

Abstract Abstract
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are severe syndromes resulting from the diffuse damage of the pulmonary parenchyma. ALI and ARDS are induced by a plethora of local or systemic insults, leading to the activation of multiple pathways responsible for injury, resolution, and repair or scarring of the lungs. Despite the large efforts aimed at exploring the roles of different pathways in humans and animal models and the great strides made in understanding the pathogenesis of ALI/ARDS, the only viable treatment options are still dependent on ventilator and cardiovascular support. Investigation of the pathophysiological mechanisms responsible for initiation and resolution or advancement toward lung scarring in ALI/ARDS animal models led to a better understanding of the disease’s complexity and helped in elucidating the links between ALI and systemic multiorgan failure. Although animal models of ALI/ARDS have pointed out a variety of new ideas for study, there are still limited data regarding the initiating factors, the critical steps in the progression of the disease, and the central mechanisms dictating its resolution or progression to lung scarring. Recent studies link deficiency of intersectin-1s (ITSN-1s), a prosurvival protein of lung endothelial cells, to endothelial barrier dysfunction and pulmonary edema as well as to the repair/recovery from ALI. This review discusses the effects of ITSN-1s deficiency on pulmonary endothelium and its significance in the pathology of ALI/ARDS.
Keywords: intersectin-1s, endothelium, lung injury, apoptosis, permeability
Introduction
Acute lung injury (ALI) and its severe form, acute respiratory distress syndrome (ARDS), are syndromes of acute hypoxemic respiratory failure resulting from a variety of direct and/or indirect injuries to the gas exchange unit of the lungs.1 Clinically, ALI is characterized by (1) significant arterial hypoxemia (partial pressure of arterial O2/fraction of inspired O2 [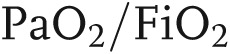 ]; 300 mmHg in ALI and <200 mmHg in ARDS), which is refractory to oxygen supplementation, (2) patchy bilateral lung infiltrates suggestive of edema, (3) decreased pulmonary compliance, (4) the absence of measurable evidence of cardiac failure, and (5) lack of an alternative justification for these findings.2,3 ALI/ARDS is the result of a variety of causes that trigger stereotyped physiopathological responses, making the modeling of the illness difficult. A variety of insults can initiate and induce ALI/ARDS; the American-European Consensus Conference in 1994 divided these insults into (1) direct (pulmonary or primary) injuries of lung parenchyma, such as aspiration of gastric content and smoke inhalation, and (2) indirect (extrapulmonary or secondary) injuries, such as sepsis, trauma, and blood transfusions, mainly a result of an acute systemic inflammatory response.4 Recently, some studies have shown differences in clinical sequelae between the two modes of injury, because others have not demonstrated any significant differences in outcomes and response to therapy in ARDS due to direct versus indirect causes. Direct lung injury was associated with higher mortality rate, increased ventilator requirements, and pulmonary consolidation, whereas indirect lung injury was associated with interstitial edema and alveolar collapse.5 Regardless of the initial cause of the injury, the mortality is high. Incidence of ALI is reported as 17–34 to 78.9 per 100,000 people per year.6,7 More recent estimates indicate around 190,000 of ALI cases per year in the United States, with 75,000 associated deaths per year.8 Unfortunately, the incidence figures are not very consistent, likely because of problems with reliability of diagnosis and also because ALI is a critical-care illness, making its epidemiology directly linked to availability of intensive care unit resources.9 The main treatment option remains ventilator and cardiovascular support, based on recognition of clinical signs.3 To facilitate case recognition and more consistent treatment strategies based on severity, the American-European Consensus Conference used epidemiological, physiological, and clinical data to revise and update the ARDS definition.10 The new “Berlin” definition of ARDS includes (1) a criterion of less than 7 days to define acute onset, (2) clinical judgment for characterizing hydrostatic pulmonary edema, unless there is no apparent ARDS risk factor, (3) three categories of ARDS severity—mild (201–300 mmHg), moderate (101–200 mmHg), and severe (≤100 mmHg)—based on the
]; 300 mmHg in ALI and <200 mmHg in ARDS), which is refractory to oxygen supplementation, (2) patchy bilateral lung infiltrates suggestive of edema, (3) decreased pulmonary compliance, (4) the absence of measurable evidence of cardiac failure, and (5) lack of an alternative justification for these findings.2,3 ALI/ARDS is the result of a variety of causes that trigger stereotyped physiopathological responses, making the modeling of the illness difficult. A variety of insults can initiate and induce ALI/ARDS; the American-European Consensus Conference in 1994 divided these insults into (1) direct (pulmonary or primary) injuries of lung parenchyma, such as aspiration of gastric content and smoke inhalation, and (2) indirect (extrapulmonary or secondary) injuries, such as sepsis, trauma, and blood transfusions, mainly a result of an acute systemic inflammatory response.4 Recently, some studies have shown differences in clinical sequelae between the two modes of injury, because others have not demonstrated any significant differences in outcomes and response to therapy in ARDS due to direct versus indirect causes. Direct lung injury was associated with higher mortality rate, increased ventilator requirements, and pulmonary consolidation, whereas indirect lung injury was associated with interstitial edema and alveolar collapse.5 Regardless of the initial cause of the injury, the mortality is high. Incidence of ALI is reported as 17–34 to 78.9 per 100,000 people per year.6,7 More recent estimates indicate around 190,000 of ALI cases per year in the United States, with 75,000 associated deaths per year.8 Unfortunately, the incidence figures are not very consistent, likely because of problems with reliability of diagnosis and also because ALI is a critical-care illness, making its epidemiology directly linked to availability of intensive care unit resources.9 The main treatment option remains ventilator and cardiovascular support, based on recognition of clinical signs.3 To facilitate case recognition and more consistent treatment strategies based on severity, the American-European Consensus Conference used epidemiological, physiological, and clinical data to revise and update the ARDS definition.10 The new “Berlin” definition of ARDS includes (1) a criterion of less than 7 days to define acute onset, (2) clinical judgment for characterizing hydrostatic pulmonary edema, unless there is no apparent ARDS risk factor, (3) three categories of ARDS severity—mild (201–300 mmHg), moderate (101–200 mmHg), and severe (≤100 mmHg)—based on the 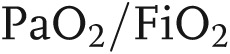 ratio, and (4) a continuous positive airway pressure value of at least 5 cm H2O. The requirement of pulmonary artery wedge pressure and the category of ALI were removed. Even if this new definition is valuable as a clinical prediction tool, it still does not resolve the heterogeneity, the complex pathology, and the cellular and molecular mechanism of ARDS. There is an unmet need for groundbreaking research to help reduce mortality and morbidity from ARDS.11 Availability of human lung tissue for scientific investigation is limited by the lack of surgical biopsies of ALI/ARDS in clinical practice. Thus, modeling the disease in animals has been the basis for many crucial advances in this field. Animal models of ALI provide an excellent tool to test in a controlled environment the data collected from critically ill patients and to investigate new pathways, fundamental for improving clinical outcomes. Widely used experimental models are based on reproducing injury to epithelial and endothelial lung barriers and acute parenchyma inflammation. Animal studies are hierarchical, from mice to primates, and differ in mimicking the complex features of human ALI; most experimental models, such as mechanical ventilation, lipopolysaccharide (LPS) injection, and cecal ligation and puncture (considered “the gold standard” for animal models of sepsis), mimic sepsis-induced ALI; oleic acid injection mimics ALI caused by lipid embolization, while acid aspiration, pulmonary ischemia/reperfusion, and surfactant abnormalities simulate different conditions (disruption of the alveolocapillary interface with neutrophilic infiltration, increased vascular permeability, etc.) associated with ALI.12 To this date, no single animal model reproduces all of the characteristics seen in patients. Although with limitations, each model is relevant for specific ALI features and helps answer mechanistic and therapeutic questions we still have about the disease. The focus of this review is to underline some of the latest attempts at modeling the disease and to understand the specific mechanisms triggered by defined factors involved in ALI/ARDS initiation, progression, and resolution.
ratio, and (4) a continuous positive airway pressure value of at least 5 cm H2O. The requirement of pulmonary artery wedge pressure and the category of ALI were removed. Even if this new definition is valuable as a clinical prediction tool, it still does not resolve the heterogeneity, the complex pathology, and the cellular and molecular mechanism of ARDS. There is an unmet need for groundbreaking research to help reduce mortality and morbidity from ARDS.11 Availability of human lung tissue for scientific investigation is limited by the lack of surgical biopsies of ALI/ARDS in clinical practice. Thus, modeling the disease in animals has been the basis for many crucial advances in this field. Animal models of ALI provide an excellent tool to test in a controlled environment the data collected from critically ill patients and to investigate new pathways, fundamental for improving clinical outcomes. Widely used experimental models are based on reproducing injury to epithelial and endothelial lung barriers and acute parenchyma inflammation. Animal studies are hierarchical, from mice to primates, and differ in mimicking the complex features of human ALI; most experimental models, such as mechanical ventilation, lipopolysaccharide (LPS) injection, and cecal ligation and puncture (considered “the gold standard” for animal models of sepsis), mimic sepsis-induced ALI; oleic acid injection mimics ALI caused by lipid embolization, while acid aspiration, pulmonary ischemia/reperfusion, and surfactant abnormalities simulate different conditions (disruption of the alveolocapillary interface with neutrophilic infiltration, increased vascular permeability, etc.) associated with ALI.12 To this date, no single animal model reproduces all of the characteristics seen in patients. Although with limitations, each model is relevant for specific ALI features and helps answer mechanistic and therapeutic questions we still have about the disease. The focus of this review is to underline some of the latest attempts at modeling the disease and to understand the specific mechanisms triggered by defined factors involved in ALI/ARDS initiation, progression, and resolution.
