

Abstract Abstract
The natural history of familial pulmonary arterial hypertension (PAH) typically involves mutations in and/or haploinsuffciency of BMPR2 (gene for bone morphogenetic protein receptor type 2) but with low penetrance (10%–15%), delayed onset (in the third or fourth decade), and a gender bias (two- to fourfold more prevalent in postpubertal women). Thus, investigators have sought an understanding of “second-hit” modalities that might affect BMPR2 anterograde trafficking and/or function. Indeed, vascular lung lesions in PAH have been reported to contain enlarged “vacuolated” endothelial and smooth muscle cells with dilated endoplasmic reticulum (ER) cisternae, increased ER structural protein reticulon 4 (also called Nogo-B), and enlarged and fragmented Golgi apparatus. We recently replicated this cellular phenotype in primary human pulmonary arterial endothelial cells and human pulmonary arterial smooth muscle cells in culture by acute knockdown of the estradiol 17β (E2)–responsive proteins signal transducer and activator of transcription 5a (STAT5a) and STAT5b using small interfering RNAs (siRNAs). We have now investigated whether functional haploinsufficiences of these molecules, alone or in combination with other modalities, might interfere with anterograde membrane trafficking using (a) the quantitative tsO45VSV-G-GFP trafficking assay and (b) assays for cell-surface localization of Flag-tagged BMPR2 molecules. The G glycoprotein of the vesicular stomatitis virus (VSV-G) trafficking assay was validated in EA.hy926 endothelial cells by showing that cells exposed to monocrotaline pyrrole displayed reduced anterograde trafficking. Thereafter, the combinatorial knockdowns of STAT5a, STAT5b, BMPR2, and/or endothelial nitric oxide synthase as well as exposure to E2 or 2-methoxyestradiol were observed to significantly inhibit VSV-G trafficking. These combinations also led to intracellular trapping of wild-type Flag-tagged BMPR2. Overexpression of the PAH disease–derived F14 and KDF mutants of BMPR2, which were trapped in the ER/Golgi, also inhibited VSV-G trafficking in trans. Moreover, probenecid, a chemical chaperone in clinical use today, partially restored cell-surface localization of the KDF but not the F14 mutant. These data identify several combinatorial modalities that inhibit VSV-G anterograde trafficking and cause mislocalization of BMPR2. These modalities merit consideration in defining aspects of the late-developing and gender-biased natural history of human PAH.
Keywords: vascular cell remodeling, membrane protein trafficking, tsO45VSV-G-GFP trafficking assay, monocrotaline pyrrole (MCTP), signal transducer and activator of transcription 5a/b (STAT5a/b), endothelial nitric oxide synthase (eNOS), estradiol 17β, probenecid
Introduction
Pulmonary arterial hypertension (PAH) is a disease with high morbidity and an unrelentingly fatal outcome.1-4 Vascular remodeling in this disease includes neointimal proliferation as well as the characteristic onion-skin obliterative and the recanalized plexiform lesions associated with pulmonary arteries.1,5 In as much as bone morphogenetic protein (BMP) signaling is considered to inhibit vascular cell proliferation, genetic evidence linking mutations in and/or haploinsufficiency of BMPR2 (gene for BMP receptor type 2) with a majority of cases of familial PAH (FPAH) and a subset of instances of idiopathic PAH (IPAH)1-6 has catalyzed an effort to understand the mechanistic bases for this relationship. Despite intensive investigations over >10 years, the processes by which mutations in and/or haploinsufficiency of BMPR2 lead to the overt disease are poorly understood.6 Many of the ∼300 PAH disease–associated BMPR2 mutants display intracellular trapping along the endoplasmic reticulum (ER)/Golgi apparatus/plasma membrane anterograde vesicular trafficking pathway.4,6 Moreover, almost all of the disease-associated BMPR2 molecules, even those that reach the cell surface, mediate reduced Smad signaling.4,6-10 It is important to note that BMP/Smad signaling as well as plasma membrane to nucleus signaling from a plethora of vasorelevant ligand/receptor pairs is itself well established to be associated with retro grade endocytic and caveolar vesicular trafficking pathways.11-14 Thus, alterations in either anterograde or retrograde membrane trafficking will affect not only the biology of BMP but also that of all other vasorelevant signaling pathways (including vascular endothelial growth factor, platelet-derived growth factor, interleukin 6, etc.; see Sieber et al.,11 Hartung et al.,12 DiGuglielmo et al.,13 Sehgal,14 and citations therein). Whether mutant BMPR2 species trapped along the anterograde trafficking pathway have inhibitory effects on trafficking of other cell-surface proteins and receptors in trans is unknown. Additional unexplained issues include why disease development, even in FPAH kindreds with known mutations in BMPR2, has a low penetrance (10%–20%) and a late onset (median age at diagnosis is in the third decade for women and the fourth decade for men).1-4,6,15,16 Thus, there have been searches for candidate “second- hit” entities that might be involved in determining penetrance and late onset.1-4,6,9
There is also a sexual dimorphism in this disease, with the incidence approximately two- to fourfold higher in postpubertal women than in men for both IPAH and kindreds of FPAH patients.6,15,16 This is the converse of what has been typically observed in various experimental models of PAH, in which female rats or mice are less susceptible to the development of PAH, and estradiol 17β (E2) has been shown to ameliorate established PAH (reviewed in Umar et al.17 and citations therein). In humans, genetic association data suggest that mutations in cytochrome P450 1B1 (CYP1B1), an enzyme involved in estrogen metabolism, is a candidate genetic cofactor contributing to development of the overt disease in kindreds of FPAH patients.15 However, although increased pulmonary vascular CYP1B1 was observed in idiopathic PAH and hypoxia elicited a greater increase in CYP1B1 in the lungs of female mice compared with male mice, different measures of hypoxic PAH were variably affected in male compared with female CYP1B1−/− mice.18 Recently, E2 was reported to decrease BMPR2 expression by 15%–20% in cultured peripheral blood lymphocytes (PBLs) derived from healthy human subjects,16 but the mechanistic link between this observation and PAH disease development remains unclear. The present study focused on the mechanisms of potential penetrance-determining second hits and of gender bias in PAH from a novel perspective—that of the involvement of the estradiol- and prolactin-responsive proteins signal transducer and activator of transcription 5a (STAT5a) and STAT5b in membrane trafficking19,20 and anterograde transit of BMPR2 through the cell.
We discovered this “nongenomic” membrane trafficking function in the cytoplasm19,20 for the “transcription factors” STAT5a and STAT5b.21-24 STAT5a/b proteins associated with structural components of the ER/Golgi and their downregulation led to a characteristic cystic ER/Golgi fragmentation phenotype in primary human pulmonary arterial endothelial cells (HPAECs) and human pulmonary arterial smooth muscle cells (HPASMCs).19,20 Importantly, this cystic ER phenotype had already been reported to occur in cells in lesions in IPAH25 and in pulmonary arterial endothelial cells in hypoxic rats26 by Smith and Heath in the late 1970s. More recently, we showed Golgi fragmentation in cells in lesions of IPAH.27 Moreover, STAT5 species are already recognized to mediate aspects of the sexual dimorphic phenotypes in terms of postpubertal development and gene expression.22,23 Additionally, the known responsiveness of STAT5a/b to E223 and the newly recognized nongenomic effects of E2 on ER membranes itself28-32 made it attractive to investigate the effects of these combinatorial modalities (STAT5a/b downregulation and E2) on trafficking of BMPR2.
