

Abstract Abstract
Over the past 2 decades, major advances in our understanding of pulmonary arterial hypertension (PAH) have led to the development of new targeted therapeutics and management strategies that have provided benefits to patients with this devastating disease. Despite such improvements, no therapies are curative, and PAH remains a progressive disease associated with high morbidity and suboptimal survival in many patients. Clinical research in PAH is currently at a crossroads. To move forward, not only are new therapies needed, but novel approaches to clinical trial design are also required. Trials should be designed to assess the longer-term benefits of investigational therapies in what has become a chronic disease. Moreover, there is a need to consider moving away from short-term trials that use markers such as the 6-minute walk distance as a measure of exercise capacity as primary end points to longer-term, event-driven trials with composite end points made up of clinically relevant measures that better reflect the ultimate goals of reducing morbidity and mortality. A shift in trial design may also be useful in overcoming some of the muted results from recent pivotal phase III studies of combination therapy by allowing the potential of these regimens to be more comprehensively assessed.
Keywords: pulmonary arterial hypertension; mortality; morbidity; surrogate markers; clinical worsening, macitentan.
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disorder with high morbidity and mortality in persons of all ages. The disease is characterized by endothelial cell dysfunction, smooth muscle cell hypertrophy, platelet dysfunction leading to in situ thrombosis, and abnormal expression of vasoactive mediators resulting in pulmonary vascular remodeling. Restriction in blood flow through the pulmonary vascular bed results from increased pulmonary vascular resistance and pulmonary arterial pressures; over time, increased workload on the right ventricle leads to right heart failure.1,2 Until the late 1990s, treatment options for patients with PAH had been limited to general supportive measures and lung or heart-lung transplantation. In the National Institutes of Health (NIH) registry of the 1980s, median survival after a diagnosis of idiopathic PAH was less than 3 years, and only one-third of patients survived for 5 years; survival in children was often less than 1 year after diagnosis.3
In the past 2 decades, however, there have been major advances in our understanding of PAH that have led to clearer categorization of PAH subtypes and the development of PAH-specific therapies that target pathways implicated in its underlying pathogenesis.2 Advances in management and the use of these therapies have resulted in significant improvements and better quality of life (QoL) for patients with PAH, although none of these therapies is considered curative.4-6
Current approved therapies for PAH target 1 of 3 main pathways (prostacyclin, endothelin, or nitric oxide) thought to be involved in the vasoconstriction, vascular endothelial cell proliferation, smooth muscle cell proliferation, and endothelial dysfunction characteristic of the disease.2,7 Phase III trials of these therapies have only shown a modest improvement in hemodynamic characteristics, when measured, but have still reported significant increases in exercise capacity, lessening of symptoms, and better QoL compared with placebo. However, rigorous long-term data for reductions in morbidity and mortality, which are the ultimate goals of treatment, are lacking and have only been inferred through open-label observational studies and registries.2 This is partially attributable to the limited capabilities of available therapies, but it also reflects clinical trial design in PAH, which has predominantly involved short-term studies using exercise capacity, as measured by the 6-minute walk distance (6MWD), as a primary end point. However, such trials provide little assurance of long-term safety and efficacy.
Contemporary outcomes with modern PAH management
Recent advances in PAH treatment have, in many ways, mirrored those seen 2 decades ago in another chronic and debilitating cardiovascular condition, chronic left ventricular heart failure. In heart failure, goals of therapy have evolved from symptomatic relief, through short-term hemodynamic improvements and intermediate-term improvement in functional capacity and exercise capacity (provided by vasodilators, nitrates, and digoxin), to the ultimate objective of long-term improvement in morbidity and survival, as seen in trials of angiotensin-converting enzyme inhibitors and beta blockers.8 Reflecting this, morbidity and mortality has been the standard response variable in heart failure studies for many years; however, softer end points, such as exercise capacity or functional class, have not been ignored, because they are still clinically relevant in the early stages of the disease and are likely to be very relevant to patients.
There is an ever-growing list of PAH-targeted therapies available that, when administered as monotherapy or as add-on combination therapy in randomized clinical trials, have shown short-term (12–16 weeks) improvements in exercise capacity, hemodynamic characteristics, functional class, and QoL. Although a meta-analysis of these trials suggested a mortality benefit, the effect was heavily driven by a minority of prostacyclin-based studies.9 Furthermore, contemporary registries provide updated and improved survival estimates, when compared with the NIH registry of the 1980s.3 A French registry demonstrated a survival rate of 67% at 3 years in patients with idiopathic, familial, or anorexigen-associated PAH.10 Similarly, the US-based Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL), which enrolled patients with PAH between 2006 and 2009 and also included associated PAH cases, estimated survival rates from time of diagnosis of 68%, 57%, and 49% at 3, 5, and 7 years, respectively (Fig. 1).6 It is important to note that these registries include incident and prevalent cases, which introduces a “survivor bias,” because prevalent cases must survive the period from diagnosis to registry enrollment to be included in the cohort, thus inflating overall survival estimates. To state this another way, patients who were too sick to survive the time from diagnosis to enrollment could never be included in the registry. More recently, a registry of incident, treatment-naive cases of largely idiopathic PAH from the United Kingdom and Ireland reported survival rates of 73% at 3 years and 61% at 5 years, reflecting survival in a population with newly diagnosed PAH.11 Furthermore, a subgroup analysis of REVEAL also provides evidence to support the notion that contemporary practices, which include use of targeted PAH therapies, have improved survival. REVEAL investigators assembled a cohort of patients with idiopathic/familial PAH, weighted to match the idiopathic/heritable cohort from the 1980s NIH registry (conducted in an era devoid of targeted PAH therapies), who subsequently initiated contemporary PAH-specific therapy within 6 months of diagnosis.6 Observed survival was considerably better than predicted survival based on the validated NIH equation; 78% versus 46%, 70% versus 35%, and 66% versus 32% at 3 years, 5 years, and 7 years after diagnosis, respectively.3,6
Figure 1.