Animal models of ALI
Animal models of ALI/ARDS have been notably useful for exploring pathophysiological mechanisms and for evaluating novel therapeutic agents while contributing significantly to the development of treatment plans for these complex syndromes. As the scientific community began to realize that humans are inherently variable in their response to causative stimuli of ALI and that pharmacologic treatments have shown little or no benefit,13 it was claimed that one of the reasons for this unfortunate condition, at least for sepsis-induced ALI/ARDS, is the redundancy in the pathways driving the systemic inflammatory response syndrome.14 Clinical studies revealed significant variability among individuals in the expression of blood cytokines in response to LPS;15 however, detailed investigations revealed that this variability in inflammatory responses to LPS is caused by polymorphisms affecting the Toll-like receptor-1, leading to excessive inflammation and increased risk for poor outcomes in patients with sepsis.15,16 Against this background, modeling the acute and chronic changes of ALI in animals, in order to understand and establish the molecular mechanism of disease pathogenesis, has been a major challenge. Over time, many animal models of ALI have been developed (Table 1), the advantages and disadvantages in each case meticulously characterized and broadly reviewed.1,12 When the limitations are recognized and the necessary caution is used, studies performed on animal models are very useful for identifying, evaluating, and establishing key physiological mechanisms, the extent of involvement of different molecular pathways, and their subtle individual adaptation, while allowing the development of new hypotheses and therapies for treatment of human disease. The animal models highlighted in Table 1 have been extensively reviewed over time;1-3,12,31 thus, we discuss here briefly the emerging human models.
Table 1.
Animal models of ALI
| Model | General/technical considerations |
|---|---|
| LPS17 | High reproducibility, some resemblance to the human disease, strain specific |
| Oleic acid18 | Reproducible, mechanisms of action not clear |
| Bacterial administration19, 20 | Significant biological and individual variations, resemblance to toxemia |
| Cecal ligation and puncture21 | Similarities to human disease, biological variability |
| Acid aspiration22 | Reproducible, similarities to exudative phase of human disease |
| Bleomycin23 | Reproducible, strain variability, pathophysiology still not well known |
| Hyperoxia24 | Reproducible, scant inflammatory response, direct relationship with human disease questionable |
| Lavage of surfactant25 | Some similarities to the human disease, technologically demanding |
| Ischemia/reperfusion26 | Surgery needed, elevated interstitial participation |
| Mechanical stretch injury27-30,a | Challenging, mainly in small animals; ventilation strategy determines the severity of lung injury; type of ventilator and physiological monitoring are important; main advantage: clinical relevance |
High tidal volume type of ventilation injury.
Human models of ALI
An emerging field for ALI studies is the development of human models of the disease. The foremost impact of this type of ALI model is the development and testing of specific therapies and therapeutic targets. These models could be divided into three groups: in vivo, ex vivo, and inhaled bacterial products (specifically LPS). The in vivo models include the one-lung ventilation technique, in which one lung is collapsed during thoracic anesthesia while the other one is ventilated. The method has clinical significance, since 3% of patients with lobectomy and 8% of patients with pneumonectomy develop ALI and the rate of mortality in both cases reaches 40%–60%.32,33 Two other pathological states associated with high incidence of ALI are esophagectomy, a condition in which ALI was found in almost 24% of cases,34 and cardiopulmonary bypass, in which the disease developed in 2% of cases.35 One-lung ventilation is associated with pulmonary inflammation, endothelial and epithelial dysfunction, neutrophil infiltration, and vascular activation, signs that belong to ALI of different etiologies, while some cardiopulmonary bypass causative mechanisms include endothelial dysfunction, ischemia-reperfusion, inflammation, and atelectasis.35,36 These models hold a lot of practical promise, but at the same time, like all pathologies for which the mechanisms are not well understood, they hold a lot of challenges. Because of the high incidence of ALI in esophagectomy, cohorts of patients undergoing the procedure are enrolled in a clinical trial in which inhibitors of hydroxymethylglutaryl-coenzyme A (CoA) reductase (simvastatin) are tested as potential drugs for reducing the incidence of ALI (http://www.controlledtrials.com/ISRCTN5643987).
The ex vivo models usually use donor lungs that, according to the International Society for Heart and Lung Transplant, do not meet the standards for transplantation and therefore have a modified basal physiology usually associated with comorbidities and large variations in ischemic times. For ex vivo research, the lungs are kept in a viable status with well-controlled ventilation and perfusion systems, which are demanding with respect to the surgical and perfusion expertise needed, not to mention the insufficient available data regarding the effect of organ cooling, required for lung transportation. Of the two methods used for ex vivo maintenance of the lungs—isolated perfused lungs and ex vivo lung perfusion—only the first was used to study ALI.37,38 Ex vivo lung perfusion is a technically demanding methodology used to assess the quality of donor lungs harvested in special circumstances and to recondition them in order to qualify for transplant. These methods have tremendous potential for study of different lung pathologies, ALI/ARDS included, but the cost, technicality, and competition with the transplant option make it less accessible; thus, modeling ALI in an ex vivo lung perfusion setting should be initiated.
The inhaled-LPS model induces an inflammatory response with neutrophil recruitment and mild enhancement of cytokine production,39 associated with fibrinolytic and coagulopathic defects similar to the ones described in human ALI.40 On the basis of this evidence, the model was used to assess the effect of simvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, on the inflammatory component of ALI. The treatment reduced the number and activation of neutrophils in the alveolar space, as well as the level of most cytokines in the bronchoalveolar lavage fluid.41All in vivo human models are plagued by limitations, such as ethical considerations, clinical priorities, and mandated patient safety, that sometimes make their interpretation more complicated than expected.42
Regardless of their limitations, the animal and human models of ALI will continue to be used and refined, since there are no alternatives available for analysis and therapeutic testing in the setting of a complex biological system. In parallel, it is expected that the refinement of human models (specifically, ex vivo models) of ALI will spawn new, forceful, and relevant data to be used before clinical trials on patients, while allowing for definition of more realistic primary and secondary end points.
Etiology, risk, and cellular and molecular pathophysiologic mechanisms of ALI
ALI/ARDS has many causes, with sepsis of pulmonary or nonpulmonary origin being the most common.7 The lung response to injuries starts with disruption of the alveolar-capillary interface (clinically known as the exudative phase), which transitions to a remodeling phase (clinically known as the fibroproliferative phase) independent of the initial cause;43 the separation in time of the two phases was proven to be an unrealistic criterion as long as the fibrosing alveolitis begins43 almost at the same time as the alveolocapillary damage.44,45 ALI/ARDS is an overwhelming inflammatory process that could be divided into two phases: the initial (acute) phase—characterized by epithelial and endothelial injury recorded histopathologically as diffuse alveolar damage, type I pneumocytes necrosis, neutrophil infiltration, alveolar hemorrhage, and hyaline membrane deposition46—and a fibroproliferative phase with various degrees of fibrosis, neovascularization, and, afterward, resolution.47 The fibroproliferative phase is seen as a process of lung repair caused by type II pneumocyte and fibroblast propagation that may restore morphofunctional characteristics of the lung. Thus, the pathological model of ALI includes, besides the exudative and proliferative stages, a phase of fibrosis that usually evolves toward resolution, but if persistent, it may progress to a point of no return, when the destruction of alveolocapillary basement membrane cannot be restored. The factors controlling progression to fibroproliferation instead of resolution of inflammatory process and restoration of normal pulmonary architecture are poorly understood.48 Currently, it is not possible to correlate the clinical path of ALI/ARDS patients with mechanisms controlling the cellular and molecular basis of lung repair.49 Broadly defined, the disease includes processes related to direct lung responses to microbial and other environmental insults as well as those related to indirect pulmonary insults such as extrapulmonary sepsis and trauma, shock, burn injury, and mass transfusions. On this background, a wide variety of cell types, proteins, and inflammatory mediators participate in the inflammation, immunity, and tissue restoration or fibroproliferation.
The lung consists of three main types of cell: endothelial cells, which represent up to 50% of all cells in the lung,50 the epithelium, which is the first line of defense against particulate material, and fibroblasts. Under inflammatory conditions there is a significant increase in lymphocytes and macrophages in the lung parenchyma, which together with the above cells participate in the pathogenesis and resolution of ALI.
Endothelial cells and ALI
The vascular component of the alveolocapillary barrier, formed by a tiny endothelial cell layer, is essential for maintaining adequate pulmonary homeostasis.51 Strategically located at the interface between blood and the surrounding tissues, the endothelium is endowed with constitutive and inducible features that allow it to adapt to local conditions and to respond to a plethora of injuries from blood and tissue (Table 2). The main function of the endothelial layer is to assemble a functional, semipermeable barrier that separates the blood from the surrounding tissues while allowing a bidirectional flux of water and small and large solutes, as well as blood cells.52 Specific structural features of the endothelial phenotype, such as the extremely rich vesicular population, tight and adherens junctions, focal adhesions, a particularly structured cytoskeleton, and local adaptations to meet permeability needs (caveolar diaphragms, fenestrae), have been the subject of excellent reviews.51,52,88,89,91,92 While the participation of some of these endothelial characteristic structures in the inflammatory process is well described and functionally characterized (e.g., endothelial adherens junctions), the involvement of other elements, such as the endothelial tight junctions, is less known and often controversial, in regard to both function and structural/morphological organization.55,93-95 This is significant because opening of interendothelial space, comprising both tight and adherens junctions, is the first structural element that changes in ALI, causing pulmonary edema. There will be no increase in permeability without the rearrangement of the tight junctional complex, located most apically in all endothelial cells and in some parts of the vascular tree (arteries and capillaries); in some specific vascular beds (brain, testis), tight junctional complexes can be found in more than one location along the interendothelial space.53,96-98 Endothelial participation and the mechanisms involved in exacerbated vascular permeability in ALI have been extensively reviewed.84,94
Table 2.