In the present study, we first investigated effects on anterograde membrane trafficking in endothelial cells of various potential second-hit modalities in various combinations using the quantitative anterograde trafficking assay based on the temperature-sensitive O45 mutant of the G glycoprotein of vesicular stomatis virus, tsO45VSV-G-GFP, which has been used extensively by cell biologists over the last decade.33-35 We then compared these effects on VSV-G–green fluorescent protein (GFP) trafficking to the mislocalization of wild-type (wt) and mutant BMPR2 species in response to various potential second-hit modalities. Additionally, we investigated whether we could find compounds already in clinical use in other human diseases that might rescue intracellularly trapped BMPR2 mutant species.
Material and methods
Chemicals, antibodies, and small interfering RNA (siRNA) reagents
Monocrotaline (Crotaline C2401) was purchased from Sigma-Aldrich (St. Louis, MO) and was converted to monocrotaline pyrrole (MCTP) using methods reported elsewhere.36 E2 (E2758), 2-methoxyestradiol (2-ME; M6383), and probenecid (P8761) were also from Sigma-Aldrich. The anti-VSV-G monoclonal antibody (mAb; clone I1, stock 8G5F11)33 was prepared from the hybridoma clone provided by Dr. Douglas S. Lyles (Department of Biochemistry, Bowman-Gray School of Medicine). Rabbit polyclonal antibodies (pAbs) to giantin (24586) and ATL3 (104262) were purchased from Abcam (Cambridge, MA). Rabbit pAbs to STAT5a (sc-1081x), STAT5b (sc-835x), STAT3 (sc-482), and the Flag tag (sc-807) and goat pAb to reticulon 4 (RTN4; sc-11027) were from Santa Cruz Biotechnology (Santa Cruz, CA). Murine mAbs to GM130 (Golgi matrix 130-kDa protein; 610823) were purchased from BD Biosciences (Eugene, OR). Respective Alexa Fluor 488– and Alexa Fluor 594–tagged secondary donkey antibodies to rabbit (A-11008 and A-11012), mouse (A-21202 and A-21203), or goat (A-11055 and A-11058) immunoglobulin G were from Invitrogen Molecular Probes (Eugene, OR). siRNA preparations and transfection reagents were obtained from Santa Cruz Biotechnology. These were siRNA transfection reagent (sc-29528), siRNA transfection medium (sc-36868), scrambled control-A siRNA (sc-37007), and siRNA preparations to STAT5a (human; sc-37008), STAT5b (human; sc-370010), BMPR2 (human; sc-40220), and nitric oxide synthase (NOS) 3 (human; sc-36093).
Cells and cell culture
The human endothelial cell line EA.hy926 was obtained from Dr. Michal Schwartzman (Department of Pharmacology, New York Medical College) and grown in Dulbecco’s modified Eagle medium supplemented with 10% v/v fetal bovine serum and 100 μM hypoxanthine, 0.4 μM aminopterin, and 16 μM thymidine.37,38 Primary bovine pulmonary arterial endothelial cells (BPAECs) were cultured and exposed to MCTP as described elsewhere.36,38
Plasmids and siRNAs
The tsO45VSV-G-GFP expression vector34,35 was a gift from Dr. Jennifer Lippincott-Schwartz (National Institutes of Health [NIH]). Constitutive expression vectors were constructed for Flag-tagged BMPR2 proteins (vectors were 3x C-terminal Flag tag/CMV in pCIneo, corresponding to wt; “F14,” exon 3 T354G [Cys118Trp]; and “KDF,” exon 8 C944T [Arg322X]). Plasmid transfections were carried out using PolyFect (30117; Qiagen, Valencia, CA) and the manufacturer’s protocols. Respective siRNA oligonucleotides were purchased from Santa Cruz Biotechnology (see specific catalog numbers above). Acute knockdown of target proteins was carried out using siRNA transfections per the manufacturer’s protocol, as used by usearlier.19,20 Typically, 3 μL of a 10-μM siRNA stock and 3 μL of siRNA transfection reagent (1∶1 ratio of siRNA to transfection reagent) were used per 35-mm plate or per well in a 6-well plate. STAT5a/b double-knockdown experiments were carried out using 3 μL of STAT5a siRNA and 3 μL of STAT5b siRNA together with 6 μL of siRNA transfection reagent; similarly, all combination experiments included 3 μL of the respective siRNA. Corresponding controls received matched amounts of the scrambled siRNA and transfection reagent.
Temperature-sensitive VSV-G-GFP trafficking assay
The quantitative tsO45VSV-G-GFP trafficking assay19,34,35 was carried out in EA.hy926 cells, which survive culturing at the “nonpermissive” temperature (40°C). The temperature-sensitive phenotype of VSV-G is such that it remains trapped in the ER at 40°C but transits to the Golgi and then to the plasma membrane when cells are shifted to 32°C.34,35 Briefly, thus, EA.hy926 cells were transfected with the expression plasmid for tsO45VSV-G-GFP at 37°C using the PolyFect reagent. Four to five hours later, the cultures were shifted to 40°C and kept at this nonpermissive temperature overnight. The cells were then shifted to 32°C for 60 minutes. To detect the surface VSV-G, live cells were stained with a monoclonal antibody (clone I1, stock 8G5 F11) directed against the intraluminal domain of VSV-G (which becomes exposed at the cell surface) for 10 minutes at 4°C, washed extensively, fixed with ice-cold paraformaldehyde (4%), and stained using Alexa Fluor 594–conjugated secondary anti-mAb antibody. Cells were analyzed using a Zeiss AxioImager M2 microscope system with a Zeiss W N-Achroplan 40×/NA0.75 objective equipped with a high-resolution RGB HRc AxioCam camera and AxioVision software (ver. 4.8.1) in 1,388 × 1,040-pixel high-speed color-capture mode. Digital images were acquired in the red (surface VSV-G) and green (total VSV-G-GFP) channel and were analyzed by means of ImageJ software. The images were merged, and the mean fluorescence value in each channel was measured by manually selecting the whole cell. Average pixel values in the field lacking expressing cells were taken as background fluorescence and subtracted. The ratio of surface to total fluorescence was used to calculate the extent of VSV-G transport.
Cultures were treated with MCTP 1 day in advance of transfecting the VSV-G-GFP expression vector. For evaluating effects of siRNA on trafficking, the respective siRNA combinations or the scrambled control siRNA were mixed with the VSV-G-GFP expression vector, and the siRNA transfection reagent (Santa Cruz Biotechnology) was used to introduce the nucleic acids. For evaluating effects of expression of respective BMPR2 species on VSV-G trafficking, these expression vectors were mixed with the VSV-G-GFP expression vector, and PolyFect reagent was used to introduce the DNA.
Immunofluorescence microscopy of cells in culture
Cells in culture were fixed using cold paraformaldehyde (4%) and permeabilized,19,20 and immunofluorescence assays were carried out using antibodies from specific sources and corresponding to specific catalog numbers as enumerated above (dilution range, 1∶100 to 1∶1,000) and as described elsewhere.19,20 The cells were imaged using a Zeiss AxioImager M2 motorized microscopy system with Zeiss W N-Achroplan 40×/NA0.75 or Zeiss EC Plan-Neofluor 100×/NA1.3 oil objectives equipped with a high-resolution RGB HRc AxioCam camera and AxioVision software (ver. 4.8.1) in 1,388 × 1,040-pixel high-speed color-capture mode. Controls included irrelevant primary antibodies as well as secondary antibodies alone. All data within each experiment were collected at identical imaging settings.