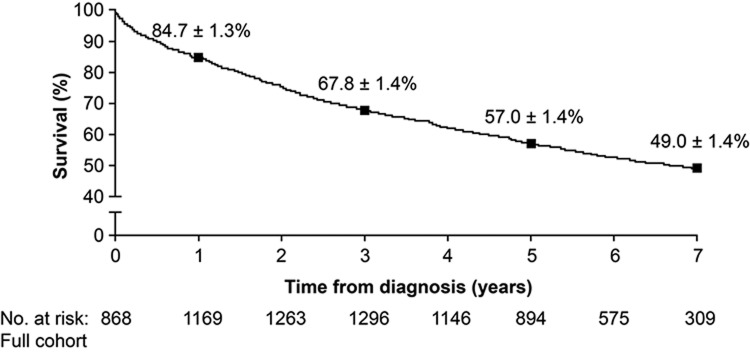
Seven-year survival from time of diagnostic right heart catheterization in the full Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) cohort. Squares indicate estimated survival estimate ± standard error at each particular time point. Reproduced with permission from the American College of Chest Physicians.6
Most contemporary registries include only adult patients, with a predominance of idiopathic PAH. Because the underlying etiology of PAH affects overall survival,12 more data from studies of specific subgroups are needed. For example, patients with PAH associated with systemic sclerosis have a particularly poor prognosis, although recent studies suggest that outcome has improved, with a contemporary 1-year survival rate of 78% compared with 45% in historical reports.4 There is also a lack of contemporary survival data for children with PAH, because interpretation of data from registries including children is complicated by a range of factors that include small patient numbers, poor characterization, and the retrospective nature of such studies.13 Before targeted therapy became available, most children died within 1 year of idiopathic PAH diagnosis; however, more recent data report that, with treatment, 3-year survival rates for pediatric idiopathic PAH are greater than 80%.14 Longitudinal follow-up of patients included in the Tracking Outcomes and Practice in Pediatric Pulmonary Hypertension (TOPP) registry, which is the first global, prospective registry of pediatric pulmonary hypertension, should provide outcome data in pediatric pulmonary hypertension.13 Obtaining data in low-prevalence PAH subgroups will, however, be challenging, because an adequate period of observation will require a prolonged duration of study because of low numbers of patients.
Despite these limitations, the aggregate evidence from contemporary registries suggests an improvement in survival in the modern treatment era, even though none of the approved therapies are curative. Better use of adjunctive therapies (e.g., greater reliance on diuretics and avoidance of nonselective vasodilators), more adept acute management of acute right heart failure, better recognition of perioperative/peripartum hazards, and more disciplined longitudinal monitoring of patients with PAH by specialized pulmonary hypertension centers has also likely contributed to the observed improved outcomes. However, there remains considerable room for improvement in terms of longer-term morbidity and mortality rates. Moreover, patients may be stable and even exhibit a good functional capacity for years with persistently elevated pulmonary artery pressures and a chronically impaired right ventricle, yet still succumb to “late” disease progression or become vulnerable to acute insults, such as sepsis,15,16 pulmonary embolism,17 and arrhythmias.18 As a result, patients with PAH still warrant vigilant and systematic monitoring of their condition. To further improve outcomes and advance the field, not only are new therapeutic agents needed, but study designs and end points need to be redefined, particularly the primary end point used to judge the relative benefits of different treatment approaches on the ultimate longer-term goals of reducing morbidity and mortality across a range of patients with PAH.
Challenges of clinical research in PAH
Conducting effective clinical trials in PAH has always been challenging because of the rarity of the disease, its heterogeneous biology, and the rapidly changing landscape of therapies. Current European registries have estimated a prevalence of 5–8 cases per million for idiopathic PAH and 15–26 cases per million for the larger and more heterogeneous category of PAH that also includes, for example, PAH associated with connective tissue disease, toxins, and congenital systemic-to-pulmonary shunts.4,19 As a result, large-scale studies involving thousands of patients that have been conducted in other disease states are not realistically feasible in PAH. Furthermore, the increasing number of PAH-specific treatments and the number of competing clinical studies have further limited the number of appropriate PAH subjects for clinical trials, making it extremely difficult to recruit subjects, particularly treatment-naive subjects, in many areas of the world. As a result, enrollment goals in numerous recent PAH trials have been challenging to meet.
The heterogeneous nature of patients with PAH also makes designing clinical trials difficult. Expression of the condition may vary significantly, because the PAH category encompasses individuals from childhood (i.e., those with idiopathic PAH who typically lack other medical problems or those with PAH associated with congenital heart defects) to those in their sixth or seventh decade of life (i.e., those with scleroderma-associated PAH, who often have comorbidities and/or other manifestations of their systemic disorder). Accordingly, disease-related parameters used to monitor patients that have often served as end points in clinical trials (e.g., 6MWD and World Health Organization [WHO] modified functional class) are susceptible to factors other than cardiopulmonary issues and therefore may be unreliable. Furthermore, the baseline characteristics of patients can also vary considerably as a result of differing demographic characteristics, comorbid conditions, and baseline conditions.
Although the influence of PAH subject heterogeneity in a specific trial can be minimized through randomization, the population’s heterogeneity can still affect clinical research by applying unique and hidden pressures on certain segments of the population, which could influence ultimate study outcome. As an example, the resilience of the right ventricle to increased afterload appears to differ by PAH subgroup. The right ventricles of scleroderma patients appear to be particularly susceptible and prone to failing, whereas the right ventricles of patients with congenital systemic-to-pulmonary shunts (Eisenmenger physiology) are remarkably durable even in the face of severe pulmonary hypertension.20,21 As a result, an investigational agent’s impact on the pulmonary vasculature may have a variable clinical effect based on these inherent differences in right ventricular adaptation. This heterogeneity of PAH subtypes is likely to result from unique biologic pathways and their relative importance to either the pulmonary vasculopathy or right ventricular adaptation and could influence natural history and response to a particular intervention.22,23 Finally, this heterogeneity of the PAH population limits comparison of outcomes across trials, because enrollment is unlikely to be consistent with regard to PAH subtypes, baseline disease severity, and/or the use of background PAH-specific therapies.
Another challenging aspect of PAH clinical research is the impact of approved PAH-specific background therapies. Drugs in the endothelin receptor antagonist, phosphodiesterase type 5 inhibitor, (PDE-5i) or prostanoid classes have become the mainstays of PAH treatment. Since their introduction, slower rates of disease progression and delays in clinical worsening have been reported and are challenging the ability to demonstrate efficacy of novel drugs when added to ongoing treatment regimens.24-27 Accordingly, new treatments must demonstrate a measurable and clinically meaningful incremental benefit in placebo-controlled investigations. In the future, the blinded phase of “add-on” trials will probably need to be extended beyond the usual 12–24 weeks for the incremental benefits of new therapies over background therapies to be observed.28 Furthermore, clinical trials need to account for the use of advanced prostanoids that “rescue” patients whose condition is deteriorating and, depending on study design, may potentially avoid or delay predefined study outcomes. One way to balance this and not jeopardize study integrity or subject safety is to develop a priori practical “escape criteria” that could be incorporated into study end points. Because of these stark realizations, it is important that any studies involving patients with PAH generate clinically useful data by ensuring that appropriate end points and durations are used.
Candidate surrogate markers in PAH
An end point used in a clinical trial should be accurate, precise, reliable, and measurable. In a life-threatening condition, such as PAH, the most definitive outcome is mortality. However, the use of mortality as a primary end point in clinical trials of PAH has traditionally been impractical. This is primarily because of the large sample size and long study duration that may be required to show a significant survival advantage, factors that pose inherent challenges in an orphan disease in which the patient population is limited. As a result, other important clinical outcomes have been studied in PAH trials, including hospitalization for worsening PAH; undergoing a lung or heart-lung transplant; creating a palliative right-to-left anatomic shunt (e.g., atrial septostomy or Potts shunt); escalating to continuous intravenous or subcutaneous parenteral prostanoid therapy, and/or disease progression (prospectively defined by specific criteria), all of which are undesirable for patients. Accordingly, reduction of these events is a laudable goal for any investigational PAH agent and is frequently represented as a composite end point, called “clinical worsening,” in short-term clinical trials.