Major functions of pulmonary endothelium
| Function | |
|---|---|
| 1. | Barrier function52-55 |
| 2. | Regulation of coagulation and thrombolysis56,57 |
| 3. | Drug removal and biotransformation58-61 |
| 4. | Active interactions with cellular components of blood, with bacteria, viruses, or any particulate compounds found in the blood or in the tissue62-65 |
| 5. | Expression of a variety of receptors and signaling molecules66-70 |
| 6. | Expression of definite enzymes such as NO synthase, ACE, endothelin converting enzyme, lipoprotein lipase71-73 |
| 7. | Synthesis of vasoactive autacoids involved in establishing and controlling the vascular tone as well as the blood flow74-76 |
| 8. | Binding and interacting with immune complexes and immunocompetent cells upon activation77-79 |
| 9. | Production of ROS and RNS, cytokines, growth factors80-83 |
| 10. | Expression of adhesion molecules84-87 |
| 11. | Expression of specific structural elements (fenestrations, diaphragms, interendothelial sealing, supramolecular complexes)88-90 |
ACE: angiotensin-converting enzyme; RNS: reactive nitrogen species; ROS: reactive oxygen species.
In pathological conditions, dysfunctional endothelial cells show impaired caveolae and clathrin-coated vesicle endocytosis and altered intracellular trafficking and signaling of cell surface receptors such as transforming growth factor beta receptor (TGFβR) and bone morphogenetic protein receptor-2 (BMPR2), both implicated in the pathogenesis of ALI/ARDS.99-102 However, the consequences of endothelial cell endocytic dysfunction in the context of human lung disease in general, and in ALI/ARDS in particular, are understudied. Endocytic dysfunction and nonproductive assembly of endocytic machinery may alter canonical signaling pathways, with detrimental consequences for endothelial cell function.103,104 Frequently, dysfunctional endothelial cells show increased occurrence of pleomorphic endocytic/transcytotic structures that function as morphological intermediates of alternative transport pathways to compensate for deficient vesicular trafficking.11,105 Our recent work indicates that in mouse lung endothelial cells deficient of intersectin 1-s (ITSN-1s), a general endocytic protein required for endothelial cell survival, these enlarged endocytic structures and impaired endocytosis alter the signaling of Alk5, a broadly expressed type 1 TGFβR;106 as result, downstream signaling of Alk5 is switched from the canonical Smad2/3 signaling to Erk1/2 MAPK, resulting in proliferation of endothelial cells and microvascular and lung remodeling.107 Endothelial cells are not only a target but also an active player in ALI/ARDS. While endothelial cells alone are insufficient to cause ALI,17 their injury/dysfunction and activation, as well as interaction with alveolar epithelium, are critical not only for the onset of ALI/ARDS but also for repair and remodeling of the injured lung. Endothelial cell heterogeneity, the complexity of these cells’ interaction with other pulmonary cells, and the lack of complete understanding of the molecular mechanisms involved in endothelial response in ALI/ARDS make lung endothelium an attractive field for exploration, with high chances of improving detection, amelioration, and therapy for these severe syndromes.
Apoptosis in ALI/ARDS: ITSN-1s regulation of endothelial cell apoptosis
Apoptosis has been documented in the lung during pathogenesis of ALI/ARDS.108 Two hypotheses—“neutrophilic apoptosis” and “epithelial apoptosis”—connect apoptosis to the pathogenesis of ALI in both experimental models of ALI/ARDS and humans.109 The loss of cells from the alveolocapillary unit may result from apoptosis of epithelia, endothelia, and neutrophils, and this is an important event for the initiation and the development of ALI/ARDS.110 Apoptosis and the clearance of apoptotic cells can be detrimental or beneficial, depending on cell type, circumstances, and timing.111 For example, inhibition of neutrophil apoptosis has been shown to contribute to basement membrane destruction and increased permeability of the alveolocapillary barrier.112 In contrast, phagocytosis of dead neutrophils inhibits proinflammatory cytokine production and leads to the resolution of ALI/ARDS.113 However, ALI/ARDS can occur without neutrophil activation as well.114 Early apoptosis of epithelial and endothelial cells, leading to loss of alveolocapillary barrier integrity, with consequent pulmonary edema and acute hypoxemia, is pivotal for human ALI/ARDS.22 Accumulation of apoptotic cells in the lung due to deficient clearance, combined with enhanced endothelial/epithelial cell death at the same site, is even more deleterious to ALI/ARDS progression.115 Growing evidence indicates, however, that apoptosis may be helpful during ALI resolution; apoptotic/activated cells may release growth factors and circulating microparticles that contribute to upregulation of proliferative signaling pathways, apoptosis resistance, or replacement by progenitor cells.116-121 The replacement of dead cells may lead to neovascularization, repair of the injured lungs, and microvascular/lung tissue remodeling.122 Lung epithelial and endothelial cell apoptosis and microvessel loss, followed by endothelial cell proliferation and abnormal neovascularization, are pathological traits seen in the lungs of ALI/ARDS patients.123 Circulating microparticles can transfer genetic material and proteins from the donor cells (cells generating the microparticles) to a wide range of target cells, by several mechanisms: (1) internalization and lysosomal processing of microparticles, (2) fusion-mediated transfer of surface receptors, proteins, and lipids, (3) outside-in signaling via ligand-receptor internalization, and (4) temporary fusion with the target cell, followed by complete or selective transfer of microparticle content.116 The microparticles are heterogeneous, in terms of cellular origin, biochemical makeup, and size; elevated levels of microparticles have been associated with sepsis and ALI.124 They may have both positive and harmful effects in ALI/ARDS, with the potential of ameliorating or worsening several processes with well-documented contributions to the pathology of ALI/ARDS, such as vascular permeability, coagulation, and inflammation.116
Studies on animal models have shown that ALI triggers growth factor–mediated interactions between the alveolar, endothelial, and mesenchymal cells, aimed at restoring lung integrity.125 Vascular endothelial growth factor (VEGF), transforming growth factor beta (TGFβ), epithelial growth factor (EGF), and basic fibroblast growth factor (bFGF) are known mediators of cell proliferation and differentiation, angiogenesis, apoptosis, vascular permeability, and tissue repair126-130 Studies have shown that plasma VEGF levels rise and intrapulmonary levels fall in the early stages of lung injury, with normalization of both during patients’ recovery.131 The actions of TGFβ in ALI/ARDS have been evaluated mostly during the late phases mediating mesenchymal cells hyperproliferation and synthesis of collagen fibers, lung remodeling, and fibrosis.130
In vivo long-term ITSN-1s knockdown (KDITSN), via repeated delivery of siRNAITSN (small interfering RNA)/cationic liposome complexes, induced extensive apoptosis of mouse lung epithelial and endothelial cells in a process that involved downregulation of Erk1/2 MAPK signaling.107 Death of endothelial cells resulted in destruction of the mouse pulmonary microvessels and loss of alveolar septa, pathophysiological hallmarks of ALI/ARDS.132 After only 7 days of KDITSN, the expression of several growth factors (i.e., TGFβ, VEGF) whose downstream signaling includes Erk1/2 MAPK was increased. As result, Erk1/2 signaling was restored, and the remaining endothelial cells exhibited phenotypic changes including hyperproliferation and apoptosis resistance against KDITSN, leading to increased microvessel density, repair, and remodeling of the injured lungs.107
The literature is limited regarding ITSN-1 involvement in pathological conditions. So far, the existing data related to ITSN-1s participation in pathophysiological processes have been accumulated by studying Down syndrome and Alzheimer’s disease, in which the ITSN-1 gene is upregulated.133 A clear association between endocytic abnormalities and the pathophysiological mechanisms of Down syndrome has been established in neurons.134 Enlarged early endosomes, marked variation in their size, and an increase of the total number were reported.135 While in neurons, ITSN-1 overexpression induces severe impairment of endocytosis,133,136 in fibroblasts isolated from Down syndrome patients, reports indicate an increase in internalization rate.137 Genetic approaches to elucidate the function of mammalian ITSN-1 have yielded contradictory results, ranging from complete lethality to unaffected viability; the gene trap insertion strategy applied to ITSN-1 locus is lethal,138 while the homologous recombination of the same locus resulted in viable and fertile mice with defects in the endocytic pathway.139 Similar problems, perhaps compounded by the possibility of obtaining phenotypes from “null” alleles, explain the loss of function of ITSN and lethality in Drosophila,140 while ablation of the ITSN gene in Caenorhabditis elegans results in live worms.141
Because ITSNs participate in the activation of different mitogenic kinases,107,142 their involvement in cell growth, proliferation, and cancer is gaining attention. Low levels of ITSN-2 expression were associated with poor prognosis of breast cancer patients after adjuvant chemotherapy.143 Low levels of ITSN-1 are reported in several cancers, lung cancer included (http://www.proteinatlas.org/ENSG00000205726/summary).144
Overexpression of ITSN-1 induces oncogenic transformation of rodent fibroblasts.145,146 Future work is needed to investigate the role of ITSN-1s in the context of human tumorigenesis and to delineate the mechanisms and pathways modulated by the members of ITSN family.
Our recent data suggest that ITSN-1s deficiency in mouse lungs, resulting in epithelial and endothelial cell apoptosis and alveolar damage, causes increased paracellular permeability and patchy protein-rich interstitial edema,105 altogether resembling common features of ALI in humans. The peak in airway space enlargement due to alveolar cells apoptosis (epithelium and endothelium) in mouse lungs, as indicated by the 40% increase in mean linear intercept value, was reached at day 10 of ITSN-1s deficiency. Activated caspase-3 in mouse lungs was detected at day 3 of KDITSN and reached the highest levels after 10 days of KDITSN by reference to control lungs.107 Therefore, the alveolar damage may be a multifactorial process: the initial loss of the capillary bed, essential for growth and stability of alveolar septa and activation of the elastolytic activity of apoptotic cells, all potentiated by inhibition of ITSN-1s expression in the epithelial cells, contributed to alveolar damage, loss of alveolar membrane integrity, and pulmonary edema, as also described in other settings.147 Studies from patients who died of ALI have shown pneumocytes with DNA fragmentation and activation of the pro-apoptotic Bax protein,148,149 two mechanisms triggered by ITSN-1s deficiency in human lung microvascular endothelial cells.150
There is evidence that reactive oxygen species (ROSs) generated by inflammatory cells, as well as epithelial and endothelial cells, are responsible for lung damage and abnormal repair.151 Mitochondrial dysfunction and ROS production by mitochondria can contribute to apoptotic cell death150,152 and thus participate in cellular processes involved in the maintenance of lung integrity. KDITSN in lung endothelial cells increased ROS production significantly,150 and therefore it is not surprising that affecting ITSN-1s expression and function is involved in the development of ALI/ARDS.