Cell extracts and Western blotting
Whole-cell extracts as well as cytoplasmic extracts were prepared from endothelial cells as described elsewhere.19,20,36 Methods for Western blotting were as reported elsewhere (Lee et al.,19 Lee et al.,20 Sehgal et al.,36 and citations therein).
Quantitative image analyses
Fluorescence intensity in imaging data (e.g., GFP in green or Alexa Fluor 594 in red) was quantitated using the McMaster Biophotonics Facility version of NIH ImageJ software and respective utility plug-ins (now available as free downloads from http://digital.bsd.uchicago.edu/ImagejandFijiHelp.html), as reported elsewhere.38 Golgi apparatus size per cell and its fragmentation was enumerated using Otsu thresholding and area quantitation, also using ImageJ as described elsewhere.38 Trafficking of Flag-tagged BMPR2 species to the plasma membrane surface or their complete or partial blocking within the cell cytoplasm was quantitated as histograms indicating the proportion of cells enumerated in each of the surface, partially blocked, and completely blocked categories. Statistical testing was carried out using the Student t test or multigroup analysis of variance.
Results
MCTP blocks tsO45VSV-G-GFP trafficking to the surface of endothelial cells
The tsO45VSV-G-GFP membrane trafficking assay was adapted to endothelial cells. As a prerequisite for using the tsO45VSV-G-GFP trafficking assay, we noted that primary BPAECs as well as human EA.hy926 cells were able to withstand overnight incubation at the nonpermissive temperature (40°C) but that primary HPAECs and HPASMCs were not. Because of the convenience of growing EA.hy926 endothelial cells as an immortalized cell line, we elected to use these cells in most of the present experiments. Figure 1 illustrates the expression of VSV-G-GFP in EA.hy926 cells at the nonpermissive temperature (40°C), followed by its trafficking to the Golgi apparatus within 20 minutes of shiftdown to the permissive temperature (32°C; arrows in Fig. 1A, top row) and subsequent appearance of the VSV-G at the cell surface (Fig. 1A, bottom row), as assayed by surface binding of the 8G5 mAb to live cells in the cold (4°C; to stop endocytosis of the mAb19,34). Analyses of the extent of trafficking in terms of the ratio of cell-surface red immunofluorescence to total VSV-G-GFP green fluorescence provided a quantitative measure of the kinetics of VSV-G trafficking from the ER to the Golgi apparatus and from there to the plasma membrane (Fig. 1B; Hirschberg et al.34 and Lippincott-Schwartz et al.35). Clear trafficking to the cell surface was evident by 60 minutes with increasing cell-surface delivery at least up to 180 minutes (Fig. 1B; Hirschberg et al.34 and Lippincott-Schwartz et al.35). For most of the present experiments on the quantitative effects of various modalities on trafficking, we selected the 60-minute (32°C) time point as reflective of the rate of anterograde trafficking without significant retrograde or endocytic transit.34,35
Figure 1.
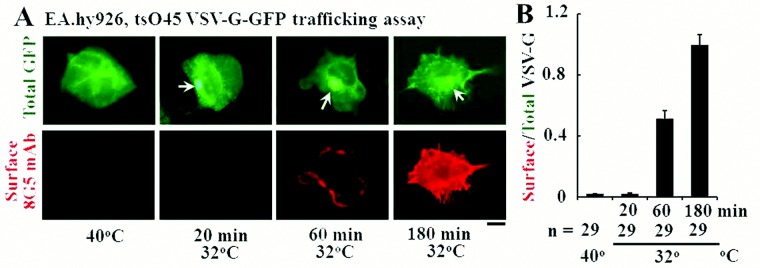
Characteristics of the quantitative anterograde trafficking assay using tsO45VSV-G-GFP in human endothelial cells (EA.hy926 cell line). The endothelial cells grown in 35-mm plates at 37°C were transfected with an expression vector for tsO45VSV-G-GFP at the same temperature. Five hours later, the cultures were shifted to 40°C. After incubation at this nonpermissive temperature for 16–18 hours, the cultures were shifted down to 32°C. At 20, 60, and 180 minutes after shiftdown to the permissive temperature, the cultures were rapidly cooled to 4°C by rinsing with ice-cold phosphate-buffered saline, and the intact live cells reacted with 8G5 F11 monoclonal antibody (mAb) to G glycoprotein of the vesicular stomatitis virus (VSV-G) for 10 minutes in the cold. The cultures were then fixed with paraformaldehyde, reacted with Alexa Fluor 594–tagged donkey anti–mouse antibody, and washed extensively; single cells were then imaged in both the green (for green fluorescent protein [GFP]) and the red (for cell-surface VSV-G) channels, and the respective surface red/total green ratios were derived using ImageJ software. A illustrates representative cell images at different times in the trafficking assay (arrows indicate Golgi apparatus). B summarizes the single-cell-based quantitation (mean ± standard error; n = number of single cells enumerated). Scale bar = 10 μm.
This VSV-G-GFP membrane trafficking assay was validated by testing whether exposure of endothelial cells to MCTP inhibited trafficking (Fig. 2). It has been previously shown by us that exposure of endothelial cells to MCTP led to marked accumulation in the enlarged Golgi apparatus of multiple tether and membrane proteins that mediate anterograde trafficking.36 Endothelial cells (BPAECs in Fig. 2A, 2B; EA.hy926 cells in Fig. 2C, 2D) were exposed to MCTP for 1 day at 37°C and then transfected with the tsO45VSV-G-GFP expression plasmid together with a shiftup to 40°C. One day later, the cultures were shifted down to 32°C, and the extent of VSV-G trafficking to the cell surface was evaluated after 60 minutes. The data in Figure 2 confirm clear inhibition of anterograde vesicular membrane trafficking in MCTP-treated BPAECs (Fig. 2A, 2B) and EA.hy927 cells (Fig. 2C, 2D).
Figure 2.

Exposure of endothelial cells to monocrotaline pyrrole (MCTP) inhibits trafficking of the anterograde G glycoprotein of the vesicular stomatitis virus (VSV-G)–green fluorescent protein (GFP). Replicate 35-mm cultures (3–4 per variable) of bovine pulmonary arterial endothelial cells (BPAECs; A, B) or EA.hy926 cells (C, D) were exposed once to MCTP (groups designated M) or left untreated (groups designated C). One day later, the cultures were transfected with the tsO45VSV-G-GFP expression vector, and the trafficking assay was carried out as indicated in the legend for Figure 1 with a shiftdown to 32°C for 1 hour. A and C illustrate representative cell images in the different control and experimental groups, and B and D summarize the respective quantitation, with the mean surface/total GFP value in the untreated control (C) group at 1 hour after shiftdown taken as 100%. Data are expressed in terms of single cells enumerated (mean ± SE using the indicated n). Asterisks indicate 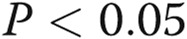 compared with the control (1-hour) group. Scale bars = 10 μm. mAb: monoclonal antibody.
compared with the control (1-hour) group. Scale bars = 10 μm. mAb: monoclonal antibody.