The use of surrogates in clinical trials has the potential to overcome some of the limitations associated with measuring “harder” end points, such as mortality or lung or heart-lung transplantation. A surrogate end point can be defined as a marker that acts as a substitute for a clinically significant outcome. Surrogates should be reliable measures that are in the causal pathway to the ultimate clinical outcome (e.g., mortality). In addition, changes to a surrogate marker should be linked to changes in the clinical outcome.29 Without meeting this second criterion, a candidate measure behaves as a correlate, rather than a surrogate; this is an important distinction, because only a surrogate can be substituted for the targeted clinical outcome.30 To take an example from cardiovascular medicine, parameters such as cardiac output, ejection fraction, and exercise capacity are correlated with long-term survival of patients with congestive heart failure. However, treatment-induced improvements in those measurements are not reliable predictors of the effect on mortality, and thus they are correlates and not surrogates of mortality in congestive heart failure.30 Furthermore, surrogate measures may also be patient-centric by directly reflecting how a patient feels. This is valued by patients, healthcare professionals, and regulatory agencies but is not a requirement for a surrogate measure.29 The use of surrogate markers has a number of potential advantages, including a smaller sample size (particularly important in rare diseases such as PAH) and possibly shorter-duration trials, thereby improving a study’s efficiency and facilitating earlier clinical availability of effective therapies. However, the selection of valid and appropriate surrogates as primary end points in clinical trials is critical, and the use of inappropriate surrogates may result in incomplete, inadequate, or misleading analyses and conclusions.29
Given the understanding that hemodynamic measurements, such as right atrial pressure and cardiac index, are important predictors of survival in PAH3 and are relied upon heavily in clinical practice, it would be explicable for hemodynamic characteristics to serve as surrogate markers in PAH. However, hemodynamic characteristics have rarely been used as primary end points in PAH clinical trials, especially large-scale phase III studies, for a myriad of reasons. The technical challenges of right heart catheterization, the unwillingness of patients to undergo invasive testing, and trial sponsors’ reluctance to incur costs have all been significant deterrents toward greater reliance on hemodynamic characteristics in clinical trials. Furthermore, regulatory agencies’ preferences for simple, noninvasive, and more patient-centric measures have further stifled enthusiasm for using hemodynamic parameters as primary end points in PAH trials. As a result, other simpler, less expensive, and more patient-focused measures have been used.
6MWD as primary end point and potential surrogate marker in PAH clinical trials
The 6-minute walk test (6MWT) is a self-paced, submaximal exercise test, and its principal output measure, the 6MWD, has been used to assess exercise capacity in patients with a range of chronic cardiopulmonary conditions. In fact, the 6MWD correlates with peak oxygen consumption31 and is also reflective of activities of daily living.32 6MWD has been used widely as a primary end point in clinical PAH trials largely because baseline 6MWD has been shown to predict mortality and morbidity in conditions such as heart failure33,34 and chronic obstructive pulmonary disease.35,36 As a result of its simplicity, low cost, minimal technical needs, reproducibility, and patient-centric nature, the widely used 6MWT has many attractive features for being a primary outcome measure in clinical trials. However, its robustness as a potential surrogate end point for morbidity and mortality has recently been challenged.
The 6MWD has been shown to correlate with QoL, functional class, and oxygen consumption in several analyses in the cardiopulmonary domain.31,32,37 In idiopathic PAH, Miyamoto et al.37 demonstrated that baseline 6MWD discriminated survival patterns. As such, the 6MWD has been adopted as a treatment goal and potential surrogate marker in clinical practice as well as serving as the primary end point in many PAH clinical trials.1,38-48 Considering the aggregate experience from the pivotal monotherapy trials in PAH, absolute improvements in 6MWD after short-term exposure to active agents have generally ranged from 30 to 45 m, representing an 8%–13% improvement from baseline 6MWD (Fig. 2).24,26,39-42 To date, the majority of studies using 6MWD have involved monotherapy, but there is currently considerable interest in improving patient outcomes with combination therapy of 2 or more agents from different therapeutic classes. However, the placebo-adjusted improvements in 6MWD in completed combination therapy trials have been less impressive than those seen in the monotherapy studies (Fig. 3),43-48 even though other measures, including rates of clinical worsening and hemodynamic characteristics, have improved,43-45 which suggests that 6MWD may have shortcomings as an end point in combination therapy trials.
Figure 2.
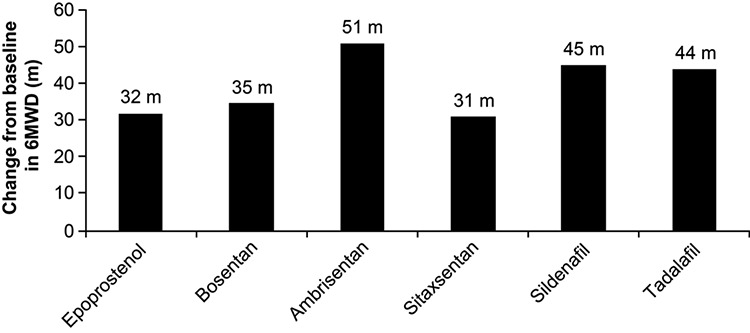
Mean change in 6-minute walk distance (6MWD) from baseline to end of study in clinical trials of pulmonary arterial hypertension treated with monotherapy. Epoprostenol, continuous infusion, 12 weeks;24 bosentan, 125 mg, 16 weeks;39 ambrisentan, 10 mg, 12 weeks;26 sitaxentan, 100 mg, 18 weeks;40 sildenafil, 20 mg, 12 weeks;41 tadalafil, 40 mg, 16 weeks.42 Only unadjusted data from active treatment arms are shown.
Figure 3.
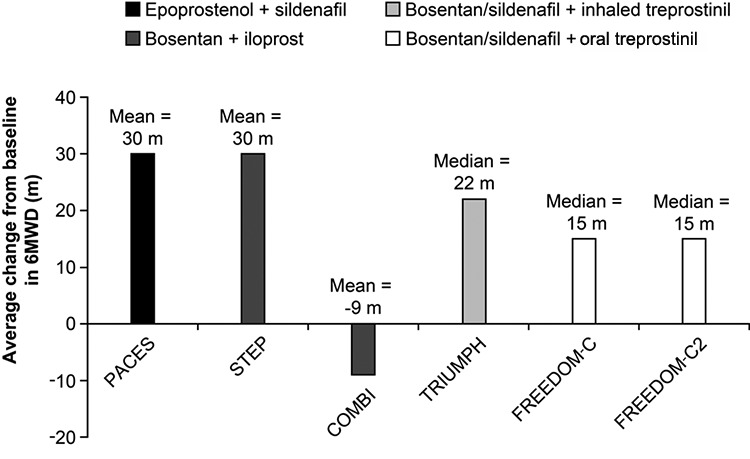
Change in 6-minute walk distance (6MWD) from baseline to study end in clinical trials of pulmonary arterial hypertension treated with combination therapy. PACES, 16 weeks;43 STEP, 12 weeks;44 COMBI, 12 weeks;45 TRIUMPH, 12 weeks;46 FREEDOM-C, 16 weeks;47 FREEDOM-C2, 16 weeks.48 Only unadjusted data from active treatment arms are shown.