Until recently, the role of ITSNs in lung pathology has not been addressed. This unique KDITSN mouse model, without lethality, connects ITSN-1s to ALI/ARDS and offers the proper substrate to investigate the effects of a disrupted endothelial barrier with intense endothelial and epithelial cell apoptosis on mouse lung vascular and alveolar architecture. Understanding the molecular mechanisms involved in upregulation of proliferative signaling, apoptosis resistance, and stimulation of dead-cell replacement might provide potential avenues for therapeutic intervention.
ITSN-1s regulation of transcellular and paracellular permeability
An intact lung microvascular endothelium is essential for maintaining adequate pulmonary and systemic homeostasis.52 The endothelium forms a tightly regulated barrier that not only separates the blood content from the surrounding tissues but also filters it before it reenters the systemic circulation. The endothelium is an active organ endowed with important physiological properties: it promotes an antiaggregation surface; creates and maintains the endothelial barrier; synthesizes, metabolizes, and uptakes vasoactive compounds that modulate vascular tone; and regulates ventilation–perfusion matching, hemofluidity, and interactions with blood-borne cells (Table 2). Most endothelial functions are constitutive; some of them are, however, induced upon endothelial activation as a result of exposure to inflammatory stimuli. In ALI/ARDS of different etiologies (sepsis, trauma), the endothelium is the initial site of injury in the lung, leading to increased microvascular permeability and pulmonary edema; the mechanisms responsible for opening and resealing the barrier and endothelium participation in these events have been the subject of many excellent reviews.84,92-94 Protein and solute transport across the endothelium involves a highly selective and organized transcellular pathway carried dominantly via caveolae and, to a lesser extent, via clathrin-coated vesicles.52,92,153,154 Caveolae density, expressed as units per square micrometer of lung endothelial cell surface, is a relatively stable endothelial parameter demonstrated to vary only in diabetes.155 It was assumed that the presence of caveolin-1 was sufficient for the formation of caveolae,156 but later it was shown that the proteins belonging to the cavins family were also necessary for caveolae biogenesis.157,158 As caveolae biogenesis is an emerging field, the findings reported in the KDITSN mouse, demonstrating that the caveolae density in endothelia is reduced, most probably as a consequence of impaired dynamin-2 recruitment to the endocytic site and deficient caveolar fission, are relevant.159 Caveolae are associated with a plethora of cellular processes (i.e., endocytosis, cell proliferation, differentiation, and apoptosis, as well as cell migration) recently linked to the pathogenesis of idiopathic pulmonary fibrosis, lung cancer, inflammation, and vascular dysfunction.160-164 Thus, our observations not only enhance the understanding of caveolar function but also link ITSN-1s to caveolae release from endothelial plasma membrane and thus to controlling their number. Moreover, the observations implicate ITSN-1 in the pathogenesis of diseases in which these prominent endothelial structures participate. It has been demonstrated that the reduced number of caveolae affects the caveolin-1/endothelial nitric oxide synthase (NOS) interaction and increases the production of NO and vascular permeability.165,166 Clinical studies and animal models suggest that NO is involved in the pathogenesis of lung injury.167 Acute ITSN-1s deficiency 72 hours after siRNAITSN delivery caused a decrease in caveolae number and dysfunction of the endothelial barrier in mouse lungs; open interendothelial junctions, with more than 30-nm-wide intercellular gaps and heavily labeled by 18–20-nm-diameter tracer particles, were often recorded (Fig. 1).105 As result, patchy protein-rich interstitial edema, a common feature of ALI in humans, was often detected (Fig. 2A).105 ITSN-1s deficiency, resulting in alterations of endothelial barrier integrity and enhanced vascular permeability, may be a consequence of increased NO production; the participation of NO in this process has not been explored yet, and thus the precise steps, the molecules involved, and how ITSN-1s regulates pertinent signaling pathways remain to be established. Moreover, the interactions of ITSN-1s with CdGAP, downstream cdc42 activation, and stress fiber/actin bundle formation168,169 may be part of the molecular mechanisms engaged by ITSN-1s deficiency to alter the interendothelial junctions and to enhance vascular permeability.
Figure 1.
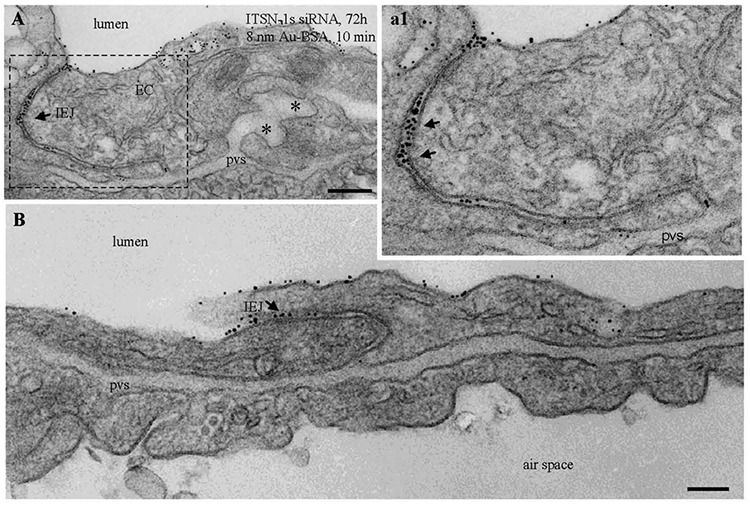
Increased endothelial permeability and impaired interendothelial junctional integrity in mouse lung endothelium acutely depleted of intersectin-1s (ITSN-1s); representative electron micrographs showing open interendothelial junctions (IEJs) labeled throughout their length by 8-nm gold albumin (Au-BSA) particles. Arrows in A (magnified in a1) point to three or four Au-BSA particles located close to each other in the same plan, indicative of the wide opening of the IEJ. Gold particles are also associated with the abluminal exit of IEJs. Note also the limited number of caveolae and the dilation of the pericapillary space (asterisks). pvs: perivascular space; siRNA: small interfering RNA. Scale bars: 200 nm (A), 100 nm (B).
Figure 2.
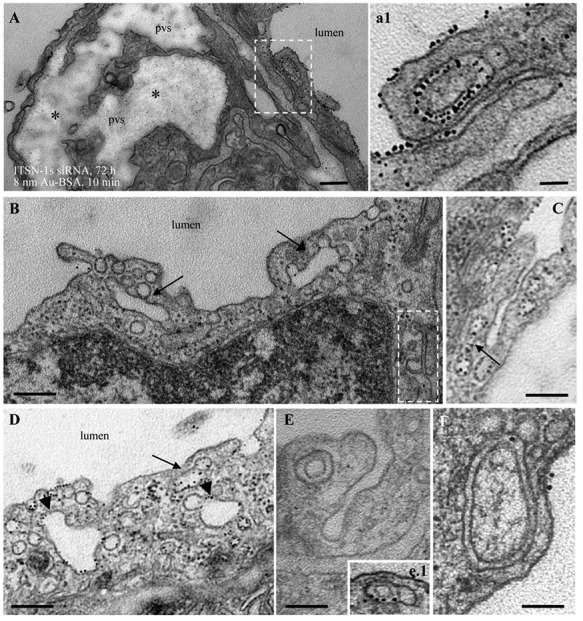
Acute perturbation of intersectin-1s (ITSN-1s) expression induces pleomorphic endocytic/transcytotic intermediates; representative electron microscopic (EM) images of membranous rings (A, a1 [area outlined in A], E, e.1, F), tubular elements (B–D, arrows), and enlarged endosomes (D, arrowheads), loaded with 8-nm gold albumin (Au-BSA) and associated with caveola-like morphology. Two tubular structures open to the lumen are shown in C and E. The EM image shown in E was selected from a mouse lung specimen not perfused with 8-nm Au-BSA. Note also the severe dilation of the perivascular space (pvs) and the proteinaceous edema (A). Asterisks have the same meaning as in Figure 1. siRNA: small interfering RNA. Scale bars: 250 nm (A, B); 200 nm (C, D); 100 nm (a1, E); 150 nm (F).
The extent to which the endothelial vesicular trafficking is affected by KDITSN is severe,105 and hence, compensatory mechanisms were developed to keep the cells alive and to maintain lung homeostasis; this was an unforeseen effect of KDITSN. Under these circumstances, upregulation of alternative endocytic pathways and their morphological intermediates (enlarged endocytic structures, membraneous rings, and tubules open to the cell surface or apparently with no communication with the extracellular milieu, all heavily labeled by tracer particles) compensate for deficient caveolae internalization and transport across the lung endothelium (Fig. 2A, 2a1, 2B [arrows and outlined area], 2D).105 Interestingly, the functionality of this alternative transport increased at 24 days of KDITSN, leading to partial resealing of interendothelial junctions and reduced interstitial edema. At this time point, mice still displayed increased microvascular permeability, a sequela of ALI, but neither the gold albumin (18–20-nm diameter) nor the DNP-BSA (dinitrophenol–bovine serum albumin; 6–10-nm diameter) tracers were able to penetrate the interendothelial junctions, meaning that resealing mechanisms were returning the endothelial barrier to its perm-selective status.105 Patchy areas of mild septal edema were still present, implying that the resealing did not return the structure of the junctional complexes to their initial status, because of continuous ITSN-1s deficiency; additional studies are needed to fully define this intermediary state of interendothelial junction opening (>3 nm but <6 nm). KDITSN in mouse lungs had a dual outcome: (1) acute (3 days) KDITSN-induced impairment in caveolae transport function, significant disruption of junctional integrity, and acute pulmonary edema and (2) chronic (24 days): KDITSN upregulated alternative transport pathways to compensate for deficient caveolae transport function and partially restored the endothelial barrier integrity, maintaining, however, a mild edema.105 Thus, ITSN-1s is positively one of the factors needed for barrier integrity, and prolonged ITSN-1s deficiency and upregulation of compensatory endocytic pathways may be part of a protective mechanism against lung injury associated with severe disruption of endothelial barrier function; this may apply in the case of acute inflammatory reaction, when the ITSN-1s protein is cleaved by the cytotoxic protease granzyme B (GrB),170 inducing a long-term ITSN-1s deficiency. Evidence indicates that the bronchoalveolar fluid of ARDS patients contains highly upregulated messenger RNA levels for GrB and perforin.171 Moreover, Bem and colleagues172 demonstrated that combined GrA/GrB deficiency participates in progression of ALI/ARDS in pneumovirus-infected mice. The absence of GrA and GrB results in delayed neutrophil recruitment, diminished activation of caspase-3, and reduced lung permeability. More work is needed to identify the targets of these cytotoxic proteases with proinflammatory and pro-apoptotic activity and the physiological significance of their cytotoxic activity. While the extent of pulmonary edema depends on several factors, it is attractive to speculate that ITSN-1s cleavage by GrB, resulting in decreased ITSN-1s expression, may be part of the pathophysiology of increased permeability and pulmonary edema in ALI/ARDS. ITSN-1s, because of its ability to regulate caveolae release from the endothelial plasma membrane during transendothelial transport as well as actin cytoskeleton remodeling and stress fiber formation associated with interendothelial junctions, mediates a unique cross talk between transcellular and paracellular transport pathways, with a critical role for barrier integrity and lung homeostasis.