Inhibition of tsO45VSV-G-GFP trafficking on combinatorial knockdown of STAT5a, STAT5b, endothelial NOS (eNOS), and/or BMPR2
The VSV-G trafficking assay was used to evaluate the effects of functional haploinsufficiency of STAT5a, STAT5b, eNOS, and BMPR2 singly or in combination. The respective proteins were downregulated using the siRNA approach. The Western blots compiled in Figure 3 indicate the effectiveness of the respective siRNAs introduced into EA.hy926 cells to reduce levels of the indicated target proteins (range, 60%–80% reduction). The trafficking data in Figure 4A and 4B confirm the previous observation that the combination of STAT5a/b downregulation reduced VSV-G trafficking.19 We then investigated the effects of combining eNOS or BMPR2 downregulation with downregulation of STAT5a or STAT5b. The data in Figure 5A and 5B show that knockdown of endogenous eNOS significantly enhanced the trafficking defect produced by STAT5a or STAT5b downregulation alone. Similarly, the data in Figure 5C and 5D show that functional haploinsufficiency of endogenous BMPR2 significantly enhanced the trafficking defect produced by STAT5a or STAT5b downregulation alone.
Figure 3.

Western blot analyses showing the knockdown of signal transducer and activator of transcription 5a (STAT5a), STAT5b, endothelial nitric oxide synthase (eNOS), and bone morphogenetic protein receptor type 2 (BMPR2) using respective small interfering RNAs (siRNAs). EA.hy926 cells were transfected with the respective siRNA preparations as indicated or with the scrambled control siRNA (Scr), and whole-cell extracts were prepared 2 days later. Western blots, prepared using matched protein amounts in each panel, were probed for the indicated protein and for β-actin. In D, the lane designated “STAT5a/b” represents a cell extract from a culture transfected with both STAT5a and STAT5b siRNAs. In E, “*BMPR2” represents a duplicate experiment from the one shown as “BMPR2”; the actin lanes in E correspond to lanes marked “BMPR2.”
Figure 4.

Inhibition of anterograde trafficking of the G glycoprotein of the vesicular stomatitis virus (VSV-G) by the combined knockdown of signal transducer and activator of transcription 5a (STAT5a) and STAT5b. Replicate cultures of EA.hy926 cells in individual 35-mm plates were cotransfected with the tsO45VSV-G-GFP expression plasmid and the various small interfering RNAs (siRNAs) as indicated. Five hours later, the cells were shifted up to the nonpermissive temperature (40°C). After overnight incubation (16–18 hours), the cells were shifted down to 32°C for 1 hour, and the extent of VSV-G–green fluorescent protein (GFP) trafficking to the cell surface was quantitated as characterized in Figures 1 and 2. A illustrates representative cell images in the control group and in the experimental group, which received both STAT5a/b siRNAs. B summarizes the respective quantitation, with the mean surface/total GFP value in the untreated control (Scr) group at 1 hour after shiftdown taken as 100%. Data are expressed in terms of single cells enumerated (mean ± standard error using the indicated n). The asterisk indicates 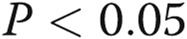 compared with the control (1-hour) group. Scale bar = 10 μm. mAb: monoclonal antibody.
compared with the control (1-hour) group. Scale bar = 10 μm. mAb: monoclonal antibody.
Figure 5.

Combinatorial knockdowns of endothelial nitric oxide synthase (eNOS) or bone morphogenetic protein receptor type 2 (BMPR2) together with that of either signal transducer and activator of transcription 5a (STAT5a) or STAT5b inhibited anterograde trafficking of the G glycoprotein of the vesicular stomatitis virus (VSV-G). Replicate cultures of EA.hy926 cells in 35-mm plates were cotransfected with the indicated small interfering RNAs (siRNAs) together with the VSV-G–green fluorescent protein (GFP) expression construct, and the extent of anterograde VSV-G trafficking to the cell surface was assayed 1 day later as indicated in the legend for Figure 4. A and C summarize the respective quantitation with the mean surface/total GFP value in the untreated control (Scr) group at 1 hour after shiftdown taken as 100%. Data are expressed in terms of single cells enumerated (mean ± standard error using the indicated n). B and C show illustrative examples of the single-cell trafficking images. Scale bar = 10 μm. Asterisks indicate 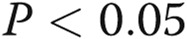 compared with the control group, and pound signs indicate
compared with the control group, and pound signs indicate 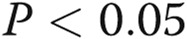 compared with the STAT5a-alone group.
compared with the STAT5a-alone group.
Downregulation of STAT5a/b inhibited trafficking of wt BMPR2 to the cell surface
We then investigated whether knockdown of STAT5a/b affected the subcellular localization of wt BMPR2. In these experiments, EA.hy926 cells were transfected with an expression vector for Flag-tagged wt human BMPR2 together with siRNAs for STAT5a/b. One day later, the cells were fixed and evaluated for the localization of the exogenously expressed BMPR2 using an anti-Flag antibody as the probe and compared it with that of an ER marker, RTN4 (also called Nogo-B). The data in Figure 6A show illustrative examples of the normal localization of wt BMPR2 at the plasma membrane (leftmost column), and the intracellular trapping of wt BMPR2 in the ER in cells cotransfected with STAT5a/b siRNAs (right two columns). This change in phenotype was quantitated using a scoring scheme for plasma membrane localization, partial intracellular block, and complete block (Fig. 6B). The data show a dramatic shift in the subcellular localization of wt BMPR2 away from the plasma membrane in cells with STAT5a/b knockdown (Fig. 6B).
Figure 6.

Double knockdown of signal transducer and activator of transcription 5a (STAT5a) and STAT5b inhibited trafficking of wild-type (wt) Flag–bone morphogenetic protein receptor type 2 (BMPR2) to the cell surface. Replicate cultures of EA.hy926 cells in 35-mm plates were cotransfected with the indicated small interfering RNAs (siRNAs; either scrambled or a combination of STAT5a/b) together with the wt Flag-BMPR2 expression construct at 37°C. One day later, the cultures were fixed, permeabilized using 0.1% Triton X-100, and probed for the subcellular localization of BMPR2 (“Flag”) and the endoplasmic reticulum (ER) marker reticulon 4 (RTN4, also called Nogo-B). Cells showing predominantly cell-surface localization (as in A, leftmost column), a partial trafficking block (as in A, middle row), or a complete block in trafficking (as in A, rightmost column) were enumerated. Scale bar = 10 μm. B summarizes the distribution histogram of the different phenotypic classes derived from 3 replicate cultures (mean ± standard error; n = total number of cells scored). The asterisk indicates 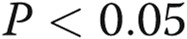 using multigroup analysis of variance in comparison with the control (Scr) histogram.
using multigroup analysis of variance in comparison with the control (Scr) histogram.
Intracellular trapping of the KDF and F14 mutants of BMPR2
The PAH-associated mutants of BMPR2 designated KDF and F14 have been previously reported to be trapped in intracellular locations away from the plasma membrane.4,39,40 The data in Figure 7A confirm these prior observations. The wt species is observed at the cell periphery, consistent with a plasma membrane localization. In contrast, the KDF species has a predominantly Golgi-like localization, and F14 is localized at the Golgi but also has a diffuse pattern consistently overlapping the RTN4 ER marker. Figure 7B recapitulates these altered distributions but also shows the presence of BMPR2 species at the Golgi. Intriguingly, overexpression of the wt and KDF mutants led to a more compact Golgi apparatus (Fig. 7B, arrows).
Figure 7.