The clinical relevance of 6MWD in PAH
An unresolved issue for 6MWD as a trial end point is what constitutes a meaningful improvement in 6MWD in response to treatment. It is essential to distinguish between changes that are clinically important or meaningful to the patient and those that result in little perceived benefit to the patient despite being statistically significant in clinical trials. A number of studies have addressed the issue of minimally important difference (MID), defined as a measure of the smallest difference in 6MWD that signifies a clinically important change. Using data from patients in the Sildenafil Use in Pulmonary Hypertension (SUPER) trial (sildenafil versus placebo in treatment-naive patients with PAH) and a distribution-based method of determining the MID, Gilbert et al.49 calculated an MID in the 6MWD of 41 m, with a range of 18.7–74.2 m. More recently, Mathai et al.50 used data from the Pulmonary Arterial Hypertension and Response to Tadalafil trial42 (tadalafil versus placebo in treatment-naive patients or as add on to bosentan-treated patients) to determine MID by an “anchor-based” method, in which changes in the 6MWD were anchored to changes in the clinically relevant physical component score of the short-form 36 QoL instrument as well as several distributional methods similar to those used in the Gilbert analysis. Mathai et al.50 determined an anchor-based MID of 38.6 m and a consensus value (anchor-based plus distribution method) of 33 m. It is important to remember that these analyses were calculated from 2 specific studies and that more generalized analyses are required to confirm these levels in PAH. Nevertheless, the similarity of the findings for MID in 6MWD between Gilbert’s and Mathai’s work raises doubts about the clinical relevance of the change in 6MWD reported in several recently completed combination therapy trials (Fig. 3),43-48 including those with statistically significant values for their respective primary or secondary end points.
In most studies, 6MWD has been assessed as an absolute change from baseline,24-26,39-41 although several studies, including observational reports, have tried to establish threshold values for the 6WMD, which could serve as benchmarks for clinical decision-making and assessing prognosis.37,51,52 It is important to note, however, that these studies were undertaken during an earlier era when fewer therapeutic options were available and that the suggested thresholds may have less relevance in the modern treatment era. In addition, none of the benchmark 6MWD values have been confirmed in prospective studies, and consequently, more evidence is required to establish whether the correct benchmark has been identified. Although the 6MWT still has an integral role in clinical practice, the lack of consensus over a threshold 6MWD to reliably discriminate longer-term outcomes does challenge its value and ability to serve as a primary end point in clinical investigations.
Importantly, there are also data to suggest that 6MWD may be a less sensitive surrogate marker of clinical outcome in patients with PAH earlier in the course of the disease, because these patients may have a longer walking distance at baseline. Degano and colleagues reported that, in patients with a baseline 6MWD ≤450 m, improvement in 6MWD was associated with better survival; however, there was no similar association in patients with baseline 6MWD >450 m.53 This may be due, at least in part, to a “ceiling effect” in 6MWD; that is, a level at which 6MWD is not significantly affected by the disease and, therefore, is less likely to predict long-term outcomes.54 One explanation of this lies in the test itself; there is a stage at which patients are walking as fast as is mechanically possible and would need to begin to run to maintain a linear increase in speed. This ceiling effect is likely to be more relevant to less severely ill patients, and this phenomenon is not observed in many patients in PAH trials, which generally include patients in functional classes III or IV. The ceiling effect of the 6MWD may be overcome by using more advanced maximal effort exercise tests, such as cardiopulmonary exercise testing (CPET) and its measurements (e.g., peak oxygen consumption or the ratio of minute ventilation to carbon dioxide production at anaerobic threshold). However, CPET is more complicated to perform than the 6MWT, requires exercise equipment and measurement systems, and needs to be performed and assessed by experienced personnel. With the appropriate level of site training, the complexity of CPET can be overcome by study personnel, and it can provide reliable data. Although CPET has been used in some trials, including the Sitaxentan therapy for pulmonary arterial hypertension (STRIDE-1)55 and in a pilot study of bosentan as an add-on therapy to iloprost or beraprost,45 it has been less widely used than 6MWT, and it is unclear whether reported data can be generalized to a wider PAH population.
Limitations of the 6MWD as a potential surrogate marker in PAH
As mentioned earlier, a critical element for a valid surrogate marker is the expected relationship between changes in a surrogate and the outcomes for which the surrogate is substituting. In PAH trials, the connection between treatment-associated 6MWD changes and improvement in relevant clinical outcomes has been assumed but has not been proven, despite the 6MWD being adopted as the primary end point for most PAH pivotal trials to date. In fact, 2 recent publications have cast doubt on the validity of the 6MWD as a surrogate marker of significant clinical outcomes in PAH and shaken the confidence of many experts in the field.56,57
Gabler et al.56 pooled data from 10 randomized controlled trials of PAH-specific therapies (2,404 patients) in an attempt to validate 6MWD as a surrogate end point by establishing whether changes in 6MWD after 12 weeks of treatment could predict the relationship between treatment assignment (active therapy vs. placebo) and occurrence of a predefined clinical event (death, lung or heart-lung transplantation, atrial septostomy, hospitalization due to worsening PAH, withdrawal for worsening of right heart failure, or addition of other PAH medications). Although change in 6MWD from baseline to week 12 met the criteria for being a mediator of the relationship between treatment assignment and development of a clinical event, the association was relatively weak, and a large proportion of the treatment effect on clinical events was not explained by changes in 6MWD. Gabler et al.56 concluded that 6MWD may not be an adequate surrogate end point for clinical events.
Similarly, Savarese et al.57 investigated whether improvement in 6MWD was associated with clinical outcomes in 22 PAH studies involving over 3,100 patients. In their meta-analysis, active treatments (prostanoids, endothelin receptor antagonists, or phosphodiesterase type 5 inhibitors used as monotherapy or in combination) resulted in significant risk reduction for the composite outcome of clinical events, including mortality, hospitalization, lung or heart-lung transplantation, or the initiation of rescue therapy at a mean follow-up of 14.5 weeks. But meta-regression failed to demonstrate a significant relationship between the change in 6MWD and the composite clinical end point, again casting doubt on the validity of the 6MWD as a surrogate for clinical outcomes.