Resolution of ALI: ITSN-1s involvement in alveolar and endothelial repair
Although cellular turnover is relatively slow in normal developed lungs, post-ALI rapid repair of the denuded and injured alveolar surface is crucial to survival. This implies that quiescent endothelial and alveolar type II cells regain the capacity to proliferate and differentiate. The signaling pathways for lung remodeling, repair, and regeneration are not well defined but possibly recapitulate the ontogeny.173 Lung development requires coordinated interactions among epithelial, endothelial, and mesenchymal cells, and it is likely that alveolar repair does so as well. For example, VEGF, a major angiogenic and permogenic factor, is partly produced by lung mesenchymal and alveolar type II cells, showing the interdependence of cells with regard to vascular and alveolar growth and maintenance. Inadequately regulated healing response after ALI/ARDS causes pulmonary fibrosis. Patients surviving the initial phase of ALI can progress to restoration of the normal pulmonary architecture or to accumulation of fibrotic tissue and lung dysfunction. The outcomes of persistent and progressive lung injury include multiple organ failure, fibrosing alveolitis, and pulmonary vascular obliteration with pulmonary hypertension and death.132,174 The genetic, cellular, and molecular factors that contribute to each of these outcomes remain largely unknown.112 It was postulated that the preferential growth of fibroblasts toward fibrosis may be a direct result of cell necrosis, particularly of the epithelium.123,175 Also, disrupted or delayed repair of alveoli, interstitium, or the alveolocapillary membrane is sufficient to promote excessive proliferation and collagen deposition.176 Some mechanistic insights about the lung fibrotic process were obtained from the bleomycin-induced ALI/ARDS rodent model.23 Although extensively used, the bleomycin has been seen as “overwhelming stimulus” with little relevance to human post-ALI/ARDS pulmonary fibrosis.12 Other animal models of ALI, such as high-dose LPS, cecal ligation and puncture, and oleic acid, have a high mortality, without allowing the study of lung-repairing process.
Long-term KDITSN in mouse lung allowed us a detailed analysis of the time course of lung injury and repair and provided mechanistic insights into the signaling pathways responsible. Mouse lung endothelial cell death, as caused by KDITSN, was a prerequisite for the subsequent endothelial cell proliferation, repair, and remodeling process. Within days, these normally quiescent cells with a low turnover rate began to hyperproliferate, indicating that the Erk1/2 MAPK survival signaling, lost because of KDITSN, had been reestablished. Expression of TGFβ, a cytokine that regulates diverse and often contradictory functions in a milieu- and cell type–dependent manner,128 was increased. Endothelial cell death induced an increased expression and activation of TGFβ in the early stages of lung injury, peaking at day 3, when Erk1/2 MAPK signaling was significantly decreased because of KDITSN. It is well documented that TGFβ is a multifunctional cytokine involved in endothelial cell proliferation, survival, and maintenance of vascular integrity128 and that Ras/Erk1/2 MAPK is a major signaling pathway downstream of Alk5, the broadly expressed TGFβ-R type 1.106 Apparently, upregulation of BMP-2/4 proteins by chronic KDITSN worked synergistically with TGFβ and VEGF to induce endothelial cell proliferation via activation of Erk1/2 MAPK pathway.107 Paracrine and autocrine growth factors released by apoptotic endothelial cells caused increased survival, proliferation, and alteration of their phenotype. In order to reestablish lung tissue homeostasis, KDITSN switched the TGFβ/Alk5 signaling from the typical Smad2/3 activation toward the less common Ras/Erk1/2 MAPK pathway, with protective effects on endothelial cells and lung vasculature causing increased microvessel density (Fig. 3), endothelial phenotypical changes, and alveolar repair.107 Stimulation, via growth factors, of the Erk1/2 MAPK pathway also inactivated the pro-apoptotic Bad protein by phosphorylation of Ser112 and Ser155 residues, conferring on endothelial cells hyperproliferative and apoptosis-resistance properties. Prolonged inhibition of ITSN-1s led to increased microvessel density, critical for repair and remodeling of the alveolar capillary membrane.107
Figure 3.
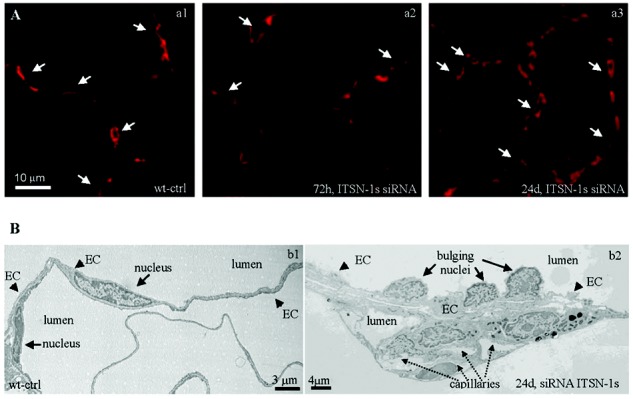
Chronic intersectin-1s knockdown (KDITSN-1s) in mouse lungs induces microvascular remodeling. A, Micrographs of GS-1 lectin staining of paraffin-embedded sections show microvessel profiles (arrows) within the alveolar walls in wild-type control (wt-ctrl) mice (a1), mice treated with small interfering RNA (siRNAITSN) for 3 days (a2), and mice treated with siRNAITSN for 24 days (a3). Scale bar: 10 μm. B, Ultrastructural features of microvascular remodeling in KDITSN mouse lungs (24 days). b1, Two vessel profiles in wt-ctrl mouse lungs display elongated endothelial cell (EC) nuclei. Note the relatively uniform thickness of the ECs throughout the vessel perimeter. b2, Segment of a midsized vessel in KDITSN mouse lung shows a distorted endothelium and several nuclei protruding into the lumen (arrows). New pulmonary microvessels (dashed arrows) with narrow openings are abundant and located in very close proximity to each other.
KDITSN mouse is an emerging model of an adaptor protein causing lung injury and endothelial cell phenotypical changes toward proliferation and apoptosis resistance, leading to repair of the injured lung. Extensive endothelial cell apoptosis, widely open interendothelial junctions, the rich proteinaceous interstitial edema, loss of pulmonary microvessels, and the histological images showing loss of the alveolar septa provide strong evidence that lung injury occurred. Moreover, absence of intra-alveolar neutrophil infiltrate and minimal inflammatory response characterize this emerging mouse model of ALI/ARDS. Furthermore, increased expression of growth factors, compensatory endothelial and epithelial cell proliferation resulting in increased microvascular density, and lung remodeling without fibrosis provide evidence for lung tissue repair/recovery following injury. However, despite reproducing several clinically relevant phenomena, the KDITSN mouse, like any animal model of human disease, has several specific limitations. ITSN-1s deficiency induced by retro-orbital delivery of liposome/siRNAITSN complexes targets mainly the vascular endothelium, while the primary target tissue in human disease is not known. Even if ITSN-1s deficiency may be relevant in clinical settings, given ITSN-1s’s susceptibility to GrB cleavage, the human disease is more complex, with many other risk factors as well as genetic determinants. Despite limitations, this mouse mimics many salient features of ALI/ARDS and can be used to better understand the molecular mechanisms underlying these severe syndromes and to develop better intervention strategies.
The genetic dimension of ALI/ARDS
The major challenge of ALI/ARDS studies in human patients is the fact that the syndromes are the result of a plethora of molecular scenarios taking place in different cell types and physiological backgrounds dictated by age, sex, and, most of the time, coincident pathological processes.42 Besides this intrinsic level of complexity, a particular characteristic of ALI/ARDS pathogenesis is the extremely high proportion of iatrogenic induced illness; half the instances of ALI occur in patients subjected to mechanical ventilation for the following major causes: (1) transfusion of blood and/or blood products, (2) massive fluid resuscitation, (3) hospital pneumonia, and (4) normalization of blood gas parameters.177-180 When critically ill patients, most of them undergoing invasive procedures, are considered, the need for individualized therapies becomes obvious. On this pathogenic background, it was thought that phenotypic variance of ill patients might be related to variations in individual genomes, an assumption that has generated a plethora of gene expression studies aimed at identifying disease susceptibility loci. Initial research using genomic and genetic strategies to find the genes implicated in ALI pathogenesis and to generate lists of putative genes involved in the susceptibility and severity of the disease was considered sufficient to generate data aimed at an individualized treatment. These initial genetic-association studies (more than 80 performed up to 2008), evaluated in two significant articles,181,182 identified 15 or 16 genes positively associated with the susceptibility and outcomes of all-cause ALI or ARDS. Candidate genes significantly associated with ALI and/or ARDS were genes controlling several major biological processes—inflammatory response, endothelial permeability, nitric oxide biosynthesis, phospholipid metabolism, apoptosis, cell motility, transcription, etc. The complexity of phenotypic (tissular, individual, or populational) heterogeneity caused significant difficulties in identifying the most likely candidate genes related to susceptibility and severity of ALI. Inconsistencies, and sometimes conflicting results, were attributed to either inappropriate study design or the modalities used for data implementation and/or interpretation.183,184 These discrepancies motivated the formulation of standards to improve quality and to help perform meta-analysis of the data,185 while leading to the consensus that such studies should be considered tools to investigate ALI/ARDS and not definitive answers for disease pathogenesis and therapeutic options. Thus, despite adding a genetic predisposition to ALI, these studies rendered the promise of an individualized treatment very challenging.