Pulmonary arterial hypertension disease–derived mutants KDF and F14 of bone morphogenetic protein receptor type 2 (BMPR2) were mislocalized away from the plasma membrane and did not fragment the Golgi apparatus. A, B, EA.hy926 cells were transfected with expression vectors for Flag-tagged wild-type (wt), KDF, or F14 species of human BMPR2, and their subcellular localization was investigated. A and B illustrate representative images of the respective Flag-tagged BMPR2 species in comparison with a marker for the endoplasmic reticulum (ER; A; reticulon 4 [RTN4], also called Nogo-B) or Golgi apparatus (B; the tether GM130). Scale bar = 10 μm. (The wt species is predominantly at the cell surface, KDF in the Golgi with some in the ER, and F14 mainly trapped in the ER/Golgi.) Arrows in B show the more compact Golgi apparatus in Flag-BMPR2-expressing cells; arrowheads show the less compact Golgi in untransfected cells. C summarizes the area of the Golgi apparatus in respective Flag-positive cells compared with that in untransfected Flag-negative cells, derived using ImageJ software. Asterisks indicate 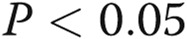 in comparison with Flag-negative cells. n = number of cells enumerated.
in comparison with Flag-negative cells. n = number of cells enumerated.
We then investigated whether overexpression of wt, KDF, or F14 BMPR2 species affected membrane trafficking in trans, that is, of other molecules that also should reach the cell surface. Figure 8A and 8B summarize data showing the modest but significant inhibitory effect of overexpression of these mutant BMPR2 species in the tsO45VSV-G-GFP trafficking assay. Thus, under the conditions of this experiment, the intracellular trapping of the KDF and F14 mutants of BMPR2 (Fig. 7A) also inhibited the trafficking of another unrelated integral membrane protein (VSV-G in this assay) through the ER/Golgi anterograde pathway to the cell surface in a dominant-negative manner (Fig. 8A). The combination of transfecting vectors for the wt, KDF, or F14 BMPR2 species together with STAT5a siRNA knockdown did not further inhibit VSV-G trafficking from that produced by STAT5a siRNA alone (data not shown).
Figure 8.

Pulmonary arterial hypertension disease–derived mutants KDF and F14 of bone morphogenetic protein receptor type 2 (BMPR2) inhibited trafficking of the G glycoprotein of the vesicular stomatitis virus (VSV-G) in trans. EA.hy926 cells were transfected with expression vectors for Flag-tagged wild-type (wt), KDF, or F14 species of human BMPR2 together with the tsO45VSV-G-GFP expression vector, and the extent of anterograde VSV-G trafficking to the cell surface was assayed 1 day later, as indicated in the legend for Figure 4. A summarizes the respective quantitation, with the mean surface/total green fluorescent protein (GFP) value at 1 hour after shiftdown in the group transfected with the wt BMPR2 expression vector taken as 100%. Data are expressed in terms of single cells enumerated (mean ± standard error using the indicated n). Asterisks indicate 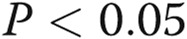 compared with the wt group. B illustrates representative images at the single-cell level. Scale bar = 10 μm.
compared with the wt group. B illustrates representative images at the single-cell level. Scale bar = 10 μm.
Effect of E2 on the subcellular localization of wt BMPR2
In light of recent reports highlighting the effects of E2 on plasma membrane and ER functions in a nongenomic manner28-32 and the known responsiveness of STAT5a/b proteins to E2,23 we investigated whether exposure of endothelial cells to E2 with or without additional knockdown of STAT5a/b might affect the subcellular localization of wt BMPR2. The data in Figure 9A and 9B first confirm the effect of STAT5a/b knockdown in causing increased intracellular trapping of wt BMPR2. Second, these data show that E2 alone also caused increased intracellular trapping of wt BMPR2. Third, E2 plus STAT5a/b knockdown was more effective in causing intracellular trapping of wt BMPR2 (Fig. 9B).
Figure 9.

Estradiol 17β (E2) alone and in combination with signal transducer and activator of transcription 5a/b (STAT5a/b) knockdown inhibited the extent of cell-surface localization of wild-type (wt) Flag–bone morphogenetic protein receptor type 2 (BMPR2). Replicate cultures of EA.hy926 cells in 35-mm plates were cotransfected with the indicated small interfering RNAs (siRNAs; either scrambled or a combination of STAT5a/b) together with wt Flag-BMPR2 expression construct and then kept in the presence or absence of E2 (10 ng/mL) for 1 day. The cultures were fixed, permeabilized using 0.1% Triton X-100, and probed for the subcellular localization of BMPR2 using the anti-Flag antibody. A illustrates representative images (scale bar = 10 μm), and B shows histograms of the phenotypic characterization of the localization of BMPR2 as defined in the legend for Figure 6. Asterisks indicate 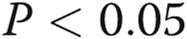 using a multigroup analysis of variance in comparison with the control (Scr) histogram, the “a” indicates
using a multigroup analysis of variance in comparison with the control (Scr) histogram, the “a” indicates 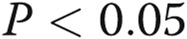 in comparison with the STAT5a/b histogram, and the circle indicates
in comparison with the STAT5a/b histogram, and the circle indicates 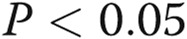 in comparison with the E2 + Scr histogram.
in comparison with the E2 + Scr histogram.
While E2 has been shown to affect STAT5 transcriptional function23 and the estrogen receptor has been shown to bind to STAT5 species,23 the effect of longer-term exposure (24 hours) of cells to E2 or the related compound 2-ME on the expression of STAT5a/b and on membrane trafficking is not known. The Western blotting data in Figure 10A and 10B show that E2 and 2-ME exposure for 24 hours led to a selective reduction in the expression of STAT5a but not STAT5b. The data in Figure 10C and 10D show that this was accompanied by reduced VSV-G membrane trafficking, consistent with the data in Fig. 10A and 10B showing the inhibitory effect of E2 on plasma membrane localization of wt BMPR2.
Figure 10.

Estradiol 17β (E2) and 2-methoxyestradiol (2-ME) decreased signal transducer and activator of transcription 5a (STAT5a) but not STAT5b protein levels and inhibited trafficking of the G glycoprotein of the vesicular stomatitis virus (VSV-G)–green fluorescent protein (GFP). A, B, Cultures of EA.hy926 cells in triplicate were exposed to E2 (10 ng/mL) or 2-ME (1.5 ng/mL) or were kept untreated (control) for 24 hours, and respective whole-cell extracts were prepared and assayed for STAT5a, STAT5b, and β-actin by Western blotting. Asterisks indicate 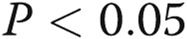 compared with the control group (
compared with the control group (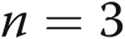 independent cultures). C, D, Cultures of EA.hy926 cells transfected with an expression vector for tsO45VSV-G-GFP were exposed to E2 (5 ng/mL) or 2-ME (1.5 ng/mL), and VSV-G trafficking to the cell surface was evaluated as in the legend for Figure 4. C shows representative images from the trafficking assay at the single-cell level (scale bar = 10 μm), and D summarizes the overall quantitation. Asterisks indicate
independent cultures). C, D, Cultures of EA.hy926 cells transfected with an expression vector for tsO45VSV-G-GFP were exposed to E2 (5 ng/mL) or 2-ME (1.5 ng/mL), and VSV-G trafficking to the cell surface was evaluated as in the legend for Figure 4. C shows representative images from the trafficking assay at the single-cell level (scale bar = 10 μm), and D summarizes the overall quantitation. Asterisks indicate 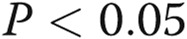 compared with the control group (n = number of cells evaluated).
compared with the control group (n = number of cells evaluated).