Limitations of these recent investigations include the study-level, and not patient-level, analyses conducted and the short time frame for assessing the relationship between changes in 6MWD and clinical events.56,57 Because the majority of PAH trials to date have been short-term investigations, typically 12–18 weeks of blinded investigation, the meta-regression could only judge the relationship in the short term. Whether the 6MWD is sufficient as a surrogate marker for longer-term clinical events (i.e., beyond the usual 16-week study duration) is unknown. This uncertainty will continue until longer-term investigations are conducted in PAH and data are available to prove or disprove the utility of the 6MWD. Until then, the lack of a more convincing short-term relationship precludes considering the 6MWD as a surrogate for clinical outcomes and challenges its use as a primary end point in pivotal PAH clinical trials.
The current role of 6MWD in clinical trials of PAH
The fact that change in 6MWD response to treatment may not be a reliable marker of clinical events does not negate its usefulness as an end point in PAH. Despite the caveats discussed previously, the use of 6MWD as the primary efficacy measure in early clinical trials of PAH-specific therapies has undoubtedly provided valuable information regarding the potential clinical efficacy of agents with novel modes of action. Moving forward, 6MWD will likely still be useful in this regard for new drugs targeting novel pathways, at least in the initial stages of clinical development, and for evaluating treatments in subpopulations of patients (i.e., those with Eisenmenger syndrome) where large-scale morbidity and mortality trials are not feasible. Improvements in function are in themselves important in patients with debilitating, chronic conditions, such as PAH, and 6MWD has been shown to correlate with QoL and a patient’s daily life physical activity.58,59 For future pivotal phase III studies in PAH, particularly those involving new drugs from established therapeutic classes, and studies of combination therapy using existing agents, 6MWD is also likely to continue being of value as a secondary end point or as part of a broader composite assessment of clinical benefit or indicator of clinical progression.
Unfortunately, PAH is lacking validated surrogate markers of clinical outcomes. Over the past decade, technical advances and increased understanding of the right ventricle have led to novel parameters of right ventricular function, which could serve as potential surrogate markers in PAH. These include parameters measured by echocardiography (e.g., stroke volume index and tricuspid annular plane systolic excursion), cardiac magnetic resonance imaging (e.g., stroke volume, right ventricular ejection fraction, right ventricular end-diastolic volume index, and left ventricular end-diastolic volume index), or biomarker analysis (e.g., N-terminal prohormone of brain natriuretic peptide).60-62 The potential of these measures will depend not only on their relationships with long-term clinical outcomes and whether they are affected by comorbid conditions but also on practical issues such as ease of use, reproducibility, and accessibility to the technique. Until then, we need to identify more direct and relevant primary outcome measures in PAH to help delineate the relative long-term benefits of novel treatments.
Shifting the focus from the 6MWD toward morbidity and mortality end points: a plea from the Dana Point World Pulmonary Hypertension Symposium
In complex chronic conditions, such as PAH, it is extremely unlikely that any single parameter will be universally predictive of a drug’s effect on mortality and morbidity. It is also important to recognize the limitation of using mortality alone as an end point in PAH trials, because a large number of patients would need to be followed up for a lengthy period of time to observe a statistically significant difference. Therefore, a composite end point made up of mortality along with other relevant nonfatal clinical end points is likely to provide a more appropriate and comprehensive measure of an intervention’s impact in a more acceptable time frame. Composite end points are not infrequently used in randomized controlled clinical trials to increase statistical efficiency by leading to higher event rates, thus requiring smaller sample sizes and/or shorter follow-up. Composite end points also provide a summary measure for a treatment effect in which several outcomes (e.g., morbidity and mortality) are important.63 As such, composite end points have been widely used as the primary outcome measure in clinical trials of morbidity and mortality, particularly in adult cardiology (e.g., heart failure).64 The vast majority of composite end points used in these trials have included mortality and a number of additional end points (most often 1 or 2) tailored to the particular clinical setting (e.g., nonfatal myocardial infarction, nonfatal stroke, and need for coronary artery bypass grafting).65
Clinical worsening, which is a composite end point that includes mortality and other disease-related end points, such as hospitalization for worsening PAH and undergoing lung or heart-lung transplantation, has already been widely used as a secondary end point to assess the treatment effect of PAH-specific therapies. Because it may be used to assess disease progression, it is considered clinically relevant to patients, clinicians, and regulatory agencies.66 However, the definition of clinical worsening has varied between trials, which makes comparison of data difficult (Table 1).24,26,28,40-44,47,55 It is also not useful to use the “need for transplant” as opposed to undergoing transplant due to the variability in listing a patient. It is also most useful for the events to be as objective as possible and adjudicated blindly and independently by committee.
Table 1.
Components of clinical worsening in pivotal/phase III clinical trials of pulmonary arterial hypertension (PAH)–specific therapies
| Variable | BREATHE-124 | EARLY25 | STRIDE-155 | STRIDE-240 | ARIES-126 | ARIES-226 | SUPER-141 | PHIRST42 | STEP44 | PACES43 | TRIUMPH28 | FREEDOM-C47 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study drug | Bosentan | Bosentan | Sitaxentana | Sitaxentana | Ambrisentan | Ambrisentan | Sildenafil | Tadalafil | Bosentan + iloprost | Epoprostenol + sildenafil | Bosentan/sildenafil + inhaled treprostinil | Bosentan/sildenafil + oral treprostinil |
| Study duration, weeks | 16 | 24 | 12 | 18 | 12 | 12 | 12 | 16 | 12 | 16 | 12 | 16 |
| Clinical worsening components | ||||||||||||
| Death | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Hospitalization for PAH | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Lung or heart-lung transplantation | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| Atrial septostomy | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| FC worsening | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||
| 6MWD | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| Initiation of additional PAH-specific therapies | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| Patients with clinical worsening (placebo/study drugb), % | NA | 14/3 | 15/2 | 16/7 (100 mg once daily) | 9/4 (5 mg) | 22/5 (5 mg) | 10/4 | 16/5 | 15/0 | 18/6 | 5/3 | 7/5 |
| P value for TTCW vs. placebo/control | <0.05 | 0.05 | NS | NS | NS | 0.05 | NS | 0.05 | <0.05 | <0.05 | NS | NS |
6MWD, 6-minute walk distance; FC, World Health Organization/New York Heart Association functional class; TTCW, time to clinical worsening; NS, not significant; NA, not available.
Withdrawn from the market.
Unless stated, data are for licensed dose.
The Fourth World Symposium on Pulmonary Hypertension (Dana Point) recommended that reducing mortality and morbidity supplant traditional exercise measures, such as the 6MWD, as the primary end point of future pivotal studies in PAH. In addition, a uniform definition for clinical worsening that could be used in future randomized controlled trials was offered.12 This included all-cause mortality, nonelective hospital stay for PAH (with predefined criteria, such as initiation of intravenous or subcutaneous prostanoids, lung or heart-lung transplantation, or atrial septostomy), and an acceptable measure of disease progression that requires worsening of more than one predefined measure across a period of time (e.g., 6MWD and functional class). An advantage of such a multifaceted definition of clinical worsening is the ability to determine a treatment’s effect beyond reliance on a single measure, such as the 6MWD, which can be influenced by comorbidities in some individuals.
