoxia inhibits Kv channel gene expression in rat distal pulmonary artery. Am J Physiol 2005;288:L1049–L1058. [DOI] [PubMed]
- 18.Yuan JX-J, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 1998;98:1400–1406. [DOI] [PubMed]
- 19.Fike CD, Kaplowitz MR, Zhang Y, Madden JA. Voltage-gated K+ channels at an early stage of chronic hypoxia–induced pulmonary hypertension in newborn piglets. Am J Physiol 2006;291:L1169–L1176. [DOI] [PubMed]
- 20.Konduri GG, Bakhutashvili I, Eis A, Gauthier KM. Impaired voltage gated potassium channel responses in a fetal lamb model of persistent pulmonary hypertension of the newborn. Pediatr Res 2009;66:289–294. [DOI] [PMC free article] [PubMed]
- 21.Caouette D, Dongmo C, Bérubé J, Fournier D, Daleau P. Hydrogen peroxide modulates the Kv1.5 channel expressed in a mammalian cell line. Naunyn-Schmiedeberg’s Arch Pharmacol 2003;368:479–486. [DOI] [PubMed]
- 22.Cogolludo A, Frazziano G, Cobeño L, Moreno L, Lodi F, Villamor E, Tamargo J, Perez-Vizcaino F. Role of reactive oxygen species in Kv channel inhibition and vasoconstriction induced by TP receptor activation in rat pulmonary arteries. Ann NY Acad Sci 2006;1091:41–51. [DOI] [PubMed]
- 23.Duprat F, Guillemare E, Romey G, Fink M, Lesage F, Lazdunski M, Honoré E. Susceptibility of cloned K+ channels to reactive oxygen species. Proc Natl Acad Sci USA 1995;92:11796–11800. [DOI] [PMC free article] [PubMed]
- 24.Li H, Gutterman DD, Rusch NJ, Bubolz A, Liu Y. Nitration and functional loss of voltage-gated K+ channels in rat coronary microvessels exposed to high glucose. Diabetes 2004;53:2436–2442. [DOI] [PubMed]
- 25.Liu Y, Terata K, Rusch NJ, Gutterman DD. High glucose impairs voltage-gated K+ channel current in rat small coronary arteries. Circ Res 2001;89:146–152. [DOI] [PubMed]
- 26.Mason HS, Latten MJ, Godoy LD, Horowitz B, Kenyon JL. Modulation of Kv1.5 currents by protein kinase A, tyrosine kinase, and protein tyrosine phosphatase requires an intact cytoskeleton. Mol Pharmacol 2002;61:285–293. [DOI] [PubMed]
- 27.Michelakis ED, Rebeyka I, Wu X, Nsair A, Thébaud B, Hashimoto K, Dyck JRB, et al. O2 sensing in the human ductus arteriosus: regulation of voltage-gated K+ channels in smooth muscle cells by a mitochondrial redox sensor. Circ Res 2002;91:478–486. [DOI] [PubMed]
- 28.Mittal M, Gu XQ, Pak O, Pamenter ME, Haag D, Fuchs DB, Schermuly RT, et al. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med 2012;52:1033–1042. [DOI] [PubMed]
- 29.Park MK, Bae YM, Lee SH, Ho W, Earm YE. Modulation of voltage-dependent K+ by redox potential in pulmonary and ear arterial smooth muscle cells of the rabbit. Pflueg Arch Eur J Physiol 1997;434:764–771. [DOI] [PubMed]
- 30.Dennis KE, Aschner JL, Milatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, Fike CD. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborns. Am J Physiol 2009;297:L596–L607. [DOI] [PMC free article] [PubMed]
- 31.Fike CD, Kaplowitz MR. Chronic hypoxia alters nitric oxide-dependent pulmonary vascular responses in lungs of newborn pigs. J Appl Physiol 1996;81:2078–2087. [DOI] [PubMed]
- 32.Fike CD, Pfister SL, Kaplowitz MR, Madden JA. Cyclooxygenase contracting factors and altered pulmonary vascular responses in chronically hypoxic newborn piglets. J Appl Physiol 2002;92:67–74. [DOI] [PubMed]
- 33.Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, et al. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem 2006;281:40485–40492. [DOI] [PubMed]