Rescue of intracellular trapping of the KDF mutant by probenecid
Probenecid is in clinical use as a uricosuric agent in patients with nephritis due to mutations in the Tamm-Horsfall glycoprotein (THG).41 This drug restored trafficking of the mutant THG protein species to the plasma membrane.41 We investigated whether probenecid might have a similar effect on BMPR2 mutants and compared it with the known chemical chaperone effect of 4-phenylbutyrate (4PBA).40 The data in Figure 11A and 11B show that probenecid improved the plasma membrane localization of the KDF mutant of BMPR2 to an extent similar to or better than that of 4PBA. This effect was mutant specific in that probenecid did not have a significant effect on the intracellular localization of the F14 mutant (data not shown).
Figure 11.

Probenecid rescues the intracellular trapping of the KDF mutant of bone morphogenetic protein receptor type 2 (BMPR2). EA.hy926 cells were transfected with Flag-tagged wild-type (wt) or the KDF species of BMPR2 and then, beginning 5 hours later, exposed to either probenecid (PB; 2 mM) or 4-phenylbutyrate (4PBA; 5 mM) or left untreated. One day later, the subcellular localization of the respective BMPR2 species was evaluated using anti-Flag antibody. A illustrates some of the representative imaging data (scale bar = 10 μm). B presents histograms of the phenotypic distribution of BMPR2 species in single cells (n = number of cells enumerated) under each experimental condition using the method described in the legend for Figure 6. Asterisks indicate indicates 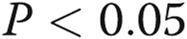 for the KDF distributions in treated cultures using a multigroup analysis of variance in comparison with that derived from the untreated culture.
for the KDF distributions in treated cultures using a multigroup analysis of variance in comparison with that derived from the untreated culture.
Discussion
The mechanisms that modulate the cell-surface residence and function of the BMPR2 receptor chain has attracted attention because of extensive genetic evidence implicating mutations in and haploinsufficiency of BMPR2 in the pathogenesis of PAH.1-4 Although such BMPR2 mutations have been genetically characterized as autosomal dominant, there is low penetrance (only 10%–15%), a delayed onset (median age, third decade in women and fourth decade in men), and a gender bias (two- to fourfold higher incidence in women than in men) in development of the overt disease. Thus, there has been a search for candidate second-hit modalities that might trigger reduced BMPR2 function and disease development. The present investigation sought to identify potential second-hit modalities that might combinatorially inhibit anterograde intracellular membrane trafficking and thus reduce delivery of BMPR2 to the cell surface. Using the quantitative tsO45VSV-G-GFP trafficking assay, the combinatorial knockdown of STAT5a, STAT5b, BMPR2, and/or eNOS as well as exposure to E2 or 2-ME were observed to significantly inhibit anterograde trafficking. These combinations also led to intracellular trapping of wt Flag-tagged BMPR2. The PAH disease–derived F14 and KDF mutants of Flag-tagged BMPR2, which were previously known to be trapped in the ER/Golgi, also inhibited VSV-G trafficking in trans, providing a mechanism for such mutants to alter the global cell-surface landscape of vascular cells. Impressively, probenecid, a drug in clinical use today as a uricosuric agent, restored trafficking of the KDF but not the F14 mutant of BMPR2.
The quantitative anterograde trafficking assay34,35 involving expression of the integral membrane protein tsO45VSV-G-GFP for 1 day at the nonpermissive temperature (40°C) using an exogenous expression vector (VSV-G remains trapped in the ER under these conditions), followed by studies of the synchronous trafficking of VSV-G from ER to Golgi to plasma membrane on shiftdown to the permissive temperature (32°C), was adapted to human endothelial cells (EA.hy926 cells). These cells withstand overnight culture at the nonpermissive temperature. The strength of this assay is the availability of a mAb (clone I1, stock 8G5F11)33 that reacts with an epitope on VSV-G that is intraluminal in the ER/Golgi but is exposed external to the cell when this protein is on the cell surface. Thus, the 8G5 mAb reactivity (10 minutes) of intact live VSV-G-expressing cells kept at 4°C, followed by washing in the cold, fixation, and display using Alexa Fluor 594–tagged secondary antibody (in red), provided quantitative evaluation of the relative fraction of VSV-G molecules at the cell surface when expressed as a ratio of the total GFP green fluorescence in the same cell. This quantitative assay has been extensively used in studies of the detailed molecular cell biology of anterograde vesicular trafficking by numerous previous investigators.35
In previous studies,10,36 we reported that exposure of PAECs to MCTP in cell culture led to the development of a dramatic megalocytosis phenotype, which included marked increase in cell size, enlargement of the Golgi apparatus, and the trapping in the Golgi of proteins that mediate vesicular trafficking (the multitude of tethers, soluble N-ethylmaleimide-sensitive attachment proteins [SNAPs], and their receptors [SNAREs] that mediate vesicular membrane fusion) as well as trapping in the Golgi of cargo proteins, such as eNOS and caveolin 1. We surmised that this phenotype would include an inhibition of anterograde membrane trafficking.10,36 Thus, as part of the process of validating the tsO45VSV-G-GFP assay, the effect of exposing endothelial cells to MCTP on anterograde trafficking was investigated. The data obtained confirm that BPAECs and EA.hy926 cells exposed to MCTP show a marked reduction in anterograde vesicular transit of the VSV-G integral membrane protein to the plasma membrane.
A focus of the present experiments was to investigate whether functional haploinsufficieny of BMPR2 (such as using siRNA knockdown) or expression of PAH-associated BMPR2 mutants might affect anterograde trafficking of other membrane or cell-surface proteins in trans. The quantitative VSV-G trafficking assay was selected for these experiments. For comparison, we included other candidate second-hit modalities, such as functional haploinsufficiencies of eNOS, STAT5a, and STAT5b (using siRNA knockdowns) as well as exposure to E2 or 2-ME singly or in combination. The data obtained show that combinatorial reduction of STAT5a or STAT5b together with reduction of either BMPR2 or eNOS inhibited VSV-G trafficking. Moreover, exposure of endothelial cells to E2 or 2-ME also inhibited VSV-G movement to the cell surface. Importantly, overexpression of the F14 and KDF mutants of BMPR2 also inhibited VSV-G trafficking in trans. That this inhibitory effect on anterograde trafficking was evident on introduction of these mutants into endothelial cells containing endogenous wt BMPR2 suggests that these mutants have a dominant-negative phenotype on trafficking. However, in contrast to MCTP or nitric oxide scavenging, which caused enlargement and fragmentation of the structure of the Golgi apparatus,10,36,38 overexpression of the F14 or KDF mutant did not cause Golgi enlargement and/or fragmentation—the effect, if anything, was to make the Golgi more compact. However, anterograde trafficking was reduced. Overall, these experiments helped identify PAH-relevant candidate second-hit modalities that could inhibit transit of membrane proteins to the cell surface when affected in a combinatorial manner.