It is worth stating that even these clinical outcomes can vary in different parts of the world and different healthcare systems, thus challenging the applicability of future study findings. For example, intravenous prostanoids and/or transplantation are not available in some parts of the world. Additionally, components of the clinical worsening end point may also need adapting for pediatric studies. The TOPP registry shows that children with PAH have clinical features that differ from those of adults, emphasizing the need for pediatric data from properly designed studies, rather than extrapolation from adult studies.13
To date, the component of composite clinical worsening in PAH trials that has been least consistent has been disease progression, which makes it a more ambiguous outcome. However, disease progression is often the component that drives the number of clinical worsening events in clinical trials, which underscores its importance. Certainly, a trade-off exists when considering how to define disease progression. Although changes in objective and reliable measures should be incorporated, the definition of disease progression should not be so restrictive that true worsening is not captured. Otherwise, a study’s efficiency and integrity as well as a subject’s safety might be jeopardized. In terms of changes in 6MWD, it remains unclear whether a specified change in 6MWD or maintaining an absolute value (e.g., 380 m threshold) is what is most important. McLaughlin et al.12 recommended a decrease in 6MWD of 15% from baseline (confirmed by a second 6MWT within 2 weeks) plus worsening functional class as a defined component of clinical worsening, which is supported by data from REVEAL.67
An additional consideration in assessing clinical end points, especially disease progression, is the potential problem of subjectivity. To help overcome this potential shortcoming, mandatory adjudication of events has also been recommended.12 The use of a panel of independent adjudicators (blinded to treatment) to assess the clinical relevance of all events classified as clinical worsening in a trial would undoubtedly improve the consistency and accuracy of event reporting and has been undertaken in some PAH studies.26 Such independent adjudication of events should be an essential element of future trials.
Event-driven morbidity and mortality trials in PAH
To date, the majority of PAH trials have been of fixed-time design, because studies have been powered to detect a significant difference in the primary end point, usually the 6MWD, at a prespecified time point (e.g., 12 or 18 weeks). This short-term design has also been aided by our understanding of the timing for 6MWD improvement after a treatment intervention. However, given the low event rates for clinical outcomes in short-term clinical trials in PAH, especially recently completed combination therapy studies (Table 2), a new trial design is needed if a composite end point capturing morbidity and mortality is to become the primary end point in pivotal PAH trials. Although a predefined time point could still be used for determining rates of clinical worsening, it is conceivable that a significant reduction in clinical worsening by an investigational agent could be missed by such an arbitrary designation, especially if the event rate in the placebo cohort is overestimated. An alternate design is to employ an event-driven study design, which makes time a variable factor and allows for the blinded portion of a trial to continue until a significant number of prespecified clinical events occur; accordingly, subjects’ exposure to blinded therapy will be open-ended and variable. The number of necessary events would be determined by the projected event rate in the placebo group and an expected reduction in the event rate in the active group. Needless to say, the use of a composite end point capturing morbidity and mortality as a primary end point and an event-driven design may result in much longer clinical trials.12 This is especially true for contemporary PAH studies conducted in countries where multiple PAH therapies are approved and where enrollment will primarily consist of subjects already receiving PAH therapies who are recruited for add-on therapy. By their very nature, such trials will be long term and will result in a much longer investigational period and duration of drug exposure, while at the same time providing extended safety information.
Table 2.
Recently completed, ongoing, and planned placebo-controlled studies including an event-driven assessment of morbidity and mortality as the primary end point
| Study name | Study drug(s) | Primary end point | Anticipated exposure (patient-years) |
|---|---|---|---|
| Studies using currently available PAH-specific therapies | |||
| AMBITION (NCT01178073) | First-line ambrisentan, tadalafil, or combination of both | Time to clinical failure | Study terminated when 105 events occurred; anticipated median treatment duration = 2.0 years; estimated total study duration = 2.75 years |
| COMPASS-2 (NCT00303459) | Addition of bosentan to baseline sildenafil | Time to first (adjudicated) morbidity/mortality event | Enrollment between 2006 and first quarter 2013 |
| Studies investigating novel agents for PAH | |||
| SERAPHIN (NCT00660179) | Macitentan | Time to first (adjudicated) morbidity/mortality event | 742 patients enrolled. |
| GRIPHON (NCT01106014) | Selexipag | Time to morbidity or mortality event (according to the definition of the Dana Point Fourth World Symposium on Pulmonary Hypertension) | Estimated to be up to 4.3 years |
| FREEDOM-Ev (NCT01560624) | Early addition of oral treprostinil to PDE-5i or ERA. | Time to first clinical worsening event | Clinical worsening assessed for up to 2.5 years |
ERA, endothelin receptor antagonist; PAH, pulmonary arterial hypertension; PDE-5i, phosphodiesterase type 5 inhibitor.
Phase III PAH clinical trials that are currently being conducted reflect the evolution of the field since the 2009 Dana Point recommendations,12 with long-term, event-driven studies using composite end points based on morbidity and mortality becoming more widely used for pivotal (phase III) studies (Table 2). These include phase III trials of combination therapy, such as the randomized, multicenter study of first-line ambrisentan and tadalafil combination therapy in subjects with PAH trial that compares first-line combination therapy with first-line monotherapy with either of the agents (AMBITION; ClinicalTrials.gov identifier NCT01178073), the trial of early combination therapy with oral prostacyclin analogue (treprostinil) on a background of PDE-5i or ERA therapy (FREEDOM-Ev; ClinicalTrials.gov identifier NCT01560624), and the effects of a combination of bosentan and sildenafil versus sildenafil monotherapy on morbidity and mortality in symptomatic patients with PAH (COMPASS-2 trial; ClinicalTrials.gov identifier NCT00303459). In terms of new drugs, studies include the GRIPHON trial (A Multicenter, Double-blind, Placebo-controlled Phase III Study to Demonstrate the Efficacy and Safety of ACT-293987 in Patients with PAH) of the orally available selective prostacyclin receptor agonist selexipag (ClinicalTrials.gov identifier NCT01106014) and the SERAPHIN (A Multicenter, Double-blind, Randomized, Placebo-controlled, Parallel Group, Event-driven, Phase III Study to Assess the Effects of ACT-064992 on Morbidity and Mortality in Patients with Symptomatic Pulmonary Arterial Hypertension) trial of the endothelin receptor antagonist macitentan (ClinicalTrials.gov identifier NCT00660179). In fact, the recent completion of the SERAPHIN trial provides hope that longer-term, event-driven studies in PAH that use reduction in morbidity and mortality as the primary end point are feasible and also provide important longer-term safety experience.68
Performance of such longer-term trials presents a number of challenges to those who run them and those who participate in them. Subject recruitment is a problem in rare diseases, such as PAH, and patient retention is a universal problem in clinical trials. Large dropout rates affect the integrity of data from such studies, and it is therefore incumbent on researchers to work closely with patients to preserve participation. This can become a major consideration in studies lasting years rather than weeks, especially as approved treatment options continue to expand and treatment paradigms evolve. Investigator fatigue can also be a problem, and the emergence of new therapies or monitoring strategies and evolving standards of care can impact enthusiasm for such lengthy trials. Furthermore, a reduction in mortality and morbidity may not be as appealing to patients as an improvement in a relevant clinical measure. To cater for both subjects’ and investigators’ psyches and to enhance an investigational agent’s overall impact, important modifiable measures that signify clinical improvement, such as 6MWD, functional class, serum markers, and especially hemodynamic characteristics, should remain as important secondary end points of trials.