Since the genetic-linkage analysis does not have the power to detect signals of common genetic variation with modest effects that are the causes of susceptibility to common complex diseases,186,187 genome-wide association (GWA) studies were considered more powerful and better suited for identifying putative risk variants for ALI. However, GWA studies are biased toward detecting the loci with the largest effects and not all the loci involved in the pathophysiology of a disease; also, GWA studies assume that the susceptibility loci identified represent a small subset of all the loci involved and that they represent the true players in the disease development. Thus, it became clear that a plethora of factors, such as phenotypic variance of ill patients, diversity of causes eliciting lung injury, possibility of locus heterogeneity, the sporadic nature of the disease, and low or incomplete gene penetrance, should be considered and included in the design of this type of study. Despite these limitations, the use of GWA studies for different diseases led to important achievements, as in the cases of type 1 and type 2 diabetes188,189 and prostate and breast cancer,190,191 while for other diseases, such as asthma,192 coronary heart diseases,193 and ALI,194 progress remains limited.
In the past 4 years or so, more than 20 GWA studies in pulmonary medicine have been published that evaluated genes associated with pulmonary function measures, the genetic basis of asthma, and susceptibility loci in chronic obstructive pulmonary disease, interstitial pulmonary fibrosis, and sarcoidosis;195 most of the data are widely accessible and can be used as basis for further research to evaluate the genetic basis for pulmonary conditions less subjected to the GWA approach so far, such as ALI/ARDS. Therefore, we have begun building a catalog of the genes associated with the disease, with the potential to advance our understanding of lung injury; in addition, it may lead to the development of more-personalized therapies and, most importantly, may help in planning the necessary changes in medical education curricula to prepare future clinicians to handle and manage genetic, genomic, and pharmacological data. At present, the most significant and conceptually agreed-on candidate genes involved in ALI/ARDS are listed in Table 3.
Table 3.
Candidate genes involved in ALI/ARDS
| Symbol | Gene |
|---|---|
| ACE | Angiotensin converting enzyme196,197 |
| CXCL2 | Chemokine ligand 2/macrophage inflammatory protein 2198 |
| GADD45A | Growth arrest and DNA-inducible alpha199 |
| IL-6 | Interleukin 6200 |
| IL-10 | Interleukin 10201 |
| LBP | Lipopolysaccharide binding protein202 |
| MLCK | Myosin light-chain kinase203 |
| MBL2 | Mannose-binding lectin204 |
| NFkB1A | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1205 |
| NRF2 | Nuclear factor (erythroid-derived 2)-like 2206 |
| PAI-1 | Plasminogen activator inhibitor207 |
| PBEF | Pre-B-cell colony enhancing factor208 |
| SOD3 | Extracellular superoxide dismutase209 |
| SP-B | Surfactant protein B210 |
| TGF-β | Transforming growth factor β211 |
| TNF-α | Tumor necrosis factor α212 |
| VEGF | Vascular endothelial growth factor126 |
Because this type of study focuses on the potential genetic and gene-gene interactions that may constitute the explanation for the disparities observed in the heterogeneous response of individuals to ALI/ARDS-generating factors and/or diseases, one should be aware that the interactions of gene and environment, along with the transitory or inherited alterations, may be as important as the genetic background alone. Years of genetic and genomic studies of susceptibility to ALI/ARDS led to the conclusion that gene expression is controlled not only by modification(s) of the gene but also by the interaction of the gene with the environment213 and by epigenetic mechanisms.214 Both ALI and ARDS are the result of a complex interplay between environmental, epigenetic, and genetic factors, along with the reality that only a fraction of individuals exposed to ALI-inducing factors (trauma, sepsis, acid aspiration) progress to develop the disease and that from this only another fraction will progress to ARDS.
The challenge for both genetic-association studies and GWA studies is the identification of the biological context in which the statistically significant putative genes act. At present, the current genotyping platforms have the power to survey only a subset of human sequence variations, as the typing of single-nucleotide polymorphisms has been limited to only 300,000–600,000, depending on the population, which after all means that they capture most of the common genetic variation in a region of interest. In other words, the reported susceptibility variants are most probably tagging the real functional variants, and thus they are not causal themselves. Consequently, all these initial discoveries must be followed by fine mapping of the regions harboring the most significant statistical signals,215 which in the absence of prior biological information is financially prohibitive.
Summary
Studies on animal and human models of ALI/ARDS are critical for a better understanding of the pathophysiology of ALI/ARDS and for the development of novel and improved therapeutic tools for treatment of these severe syndromes. While animal models bridge cell-culture studies and human trials and provide significant help in understanding the molecular events and signaling pathways involved in ALI/ARDS, the development and progression of ALI/ARDS in animals occur in a reduced time frame and in the settings of animal pulmonary physiology, quite different from that of humans. Despite limitations, animal models of ALI/ARDS share molecular etiology with human disease and add significantly to our knowledge of how endothelial barrier dysfunction, inflammatory response, nitric oxide biosynthesis, cell motility, cell adhesion, apoptosis, and so on contribute to lung injury and repair. The KDITSN mouse model discussed in detail here seems to be relevant to the pathogenesis of human ALI/ARDS. Acute endothelial and epithelial cell apoptosis, alveolocapillary damage, loss of endothelial barrier function, and pulmonary edema, followed by compensatory proliferation and a repair process, are all caused by ITSN-1s deficiency in murine lungs. The findings are relevant, considering that ITSN-1s is a general endocytic protein, a regulator of mitochondrial apoptosis, and a GrB substrate.171 Under inflammatory conditions and increased GrB levels, loss of full-length ITSN-1s protein may cause endocytic dysfunction, loss of prosurvival signaling, and apoptotic endothelial cell death. The findings implicate ITSN-1s, a key prosurvival protein of lung endothelium, and the biological processes regulated by it in the pathology of ALI/ARDS. Thus, the KDITSN mouse model may become a valuable tool to advance our understanding of ALI/ARDS pathophysiology and provide novel targets for treatment of these severe human syndromes.
Source of support: Nil.
Conflict of interest: None declared.
References
- 1.Martin TR, Matute-Bello G. Experimental models and emerging hypotheses for acute lung injury. Crit Care Clin 2011;27:735–752. [DOI] [PMC free article] [PubMed]
- 2.Windsor AC, Mullen PG, Fowler AA. Acute lung injury: what have we learned from animal models? Am J Med Sci 1993;306:111–116. [DOI] [PubMed]
- 3.Wang HM, Bodenstein M, Markstaller K. Overview of the pathology of three widely used animal models of acute lung injury. Eur Surg Res 2008;40:305–316. [DOI] [PubMed]
- 4.Bernard GR, Artigas A, Brigam KL, et al. The American-European Consensus Conference on ARDS: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 1994;149:818–824. [DOI] [PubMed]
- 5.Shimabukuro DW, Sawa T, Gropper MA. Injury and repair in lung and airways. Crit Care Med 2003;31:S524–S531. [DOI] [PubMed]
- 6.MacCallum NS, Evans TW. Epidemiology of acute lung injury. Curr Opin Crit Care 2005;11:43–49. [DOI] [PubMed]
- 7.Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med 2005;353:1685–1693. [DOI] [PubMed]
- 8.Tsushima K, King LS, Aggarwal NR, De Gorordo A, D’Alessio FR, Kubo K. Acute lung injury review. Intern Med 2009;48:621–630. [DOI] [PubMed]
- 9.Laycock H, Rajah A. Acute lung injury and acute distress syndrome: a review article. Br J Med Pract 2010;3:324.