- 34.Sampath V, Radish AC, Eis AL, Broniowska K, Hogg N, Konduri GG. Attenuation of lipopolysaccharide-induced oxidative stress and apoptosis in fetal pulmonary artery endothelial cells by hypoxia. Free Radic Biol Med 2009;46:663–671. [DOI] [PMC free article] [PubMed]
- 35.Erdos B, Simandle SA, Snipes JA, Miller AW, Busija DW. Postassium channel dysfunction in cerebral arteries of insulin-resistant rats is mediated by reactive oxygen species. Stroke 2004;35:964–969. [DOI] [PubMed]
- 36.Liu Y, Gutterman DD. Oxidative stress and potassium channel function. Clin Exp Pharmacol Physiol 2002;29:305–311. [DOI] [PubMed]
- 37.Ciorba MA, Heinemann SH, Weissbach H, Brot N, Hoshi T. Regulation of voltage-dependent K+ channels by methionine oxidation: effect of nitric oxide and vitamin C. FEBS Lett 1999;442:48–52. [DOI] [PubMed]
- 38.Núñez L, Vaquero M, Gómez R, Caballero R, Mateos-Cáceres P, Macaya C, Iriepa I, et al. Nitric oxide blocks hKv1.5 channels by S-nitrosylation and by a cyclic GMP-dependent mechanism. Cardiovasc Res 2006;72:80–89. [DOI] [PubMed]
- 39.Cox RH, Petrou S. Ca2+ influx inhibits voltage-dependent and augments Ca2+-dependent K+ currents in arterial myocytes. Am J Physiol 1999;277:C51–C63. [DOI] [PubMed]
- 40.Post JM, Gelband CH, Hume JR. [Ca2+]i inhibition of K+ channels in canine pulmonary artery: novel mechanism for hypoxia-induced membrane depolarization. Circ Res 1995;77:131–139. [DOI] [PubMed]
- 41.Lin M-J, Yang X-R, Cao Y-N, Sham JSK. Hydrogen peroxide-induced Ca2+ mobilization in pulmonary arterial smooth muscle cells. Am J Physiol 2007;292:L1598–L1608. [DOI] [PubMed]
- 42.Waypa GB, Marks JD, Mack MM, Boriboun C, Mungai PT, Schumacker PT. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary artery myocytes. Circ Res 2002;91:719–726. [DOI] [PubMed]
- 43.Kume H, Takai A, Tokuno H, Tomita T. Regulation of Ca2+-dependent K+-channel activity in tracheal myoctes by phosphorylation. Nature 1989;341:152–154. [DOI] [PubMed]
- 44.Park K, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science 2006;313:976–979. [DOI] [PubMed]
- 45.Robertson BE, Schubert R, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am J Physiol 1993;265:C299–C303. [DOI] [PubMed]
- 46.Perez-Vizcaino F, Cogolludo A, Moreno L. Reactive oxygen species signaling in pulmonary vascular smooth muscle. Respir Physiol Neurobiol 2010;174:212–220. [DOI] [PubMed]
- 47.Pourmahram GE, Snetkov VA, Shaifta Y, Drndarski S, Knock GA, Aaronson PI, Ward JPT. Constriction of pulmonary artery by peroxide: role of Ca2+ release and PKC. Free Radic Biol Med 2008;45:1468–1476. [DOI] [PubMed]
- 48.Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS, Wang YX. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCε signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med 2008;45:1223–1231. [DOI] [PMC free article] [PubMed]
- 49.Berkenbosch JW, Baribeau J, Ferretti E, Perreault T. Role of protein kinase C and phosphatases in the pulmonary vasculature of neonatal piglets. Crit Care Med 2001;29:1229–1233. [DOI] [PubMed]
- 50.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JRB, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 2010;121:2661–2671. [DOI] [PMC free article] [PubMed]
- 51.Schach C, Xu M, Platoshyn O, Keller SH, Yuan JX-J. Thiol oxidation causes pulmonary vasodilation by activating K+ channels and inhibiting store-operated Ca2+ channels. Am J Physiol 2006;292:L685–L698. [DOI] [PubMed]
- 52.Snetkov VA, Smirnov SV, Kua J, Aaronson PI, Ward JPT, Knock GA. Superoxide differentially controls pulmonary and systemic vascular tone through multiple signalling pathways. Cardiovasc Res 2011;89:214–224. [DOI] [PMC free article] [PubMed]