Conclusions
Over the past 2 decades, use of the 6MWD as a primary end point has established therapeutic options for PAH, which have evolved from nonspecific general and supportive measures to PAH-specific targeted therapies that offer improvement in functional capacity and exercise capacity and the possibility of long-term improvement in morbidity and survival. With these advances, a once universally fatal disease has been transformed into an often manageable chronic condition that, however, still has the potential for deterioration and considerable morbidity. The design and choice of end points in clinical trials needs to evolve to reflect these advances, with a shift from short-term trials with single markers as primary end points to longer-term, event-driven trials using a composite of clinically relevant end points capable of more accurately and appropriately assessing morbidity and mortality in this complex condition. Encouragingly, many newer phase III trials in PAH have evolved on the basis of expert recommendations and are using mortality and morbidity events as their primary end point. New event-driven, long-term trials in PAH may be challenging to perform but are important to provide evidence for sustained long-term benefits of new treatments in the current treatment era and carry forward the progress made against this devastating disease in the past 20 years.
Source of support: Medical writing support was provided by Liesje Quine from Elements Communications and funded by Actelion Pharmaceuticals.
Conflict of interest: In the past year, MC has received research support from Actelion Pharmaceuticals, Gilead, United Therapeutics, Novartis, Bayer, Ikaria, and AIRES. He has also served as a consultant (on a limited basis) for Actelion Pharmaceuticals, Gilead, and United Therapeutics and is on speaker’s bureaus and received honorarium from Gilead and United Therapeutics. RJB has received honoraria for consultation services from Actelion Pharmaceuticals, Bayer, Eli Lilly, Gilead, Ikaria, Pfizer, Novartis, and United Therapeutics.
References
- 1.Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30(20):2493–2537. [DOI] [PubMed]
- 2.Frumkin LR. The pharmacological treatment of pulmonary arterial hypertension. Pharmacol Rev 2012;64(3):583–620. [DOI] [PubMed]
- 3.D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med 1991;115(5):343–349. [DOI] [PubMed]
- 4.Humbert M, Sitbon O, Yaïci A, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J 2010;36(3):549–555. [DOI] [PubMed]
- 5.Condliffe R, Kiely DG, Peacock AJ, et al. Connective tissue disease–associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med 2009;179(2):151–157. [DOI] [PubMed]
- 6.Benza RL, Miller DP, Barst RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from REVEAL. Chest 2012;142(2):448–456. [DOI] [PubMed]
- 7.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004;351(14):1425–1436. [DOI] [PubMed]
- 8.Zanolla L, Zardini P. Selection of endpoints for heart failure clinical trials. Eur J Heart Fail 2003;5(6):717–723. [DOI] [PubMed]
- 9.Galiè N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J 2009;30(4):394–403. [DOI] [PMC free article] [PubMed]
- 10.Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010;122(2):156–163. [DOI] [PubMed]
- 11.Ling Y, Johnson MK, Kiely DG, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012;186(8):790–796. [DOI] [PubMed]
- 12.McLaughlin VV, Badesch DB, Delcroix M, et al. End points and clinical trial design in pulmonary arterial hypertension. J Am Coll Cardiol 2009;54(1 suppl):S97–S107. [DOI] [PubMed]
- 13.Berger RM, Beghetti M, Humpl T, et al. Clinical features of paediatric pulmonary hypertension: a registry study. Lancet 2012;379(9815):537–546. [DOI] [PMC free article] [PubMed]
- 14.Barst RJ, McGoon MD, Elliott CG, Foreman AJ, Miller DP, Ivy DD. Survival in childhood pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 2012;125(1):113–122. [DOI] [PubMed]
- 15.Oudiz RJ, Widlitz A, Beckmann XJ, et al. Micrococcus-associated central venous catheter infection in patients with pulmonary arterial hypertension. Chest 2004;126(1):90–94. [DOI] [PubMed]
- 16.Kallen AJ, Lederman E, Balaji A, et al. Bloodstream infections in patients given treatment with intravenous prostanoids. Infect Control Hosp Epidemiol 2008;29(4):342–349. [DOI] [PubMed]
- 17.Lahm T, McCaslin CA, Wozniak TC, et al. Medical and surgical treatment of acute right ventricular failure. J Am Coll Cardiol 2010;56(18):1435–1446. [DOI] [PubMed]
- 18.Tongers J, Schwerdtfeger B, Klein G, et al. Incidence and clinical relevance of supraventricular tachyarrhythmias in pulmonary hypertension. Am Heart J 2007;153(1):127–132. [DOI] [PubMed]
- 19.Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 2007;30(1):104–109. [DOI] [PubMed]
- 20.Hopkins WE. The remarkable right ventricle of patients with Eisenmenger syndrome. Coron Artery Dis 2005;16(1):19–25. [DOI] [PubMed]
- 21.Hopkins WE, Ochoa LL, Richardson GW, Trulock EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant 1996;15:100–105. [PubMed]
- 22.Kuhn KP, Byrne DW, Arbogast PG, Doyle TP, Loyd JE, Robbins IM. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med 2003;167(4):580–586. [DOI] [PubMed]
- 23.Kawut SM, Taichman DB, Archer-Chicko CL, Palevsky HI, Kimmel SE. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest 2003;123(2):344–350. [DOI] [PubMed]
- 24.Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346(12):896–903. [DOI] [PubMed]
- 25.Galiè N, Rubin LJ, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 2008;371(9630):2093–2100. [DOI] [PubMed]
- 26.Galiè N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117(23):3010–3019. [DOI] [PubMed]
- 27.Olschewski H, Simonneau G, Galiè N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002;347(5):322–329. [DOI] [PubMed]
- 28.McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol 2010;55(18):1915–1922. [DOI] [PubMed]
- 29.Snow JL, Kawut SM. Surrogate end points in pulmonary arterial hypertension: assessing the response to therapy. Clin Chest Med 2007;28(1):75–89. [DOI] [PMC free article] [PubMed]
- 30.Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med 1996;125(7):605–613. [DOI] [PubMed]
- 31.Cahalin L, Pappagianopoulos P, Prevost S, Wain J, Ginns L. The relationship of the 6-min walk test to maximal oxygen consumption in transplant candidates with end-stage lung disease. Chest 1995;108(2):452–459. [DOI] [PubMed]