- 10.Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome: the Berlin definition. JAMA 2012;307:2526–2533. [DOI] [PubMed]
- 11.Matthay MA. Treatment of acute lung injury: clinical and experimental studies. Proc Am Thorac Soc 2008;5:297–299. [DOI] [PubMed]
- 12.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol 2008;295:L379–L399. [DOI] [PMC free article] [PubMed]
- 13.Dyson A, Singer M. Animal models of sepsis: why does preclinical efficacy fail to translate to the clinical setting? Crit Care Med 2009;37:S30–S37. [DOI] [PubMed]
- 14.Roy SK, Kendrick D, Sadowitz BD, et al. Jack of all trades: pleiotropy and the application of chemically modified tetracycline-3 in sepsis and the acute respiratory distress syndrome (ARDS). Pharmacol Res 2011;64:580–589. [DOI] [PMC free article] [PubMed]
- 15.Wurfel MM, Park WY, Radella F, et al. Identification of high and low responders to lipopolysaccharide in normal subjects: an unbiased approach to identify modulators of innate immunity. J Immunol 2005;175:2570–2578. [DOI] [PubMed]
- 16.Wurfel MM, Gordon AC, Holden TD, et al. Toll-like receptor 1 polymorphisms affect innate immune responses and outcomes in sepsis. Am J Respir Crit Care Med 2008;178:710–720. [DOI] [PMC free article] [PubMed]
- 17.Wiener-Kronish JP, Albertine KH, Matthay MA. Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J Clin Invest 1991;88:864–875. [DOI] [PMC free article] [PubMed]
- 18.Schuster DP. ARDS: clinical lessons from the oleic acid model of acute lung injury. Am J Respir Crit Care Med 1994;149:245–260. [DOI] [PubMed]
- 19.Cross AS, Opal SM, Sadoff JC, Gemski P. Choice of bacteria in animal models of sepsis. Infect Immun 1993;61:2741–2747. [DOI] [PMC free article] [PubMed]
- 20.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov 2005;4:854–865. [DOI] [PubMed]
- 21.Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock: a review of laboratory models and a proposal. J Surg Res 1980;29:189–201. [DOI] [PubMed]
- 22.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000;342:1334–1349. [DOI] [PubMed]
- 23.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol 2008;294:L152–L1160. [DOI] [PubMed]
- 24.Frank L, Bucher JR, Roberts RJ. Oxygen toxicity in neonatal and adult animals of various species. J Appl Physiol 1978;45:699–704. [DOI] [PubMed]
- 25.Lachmann B, Robertson B, Vogel J. In vivo lung lavage as an experimental model of the respiratory distress syndrome. Acta Anaesthesiol Scand 1980;24:231–236. [DOI] [PubMed]
- 26.Sakuma T, Takahashi K, Ohya N, et al. Ischemia-reperfusion lung injury in rabbits: mechanisms of injury and protection. Am J Physiol 1999;276:L137–L145. [DOI] [PubMed]
- 27.de Prost N, Ricard JD, Saumon G, Dreyfuss D. Ventilator-induced lung injury: historical perspectives and clinical implications. Ann Intensive Care; 2011;1:28. [DOI] [PMC free article] [PubMed]
- 28.Dreyfuss D, Basset G, Soler P, Saumon G. Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am Rev Respir Dis 1985;132:880–884. [DOI] [PubMed]
- 29.Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 1998;157:294–323. [DOI] [PubMed]
- 30.Desai LP, Sinclair SE, Chapman KE, Hassid A, Waters CM. High tidal volume mechanical ventilation with hyperoxia alters alveolar type II cell adhesion. Am J Physiol 2007;293:L769–L778. [DOI] [PubMed]
- 31.Ward PA, Johnson KJ, Till GO. Animal models of oxidant lung injury. Respiration 1986;50(suppl. 1):5–12. [DOI] [PubMed]
- 32.Alam N, Park BJ, Wilton A, et al. Incidence and risk factors for lung injury after lung cancer resection. Ann Thorac Surg 2007;84:1085–1091. [DOI] [PubMed]
- 33.Kutlu CA, Williams EA, Evans TW, Pastorino U, Goldstraw P. Acute lung injury and acute respiratory distress syndrome after pulmonary resection. Ann Thorac Surg 2000;69:376–380. [DOI] [PubMed]
- 34.Tandon S, Batchelor A, Bullock R, et al. Peri-operative risk factors for acute lung injury after elective oesophagectomy. Br J Anaesth 2001;86:633–638. [DOI] [PubMed]
- 35.Fan X, Liu Y, Wang Q, Yu C, Wei B, Ruan Y. Lung perfusion with clarithromycin ameliorates lung function after cardiopulmonary bypass. Ann Thorac Surg 2006;81:896–901. [DOI] [PubMed]
- 36.Syed A, Fawzy H, Farag A, Nemlander A. Comparison of pulmonary gas exchange in OPCAB versus conventional CABG. Heart Lung Circ 2004;13:168–172. [DOI] [PubMed]
- 37.Frank JA, Briot R, Lee JW, Ishizaka A, Uchida T, Matthay MA. Physiological and biochemical markers of alveolar epithelial barrier dysfunction in perfused human lungs. Am J Physiol 2007;293:L52–L59. [DOI] [PMC free article] [PubMed]
- 38.Lee JW, Fang X, Gupta N, Serikov V, Matthay MA. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci USA 2009;106:16357–16362. [DOI] [PMC free article] [PubMed]
- 39.Nick JA, Coldren CD, Geraci MW, et al. Recombinant human activated protein C reduces human endotoxin-induced pulmonary inflammation via inhibition of neutrophil chemotaxis. Blood 2004;104:3878–3885. [DOI] [PubMed]
- 40.Maris NA, de Vos AF, Bresser P, et al. Activation of coagulation and inhibition of fibrinolysis in the lung after inhalation of lipopolysaccharide by healthy volunteers. Thromb Haemost 2005;93:1036–1040. [DOI] [PubMed]
- 41.Shyamsundar M, McKeown ST, O’Kane CM, et al. Simvastatin decreases lipopolysaccharide-induced pulmonary inflammation in healthy volunteers. Am J Respir Crit Care Med 2009;179:1107–1114. [DOI] [PMC free article] [PubMed]
- 42.Proudfoot AG, Hind M, Griffiths MJD. Biomarkers of acute lung injury: worth their salt? BMC Med 2011;9:132. [DOI] [PMC free article] [PubMed]
- 43.Meduri GU. The role of the host defence response in the progression and outcome of ARDS: pathophysiological correlations and response to glucocorticoid treatment. Eur Respir J 1996;9:2650–2670. [DOI] [PubMed]
- 44.Marshall RP, Bellingan G, Webb S, et al. Fibroproliferation occurs early in the acute respiratory distress syndrome and impacts on outcome. Am J Respir Crit Care Med 2000;162:1783–1788. [DOI] [PubMed]
- 45.Wesselkamper SC, Case LM, Henning LN, et al. Gene expression changes during the development of acute lung injury: role of transforming growth factor β. Am J Respir Crit Care Med 2005;172:1399–1411. [DOI] [PMC free article] [PubMed]
- 46.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med 1982;3:35–56. [PubMed]
- 47.Gattinoni L, Bombino M, Pelosi P, et al. Lung structure and function in different stages of severe adult respiratory distress syndrome. JAMA 1994;271:1772–1779. [PubMed]
- 48.Pelosi P, Rocco PR. Effects of mechanical ventilation on the extracellular matrix. Intensive Care Med 2008;34:631–639. [DOI] [PubMed]
- 49.Rocco PR, Dos Santos C, Pelosi P. Lung parenchyma remodeling in acute respiratory distress syndrome. Minerva Anestesiol 2009;75:730–740. [PubMed]
- 50.Gil J. Alveolar wall relations. Ann NY Acad Sci 1982;384:31–43. [DOI] [PubMed]
- 51.Aird WC. Phenotypic heterogeneity of the endothelium. II. Representative vascular beds. Circ Res 2007;100:174–190. [DOI] [PubMed]
- 52.Simionescu M, Simionescu N. Endothelial transport of macromolecules: transcytosis and endocytosis: a look from cell biology. Cell Biol Rev 1991;25:1–78. [PubMed]
- 53.Simionescu N. Cellular aspects of transcapillary exchange. Physiol Rev 1983;63:1536–1579. [DOI] [PubMed]
- 54.Simionescu N, Simionescu M, Palade GE. Permeability of muscle capillaries to exogenous myoglobin. J Cell Biol 1973;57:424–452. [DOI] [PMC free article] [PubMed]
- 55.Curry FR, Adamson RH. Vascular permeability modulation at the cell, microvessel, or whole organ level: towards closing gaps in our knowledge. Cardiovasc Res 2010;87:218–229. [DOI] [PMC free article] [PubMed]
- 56.van Hinsbergh VW. Endothelium: role in regulation of coagulation and inflammation. Semin Immunopathol 2012;34:93–106. [DOI] [PMC free article] [PubMed]
- 57.Arnout J, Hoylaerts MF, Lijnen HR. Haemostasis. Handb Exp Pharmacol 2006:1–41. [DOI] [PubMed]
- 58.Kamanna VS, Kashyap ML. Mechanism of action of niacin. Am J Cardiol 2008;101:20B–26B. [DOI] [PubMed]
- 59.Denis CV, Christophe OD, Oortwijn BD, Lenting PJ. Clearance of von Willebrand factor. Thromb Haemost 2008;99:271–278. [DOI] [PubMed]
- 60.Kubota Y. Tumor angiogenesis and anti-angiogenic therapy. Keio J Med 2012;61:47–56. [DOI] [PubMed]
- 61.Crabtree MJ, Channon KM. Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide 2011;25:81–88. [DOI] [PMC free article] [PubMed]
- 62.Miller F, Afonso PV, Gessain A, Ceccaldi PE. Blood-brain barrier and retroviral infections. Virulence 2012;3:222–229. [DOI] [PMC free article] [PubMed]
- 63.Harding M, Kubes P. Innate immunity in the vasculature: interactions with pathogenic bacteria. Curr Opin Microbiol 2012;15:85–91. [DOI] [PubMed]
- 64.Kozarov E. Bacterial invasion of vascular cell types: vascular infectology and atherogenesis. Future Cardiol 2012;8:123–138. [DOI] [PMC free article] [PubMed]
- 65.Magrone T, Jirillo E. The impact of bacterial lipopolysaccharides on the endothelial system: pathological consequences and therapeutic countermeasures. Endocr Metab Immune Disord Drug Targets 2011;11:310–325. [DOI] [PubMed]
- 66.Aird WC. Endothelium in health and disease. Pharmacol Rep 2008;60:139–143. [PubMed]
- 67.Kume T. Ligand-dependent Notch signaling in vascular formation. Adv Exp Med Biol 2012;727:210–222. [DOI] [PubMed]
- 68.Eichmann A, Simons M. VEGF signaling inside vascular endothelial cells and beyond. Curr Opin Cell Biol 2012;24:188–193. [DOI] [PMC free article] [PubMed]
- 69.Kuhn M. Endothelial actions of atrial and B-type natriuretic peptides. Br J Pharmacol 2012;166:522–531. [DOI] [PMC free article] [PubMed]
- 70.Parnell E, Smith BO, Palmer TM, Terrin A, Zaccolo M, Yarwood SJ. Regulation of the inflammatory response of vascular endothelial cells by EPAC1. Br J Pharmacol 2012;166:434–446. [DOI] [PMC free article] [PubMed]
- 71.Orfanos S, Catravas JD. Metabolic functions of the pulmonary endothelium. In: Yacoub M, Pepper J, editors. Annual of cardiac surgery. London: Current Science, 1993:52–59.