These candidate modalities were investigated for their effects on the cell-surface residence of BMPR2 using a semiquantitative immunofluorescence assay with the exogenously expressed Flag-tagged receptor. The combination of STAT5a and STAT5b knockdown markedly inhibited appearance of wt Flag-BMPR2 at the cell surface, as did exposure to E2. Both E2 and 2-ME selectively reduced protein levels of STAT5a but not STAT5b, concomitant with their inhibitory effects on VSV-G trafficking and reduced appearance of wt Flag-BMPR2 at the cell surface. These data provide a glimpse of potential combinatorial inhibitory interactions between the STAT5a/b knockdown and the effects of estrogens on intracellular membrane trafficking. The data also raise the possibility that the reduction in trafficking after E2 exposure might be secondary to a reduction in STAT5a. Whether there is in fact a reduction of STAT5a or STAT5b in cells in lesions of human PAH in a gender-specific manner remains to be investigated.
Moreover, as proof of principle, we discovered that probenecid could restore delivery of the KDF mutant of BMPR2, but not the F14 mutant, to the cell surface. Probenecid, a uricosuric agent in clinical use today, has been shown to restore the anterograde trafficking of mutant species of THG that remain trapped within renal epithelial cells.41 This drug helps mediate proper folding of mutant THG species and thus their delivery to the cell surface; however, the effect is mutation specific. In a similar manner, the compound kalydeco has been shown recently (and approved by the Food and Drug Administration) to restore trafficking of the G551D mutant of the cystic fibrosis receptor (CFTR), but it does not have a similar restorative effect on other CFTR mutants.42 Probenecid had a restorative effect on the cell-surface trafficking of the KDF mutant but not the F14 mutant. Sobolewski et al.40 have shown that 4PBA could partially restore trafficking of the F14 mutant of BMPR2 to the cell surface in HeLa cells. In our hands, we can confirm the restorative effect of 4PBA on the KDF and the F14 (data not shown) mutants.
The estrogen paradox
Recent discussions of the estrogen paradox17,18,43-45 have continued to point out that E2 reduces PAH in many experimental models of PAH. In contrast, human IPAH and FPAH is two to fourfold more prevalent in postpubertal females.1-4,6 In addition, as has been pointed out by Das et al.,46 the natural history of the human disease has a prolonged lead time (a delayed onset) and a low penetrance in contrast to various animal models in which the disease develops over a period of several weeks. Various investigators have puzzled over this paradox in the development of the human disease in terms of the effects of E2 at the whole-cell (increased proliferation of smooth muscle cells) or tissue (altered vascular tone) levels.17,18,43-45 However, to date there has been no consideration of the effects of E2 on subcellular mechanisms, for example, in terms of the newly discovered effects of this hormone on intracellular trafficking leading to alterations in the intracellular localization of cell-surface receptors, such as that for the dopamine transporter.28-32 The present data suggest a need for consideration of the effects of E2 regarding STAT5a levels and the cell-surface localization of BMPR2.
Limitations
A limitation of the present study is the use of the EA.hy926 cell line in the trafficking assays. This factor VIII–positive endothelial cell line was originally derived by the fusion of human umbilical vein endothelial cells with human lung carcinoma A549 cells and is continuously grown in selection medium containing hypoxanthine, aminopterin, and thymidine (HAT).37 The cells remain factor VIII positive. Importantly, these cells withstand overnight incubation at 40°C and thus can be used as cell substrates for the quantitative tsO45VSV-G-GFP trafficking assay. Primary HPAECs and HPASMCs did not withstand incubation at the nonpermissive temperature. A further advantage of EA.hy926 cells became apparent when we compared subcellular localization of exogenously expressed wt Flag-tagged BMPR2 in these cells with that in HPAECs (with both cell types kept at 37°C). Among EA.hy926 cells, most transfected cells displayed the bulk of the wt Flag-BMPR2 at the cell surface within 1 day of transfection of the expression vector. In contrast, significant intracellularly localized wt Flag-BMPR2 was observed in HPAECs 1 day after transfection of the expression vector. These observations account for our selection of EA.hy926 human endothelial cells for the present experiments. However, at this time we recognize the limitation that the extent to which the present data might be extrapolated to primary human pulmonary arterial vascular cells (endothelial and smooth muscle) in cell culture and in vivo remains open. Additionally, the question of whether delivery (or lack thereof) of the wt BMPR2 receptor to the cell surface due to trafficking defects by second-hit modalities causes commensurate functional defects in terms of signaling and transcriptional responses mediated by this receptor awaits investigation. Similarly, whether probenecid-mediated rescue of the cell-surface delivery of the KDF mutant translates into functional improvement at the single-cell or tissue level awaits exploration.
Conclusion
Overall, the present data identify several potential second-hit modalities that might be relevant in the pathogenesis of PAH in terms of their combinatorial inhibitory effects on intracellular anterograde membrane trafficking and thus the correct cell-surface delivery and residence of BMPR2 species. Moreover, alterations in anterograde membrane trafficking can be expected to cause changes not just of BMPR2 residence at the cell surface but of the cell-surface landscape globally, affecting multiple different vasorelevant receptors and signaling pathways.
Acknowledgements
We thank Douglas S. Lyles for the mouse hybridoma clone I1, stock 8G5F11, which makes the anti-VSV-G monoclonal antibody to a cell-surface-specific epitope;33 Dr. Lippincott-Schwartz for the tsO45VSV-G-GFP expression vector.34
Source of Support: This work was supported by National Institutes of Health research grants R01 HL-087176 and R03 HL-114601.
Conflict of Interest: None declared.