- 32.Solway S, Brooks D, Lacasse Y, Thomas S. A qualitative systematic overview of the measurement properties of functional walk tests used in the cardiorespiratory domain. Chest 2001;119(1):256–270. [DOI] [PubMed]
- 33.Bittner V, Weiner DH, Yusuf S, et al. Prediction of mortality and morbidity with a 6-minute walk test in patients with left ventricular dysfunction. SOLVD Investigators. JAMA 1993;270(14):1702–1707. [PubMed]
- 34.Shah MR, Hasselblad V, Gheorghiade M, et al. Prognostic usefulness of the six-minute walk in patients with advanced congestive heart failure secondary to ischemic or nonischemic cardiomyopathy. Am J Cardiol 2001;88(9):987–993. [DOI] [PubMed]
- 35.Szekely LA, Oelberg DA, Wright C, et al. Preoperative predictors of operative morbidity and mortality in COPD patients undergoing bilateral lung volume reduction surgery. Chest 1997;111(3):550–558. [DOI] [PubMed]
- 36.Casanova C, Cote C, Marin JM, et al. Distance and oxygen desaturation during the 6-min walk test as predictors of long-term mortality in patients with COPD. Chest 2008;134(4):746–752. [DOI] [PubMed]
- 37.Miyamoto S, Nagaya N, Satoh T, et al. Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension. Comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med 2000;161(2 Pt 1):487–492. [DOI] [PubMed]
- 38.Macchia A, Mariani J, Comignani PD, Tognoni G. Clinical trials using vasodilators in pulmonary arterial hypertension: where do we go from here? Rev Recent Clin Trials 2011;6(3):228–234. [DOI] [PubMed]
- 39.Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334(5):296–301. [DOI] [PubMed]
- 40.Barst RJ, Langleben D, Badesch D, et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol 2006;47(10):2049–2056. [DOI] [PubMed]
- 41.Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353(20):2148–2157. [DOI] [PubMed]
- 42.Galiè N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009;119(22):2894–2903. [DOI] [PubMed]
- 43.Simonneau G, Rubin LJ, Galiè N, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med 2008;149(8):521–530. [DOI] [PubMed]
- 44.McLaughlin VV, Oudiz RJ, Frost A, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med 2006;174(11):1257–1263. [DOI] [PubMed]
- 45.Hoeper MM, Taha N, Bekjarova A, Gatzke R, Spiekerkoetter E. Bosentan treatment in patients with primary pulmonary hypertension receiving non-parenteral prostanoids. Eur Resp J 2003;22(2):330–334. [DOI] [PubMed]
- 46.McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2010;55(18):1915–1922. [DOI] [PubMed]
- 47.Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest 2012;142(6):1383–1390. [DOI] [PubMed]
- 48.Tapson V, Jing Z-C, Xu K, et al. FREEDOM-C2: efficacy and safety of oral treprostinil diethanolamine in combination with an ERA and/or a PDE5 inhibitor in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;185(suppl):A2493.
- 49.Gilbert C, Brown MC, Cappelleri JC, Carlsson M, McKenna SP. Estimating a minimally important difference in pulmonary arterial hypertension following treatment with sildenafil. Chest 2009;135(1):137–142. [DOI] [PubMed]
- 50.Mathai SC, Puhan MA, Lam D, Wise RA. The minimal important difference in the 6-minute walk test for patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186(5):428–433. [DOI] [PMC free article] [PubMed]
- 51.Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002;40(4):780–788. [DOI] [PubMed]
- 52.Benza RL, Gupta H, Murali S, Park M, Soto FJ, Torres F. Achieving clinical thresholds of benefit with bosentan-based treatment: the COMPASS-3 trial. Am J Respir Crit Care Med 2012;185:A4780.
- 53.Degano B, Sitbon O, Savale L, et al. Characterization of pulmonary arterial hypertension patients walking more than 450 m in 6 min at diagnosis. Chest 2010;137(6):1297–1303. [DOI] [PubMed]
- 54.Frost AE, Langleben D, Oudiz R, et al. The 6-min walk test (6MW) as an efficacy endpoint in pulmonary arterial hypertension clinical trials: demonstration of a ceiling effect. Vasc Pharmacol 2005;43(1):36–39. [DOI] [PubMed]
- 55.Barst RJ, Langleben D, Frost A, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 2004;169(4):441–447. [DOI] [PubMed]
- 56.Gabler NB, French B, Strom BL, et al. Validation of six-minute-walk distance as a surrogate endpoint in pulmonary arterial hypertension trials. Circulation 2012;126(3):349–356. [DOI] [PMC free article] [PubMed]
- 57.Savarese G, Paolillo S, Costanzo P, et al. Do changes of 6-minute walk distance predict clinical events in patients with pulmonary arterial hypertension? a meta-analysis of 22 randomized trials. J Am Coll Cardiol 2012;60(13):1192–1201. [DOI] [PubMed]
- 58.Taichman DB, Shin J, Hud L, et al. Health-related quality of life in patients with pulmonary arterial hypertension. Respir Res 2005;6:92. [DOI] [PMC free article] [PubMed]
- 59.Mainguy V, Provencher S, Maltais F, Malenfant S, Saey D. Assessment of daily life physical activities in pulmonary arterial hypertension. PLoS One 2011;6(11):e27993. [DOI] [PMC free article] [PubMed]
- 60.van Wolferen SA, Marcus JT, Boonstra A, et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 2007;28(10):1250–1257. [DOI] [PubMed]
- 61.Vonk Noordegraaf A, Westerhof N. Right ventricular ejection fraction and NT-proBNP are both indicators of wall stress in pulmonary hypertension. Eur Respir J 2007;29(4):622–623. [DOI] [PubMed]
- 62.Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med 2006;174(9):1034–1041. [DOI] [PubMed]
- 63.Kliest P. Composite endpoints for clinical trials: current perspectives. Int J Pharm Med 2007;21:187–198.
- 64.Freemantle N, Calvert M, Wood J, Eastaugh J, Griffin C. Composite outcomes in randomized trials: greater precision but with greater uncertainty? JAMA 2003;289(19):2554–2559. [DOI] [PubMed]
- 65.Ferreira-González I, Busse JW, Heels-Ansdell D, et al. Problems with use of composite end points in cardiovascular trials: systematic review of randomised controlled trials. BMJ 2007;334(7597):786. [DOI] [PMC free article] [PubMed]
- 66.Galiè N, Simonneau G, Barst RJ, Badesch D, Rubin L. Clinical worsening in trials of pulmonary arterial hypertension: results and implications. Curr Opin Pulm Med 2010;16(suppl 1):S11–S19. [DOI] [PubMed]
- 67.Frost AE, Badesch DB, Miller DP, Benza RL, McGoon MD. REVEAL registry: impact of clinical worsening on three-year outcomes in pulmonary arterial hypertension. Chest 2010;138(suppl 4):837A. [DOI] [PubMed]
- 68.Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809–818. [DOI] [PubMed]