- 53.Droge W. Free radicals in the physiological control of cell function. Physiol Rev 2002;82:47–95. [DOI] [PubMed]
- 54.Janssen-Heininger YMW, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 2008;45:1–17. [DOI] [PMC free article] [PubMed]
- 55.Moudgil R, Michelakis ED, Archer SL. The role of K+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 2006;13:615–632. [DOI] [PubMed]
- 56.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, et al. An abnormal mitochondrial-hypoxia inducible factor-1α–Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats. Circulation 2006;113:2630–2641. [DOI] [PubMed]
- 57.Shimoda LA, Manalo DJ, Sham JSK, Semenza GL, Sylvester JT. Partial HIF-1α deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol 2001;281:L202–L208. [DOI] [PubMed]
- 58.Whitman EM, Pisarcik S, Luke T, Fallon M, Wang J, Sylvester JT, Semenza GL, Shimoda LA. Endothelin-1 mediates hypoxia-induced inhibition of voltage-gated K+ channel expression in pulmonary arterial myocytes. Am J Physiol 2008;294:L309–L318. [DOI] [PubMed]
- 59.Ficker E, Dennis AT, Wang L, Brown AM. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel hERG. Circ Res 2003;92:e87–e100. [DOI] [PubMed]
- 60.Cogolludo A, Moreno L, Lodi F, Frazziano G, Cobeño L, Tamargo J, Perez-Vizcaino F. Serotonin inhibits voltage-gated K+ currents in pulmonary artery smooth muscle cells: role of 5-HT2A receptors, caveolin-1, and Kv1.5 channel internalization. Circ Res 2006;98:931–938. [DOI] [PubMed]
- 61.Nanduri J, Bergson P, Wang N, Ficker E, Prabhakar NR. Hypoxia inhibits maturation and trafficking of hERG K+ channel protein: role of Hsp90 and ROS. Biochem Biophys Res Commun 2009;388:212–216. [DOI] [PMC free article] [PubMed]
- 62.Shi G, Nakahira K, Hammond S, Rhodes KJ, Schechter LE, Trimmer JS. β subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron 1996;16:843–852. [DOI] [PubMed]
- 63.Bahring R, Milligan CJ, Vardanyan V, Engeland B, Young BA, Dannenberg J, Waldschütz R, Edwards JP, Wray D, Pongs O. Coupling of voltage-dependent potassium channel inactivation and oxidoreductase active site of Kvβ subunits. J Biol Chem 2001;276:22923–22929. [DOI] [PubMed]
- 64.Strupp M, Quasthoff S, Mitrovic N, Grafe P. Glutathione accelerates sodium channel inactivation in excised axonal membrane patches. Pflueg Arch Eur J Physiol 1992;421:283–285. [DOI] [PubMed]
- 65.Yuan JX-J, Tod ML, Rubin LJ, Blaustein MP. Deoxyglucose and reduced glutathione mimic the effects of hypoxia on K+ and Ca2+ conductances in pulmonary artery cells. Am J Physiol 1994;267:L52–L63. [DOI] [PubMed]
- 66.Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M. Regulation of fast inactivation of cloned mammalian Ik (A) channels by cysteine oxidation. Nature 1991;352:711–714. [DOI] [PubMed]
- 67.Firth AL, Yuill KH, Smirnov SV. Mitochondria-dependent regulation of Kv currents in rat pulmonary artery smooth muscle cells. Am J Physiol 2008;295:L61–L70. [DOI] [PMC free article] [PubMed]
- 68.Firth AL, Gordienko DV, Yuill KH, Smirnov SV. Cellular localization of mitochondria contribute to Kv channel-mediated regulation of cellular excitability in pulmonary but not mesenteric circulation. Am J Physiol 2009;296:L347–L360. [DOI] [PMC free article] [PubMed]
- 69.Fike CD, Dikalova A, Slaughter JC, Kaplowitz MR, Zhang Y, Aschner JL. Reactive oxygen species-reducing strategies improve pulmonary arterial responses to nitric oxide in piglets with chronic hypoxia-induced pulmonary hypertension. Antioxid Redox Signal 2013;18:1727–1738. [DOI] [PMC free article] [PubMed]