References
- 1.Tuder RM, Marecki JC, Richter A, Fijlkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med 2007;28:23–42. [DOI] [PMC free article] [PubMed]
- 2.Rabinovitch M. Molecular pathogenesis of pulmonary hypertension. J Clin Invest 2008;118:2372–2379. [DOI] [PMC free article] [PubMed]
- 3.Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet 2003;361:1533–1544. [DOI] [PubMed]
- 4.Morrell NW. Role of bone morphogenetic protein receptors in the development of pulmonary hypertension. Adv Exp Med Biol 2010;661:251–264. [DOI] [PubMed]
- 5.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshoing SD, McLaughlin VV, Jessup M, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:261–272. [DOI] [PMC free article] [PubMed]
- 6.Fessel JP, Loyd JE, Austin ED. The genetics of pulmonary arterial hypertension in the post-BMPR2 era. Pulm Circ 2011;1:305–319. [DOI] [PMC free article] [PubMed]
- 7.Yu PB, Beppu H, Kawai N, Li E, Bloch KD. Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand–specific gain of signaling in pulmonary artery smooth muscle cells. J Biol Chem 2005;280:24443–24450. [DOI] [PubMed]
- 8.West J, Harral J, Lane K, Deng Y, Ickes B, Crona D, Albu S, Stewart D, Fagan K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol 2008;295:L744–L755. [DOI] [PMC free article] [PubMed]
- 9.Johnson JA, Hemnes AR, Perrien DS, Schuster M, Robinson LJ, Gladson S, Loibner H, et al. Cytoskeletal defects in Bmpr2-associated pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2012;302:L474–L484. [DOI] [PMC free article] [PubMed]
- 10.Sehgal PB, Lee JE. Protein trafficking dysfunctions: role in the pathogenesis of pulmonary arterial hypertension. Pulm Circ 2011;1:17–32. [DOI] [PMC free article] [PubMed]
- 11.Sieber C, Kopf J, Hiepen C, Knaus P. Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev 2009;20:343–355. [DOI] [PubMed]
- 12.Hartung A, Bitton-Worms K, Rechtman MM, Wenzel V, Boergermann JH, Hassel S, Henis YI, Knaus P. Different routes of bone morphogenic protein (BMP) receptor endocytosis influence BMP signaling. Mol Cell Biol 2006;26:7791–7805. [DOI] [PMC free article] [PubMed]
- 13.DiGuglielmo GM, LeRoy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-β receptor signaling and turnover. Nat Cell Biol 2003;5:410–421. [DOI] [PubMed]
- 14.Sehgal PB. Paradigm shifts in STAT signaling. Semin Cell Dev Biol 2008;19:329–340. [DOI] [PMC free article] [PubMed]
- 15.Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, Wheeler LA, Parl FF, Loyd JE, Phillips JA III. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J 2009;34:1093–1099. [DOI] [PMC free article] [PubMed]
- 16.Austin ED, Hamid R, Hemnes AR, Loyd JE, Blackwell T, Yu C, Phillips JA III, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ 2012;3:6. [DOI] [PMC free article] [PubMed]
- 17.Umar S, Rabinovitch M, Eghbali M. Estrogen paradox in pulmonary hypertension: current controversies and future perspectives. Am J Respir Crit Care Med 2012;186:125–131. [DOI] [PMC free article] [PubMed]
- 18.White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, Campbell A, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation 2012;126:1087–1098. [DOI] [PubMed]
- 19.Lee JE, Yang Y-M, Liang F-X, Gough DJ, Levy DE, Sehgal PB. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am J Physiol Cell Physiol 2012;302:C804–C820. [DOI] [PMC free article] [PubMed]
- 20.Lee JE, Yang Y-M, Yuan H, Sehgal PB. Definitive evidence using enucleated cytoplasts for a nongenomic basis for the cystic change in endoplasmic reticulum structure caused by STAT5a/b siRNAs. Am J Physiol Cell Physiol 2013;304:C312–C323. [DOI] [PMC free article] [PubMed]
- 21.Gouilleux F, Wakao H, Mundt M, Groner B. Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J 1994;13:4361–4369. [DOI] [PMC free article] [PubMed]
- 22.Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci USA 1997;94:7239–7244. [DOI] [PMC free article] [PubMed]
- 23.Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev 2008;22:711–721. [DOI] [PMC free article] [PubMed]
- 24.Hosui A, Kimura A, Yamaji D, Zhu BM, Na R, Hennighausen L. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-β and STAT3 activation. J Exp Med 2009;206:819–831. [DOI] [PMC free article] [PubMed]
- 25.Smith P, Heath D. Electron microscopy of the plexiform lesion. Thorax 1979;34:177–186. [DOI] [PMC free article] [PubMed]
- 26.Smith P, Heath D. Ultrastructure of hypoxic hypertensive pulmonary vascular disease. J Pathol 1977;121:93–100. [DOI] [PubMed]
- 27.Sehgal PB, Mukhopadhyay S, Patel K, Xu F, Almodovar A, Tuder RM, Flores SC. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am J Physiol Lung Cell Mol Physiol 2009;297:L729–L737. [DOI] [PMC free article] [PubMed]
- 28.Acconcia F, Ascenzi P, Bocedi A, Spisni E, Tomasi V, Trentalance A, Visca P, Marino M. Palmitoylation-dependent estrogen receptor α membrane localization: regulation by 17β-estradiol. Mol Biol Cell 2005;16:231–237. [DOI] [PMC free article] [PubMed]
- 29.Watson CS, Alyea RA, Hawkins BE, Thomas ML, Cunningham KA, Jakubas AA. Estradiol effects on the dopamine transporter—protein levels, subcellular location, and function. J Mol Signal 2006;1:5. [DOI] [PMC free article] [PubMed]
- 30.Maggioloni M, Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J Endocrinol 2010;204:105–114. [DOI] [PubMed]
- 31.Dominguez R, Micevych P. Estradiol rapidly regulates membrane estrogen receptor α levels in hypothalamic neurons. J Neurosci 2010;30:12589–12596. [DOI] [PMC free article] [PubMed]
- 32.Chaudhri RA, Olivares-Navarette R, Cuenca N, Hadadi A, Boyan BD, Schwartz Z. Membrane estrogen signaling enhances tumorigenesis and metastic potential of breast cancer cells via estrogen receptor-α36 (ERα36). J Biol Chem 2012;287:7169–7181. [DOI] [PMC free article] [PubMed]
- 33.Lefrancois L, Lyles DS. The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. I. Analysis of neutralizing epitopes with monoclonal antibodies. Virology 1982;121:157–167. [PubMed]
- 34.Hirschberg K, Miller CM, Wllenberg J, Siggis ED, Phair RD, Lippincott-Schwartz J. Kinetic analysis of sceretory protein traffic and characterization of Golgi to plasma membrane transport intermediates in living cells. J Cell Biol 1998;143:1485–1503. [DOI] [PMC free article] [PubMed]
- 35.Lippincott-Schwartz J, Roberts TH, Hirschberg K. Secrotory protein trafficking and organelle dynamics in living cells. Annu Rev Cell Dev Biol 2000;16:557–589. [DOI] [PMC free article] [PubMed]
- 36.Sehgal PB, Mukhopadhyay S, Xu F, Patel K, Shah M. Dysfunctions of Golgi tethers, SNAREs, and SNAPs in monocrotaline-induced pulmonary hypertension. Am J Physiol Cell Mol Physiol 2007;292:L1526–L1542. [DOI] [PubMed]
- 37.van Oost BA, Edgell CJ, Hay CW, MacGillivray RT. Isolation of a human von Willebrand factor cDNA from the hybrid endothelial cell line EA.hy926. Biochem Cell Biol 1986;64:699–705. [DOI] [PubMed]
- 38.Lee JE, Patel K, Almodovar S, Tuder RM, Flores SC, Sehgal PB. Dependence of Golgi apparatus integrity on nitric oxide in vascular cells: implications in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2011;300:H1141–H1158. [DOI] [PMC free article] [PubMed]
- 39.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet 2002;11:1517–1525. [DOI] [PubMed]
- 40.Sobolewski A, Rudrakanchana N, Upton PD, Yang J, Crilley TK, Trembath RC, Morrell NW. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: potential for rescue. Hum Mol Genet 2008;17:3180–3190. [DOI] [PubMed]
- 41.Ma L, Liu Y, El-Achkar TM, Wu XR. Molecular and cellular effects of Tamm-Horsfall protein mutations and their rescue by chemical chaperones. J Biol Chem 2012;287:1290–1305. [DOI] [PMC free article] [PubMed]
- 42.Corbyn Z. Promising new era dawns for cystic fibrosis treatment. Lancet 2012;379:1475–1476. [DOI] [PubMed]
- 43.de Jesus Perez VA. Making sense of the estrogen paradox in pulmonary arterial hypertension. Am J Respir Crit Care Med 2011;184:629–630. [DOI] [PubMed]
- 44.White K, Dempsis Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17β estradiol in the development of pulmonary arterial hypertension. Cardiovasc Res 2011;90:373–382. [DOI] [PubMed]
- 45.Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, van der Laarse A, Eghbali M. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med 2011;184:715–723. [DOI] [PMC free article] [PubMed]
- 46.Das M, Fessel J, Tang H, West J. A process-based review of mouse models of pulmonary hypertension. Pulm Circ 2012;2:415–433. [DOI] [PMC free article] [PubMed]