- 72.Sessa WC. eNOS at a glance. J Cell Sci 2004;117:2427–2429. [DOI] [PubMed]
- 73.Becher UM, Endtmann C, Tiyerili V, Nickenig G, Werner N. Endothelial damage and regeneration: the role of the renin-angiotensin-aldosterone system. Curr Hypertens Rep 2010;13:86–92. [DOI] [PubMed]
- 74.Aird WC. Endothelium as an organ system. Crit Care Med 2004;32(suppl. 5):S271–S279. [DOI] [PubMed]
- 75.Busse R, Fleming I. Vascular endothelium and blood flow. Handb Exp Pharmacol 2006:43–78. [DOI] [PubMed]
- 76.Lamas S, Rodriguez-Puyol D. Endothelial control of vasomotor tone: the kidney perspective. Semin Nephrol 2012;32:156–166. [DOI] [PubMed]
- 77.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003;101:3765–3777. [DOI] [PubMed]
- 78.Kawashima H, Fukuda M. Sulfated glycans control lymphocyte homing. Ann NY Acad Sci 2012;1253:112–121. [DOI] [PubMed]
- 79.Ley K, Reutershan J. Leucocyte-endothelial interactions in health and disease. Handb Exp Pharmacol 2006:97–133. [DOI] [PubMed]
- 80.Kuhlencordt PJ, Rosel E, Gerszten RE, et al. Role of endothelial nitric oxide synthase in endothelial activation: insights from eNOS knockout endothelial cells. Am J Physiol 2004;286:C1195–C1202. [DOI] [PubMed]
- 81.Carnesecchi S, Pache JC, Barazzone-Argiroffo C. NOX enzymes: potential target for the treatment of acute lung injury. Cell Mol Life Sci 2012;69:2373–2385. [DOI] [PMC free article] [PubMed]
- 82.Salvucci O, Tosato G. Essential roles of EphB receptors and EphrinB ligands in endothelial cell function and angiogenesis. Adv Cancer Res 2012;114:21–57. [DOI] [PMC free article] [PubMed]
- 83.Förstermann U. Nitric oxide and oxidative stress in vascular disease. Pflueg Arch Eur J Physiol 2010;459:923–939. [DOI] [PubMed]
- 84.Maniatis NA, Orfanos SE. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care 2008;14:22–30. [DOI] [PubMed]
- 85.Gorbunov NV, Das DK, Goswami SK, Gurusamy N, Atkins JL. Spatial coordination of cell-adhesion molecules and redox cycling of iron in the microvascular inflammatory response to pulmonary injury. Antioxid Redox Signal 2007;9:483–495. [DOI] [PubMed]
- 86.Shcheglovitova ON, Skliankina NN, Babaiants AA, Frolova IS, Beliaev DL, Ershov FI. Adhesion molecules expressed in vascular endothelial cells in natural immunity against viral infections [in Russian]. Vestn Ross Akad Med Nauk 2011:54–60. [PubMed]
- 87.Ling S, Nheu L, Komesaroff PA. Cell adhesion molecules as pharmaceutical target in atherosclerosis. Mini Rev Med Chem 2012;12:175–183. [DOI] [PubMed]
- 88.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 2004;84:869–901. [DOI] [PubMed]
- 89.Stan RV. Structure of caveolae. Biochim Biophys Acta 2005;1746:334–348. [DOI] [PubMed]
- 90.Tse D, Stan RV. Morphological heterogeneity of endothelium. Semin Thromb Hemost 2010;36:236–245. [DOI] [PubMed]
- 91.Palade GE, Simionescu M, Simionescu N. Structural aspects of the permeability of the microvascular endothelium. Acta Physiol Scand Suppl 1979;463:11–32. [PubMed]
- 92.Predescu SA, Predescu DN, Malik AB. Molecular determinants of endothelial transcytosis and their role in endothelial permeability. Am J Physiol 2007;293:L823–L842. [DOI] [PubMed]
- 93.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 2010;72:463–493. [DOI] [PubMed]
- 94.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 2006;86:279–367. [DOI] [PubMed]
- 95.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann NY Acad Sci 2008;1123:134–145. [DOI] [PubMed]
- 96.Simionescu M, Simionescu N. Organization of cell junctions in the peritoneal mesothelium. J Cell Biol 1977;74:98–110. [DOI] [PMC free article] [PubMed]
- 97.Bundgaard M. The three-dimensional organization of tight junctions in a capillary endothelium revealed by serial-section electron microscopy. J Ultrastruct Res 1984;88:1–17. [DOI] [PubMed]
- 98.Adamson RH, Lenz JF, Zhang X, Adamson GN, Weinbaum S, Curry FE. Oncotic pressures opposing filtration across non-fenestrated rat microvessels. J Physiol 2004;557:889–907. [DOI] [PMC free article] [PubMed]
- 99.Kranenburg AR, De Boer WI, van Krieken JH, et al. Enhanced expression of fibroblast growth factors and receptor FGFR-1 during vascular remodeling in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2002;27:517–525. [DOI] [PubMed]
- 100.Morrell NW, Yang X, Upton PD, et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-β1 and bone morphogenetic proteins. Circulation 2001;104:790–795. [DOI] [PubMed]
- 101.Voelkel NF, Cool CD. Pulmonary vascular involvement in chronic obstructive pulmonary disease. Eur Respir J 2003;22(suppl. 46):28s–32s. [DOI] [PubMed]
- 102.Sehgal PB, Mukhopadhyay S. Dysfunctional intracellular trafficking in the pathobiology of pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2007;37:31–37. [DOI] [PMC free article] [PubMed]
- 103.Mukherjee S, Tessema M, Wandinger-Ness A. Vesicular trafficking of tyrosine kinase receptors and associated proteins in the regulation of signaling and vascular function. Circ Res 2006;98:743–756. [DOI] [PubMed]
- 104.Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol 2009;10:609–622. [DOI] [PMC free article] [PubMed]
- 105.Predescu DN, Neamu R, Bardita C, Wang M, Predescu SA. Impaired caveolae function and upregulation of alternative endocytic pathways induced by experimental modulation of intersectin-1s expression in mouse lung endothelium. Biochem Res Int 2012;2012:672705. [DOI] [PMC free article] [PubMed]
- 106.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003;425:577–584. [DOI] [PubMed]
- 107.Bardita C, Predescu D, Justice MJ, Petrache I, Predescu S. In vivo knockdown of intersectin-1s alters endothelial cell phenotype and causes microvascular remodeling in the mouse lungs. Apoptosis 2013;18:57-76. [DOI] [PMC free article] [PubMed]
- 108.Schmidt EP, Tuder RM. Role of apoptosis in amplifying inflammatory responses in lung diseases. J Cell Death 2010;3:41–53. [DOI] [PMC free article] [PubMed]
- 109.Matute-Bello G, Martin TR. Science review: apoptosis in acute lung injury. Crit Care 2003;7:355–358. [DOI] [PMC free article] [PubMed]
- 110.Perl M, Lomas-Neira J, Venet F, Chung CS, Ayala A. Pathogenesis of indirect (secondary) acute lung injury. Expert Rev Respir Med 2011;5:115–126. [DOI] [PMC free article] [PubMed]
- 111.Li X, Shu R, Filippatos G, Uhal BD. Apoptosis in lung injury and remodeling. J Appl Physiol 2004;97:1535–1542. [DOI] [PubMed]
- 112.Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv 2010;23:243–252. [DOI] [PMC free article] [PubMed]
- 113.Burns AR, Smith CW, Walker DC. Unique structural features that influence neutrophil emigration into the lung. Physiol Rev 2003;83:309–336. [DOI] [PubMed]
- 114.Ognibene FP, Martin SE, Parker MM, et al. Adult respiratory distress syndrome in patients with severe neutropenia. N Engl J Med 1986;315:547–551. [DOI] [PubMed]
- 115.Cox G, Crossley J, Xing Z. Macrophage engulfment of apoptotic neutrophils contributes to the resolution of acute pulmonary inflammation in vivo. Am J Respir Cell Mol Biol 1995;12:232–237. [DOI] [PubMed]
- 116.McVey M, Tabuchi A, Kuebler WM. Microparticles and acute lung injury. Am J Physiol 2012;303:L364–L381. [DOI] [PubMed]
- 117.Tushuizen ME, Diamant M, Sturk A, Nieuwland R. Cell-derived microparticles in the pathogenesis of cardiovascular disease: friend or foe? Arterioscler Thromb Vasc Biol 2011;31:4–9. [DOI] [PubMed]
- 118.Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol 2009;297:L1035–L1041. [DOI] [PMC free article] [PubMed]
- 119.Densmore JC, Signorino PR, Ou J, et al. Endothelium-derived microparticles induce endothelial dysfunction and acute lung injury. Shock 2006;26:464–471. [DOI] [PubMed]
- 120.George JN, Pickett EB, Heinz R. Platelet membrane microparticles in blood bank fresh frozen plasma and cryoprecipitate. Blood 1986;68:307–309. [PubMed]
- 121.Guervilly C, Lacroix R, Forel JM, et al. High levels of circulating leukocyte microparticles are associated with better outcome in acute respiratory distress syndrome. Crit Care 2011;15:R31. [DOI] [PMC free article] [PubMed]
- 122.Kuwano K. Epithelial cell apoptosis and lung remodeling. Cell Mol Immunol 2007;4:419–429. [PubMed]
- 123.Dos Santos CC. Advances in mechanisms of repair and remodelling in acute lung injury. Intensive Care Med 2008;34:619–630. [DOI] [PubMed]
- 124.Simak J, Gelderman MP. Cell membrane microparticles in blood and blood products: potentially pathogenic agents and diagnostic markers. Transfus Med Rev 2006;20:1–26. [DOI] [PubMed]
- 125.Desai TJ, Cardoso WV. Growth factors in lung development and disease: friends or foe? Respir Res 2002;3:2. [DOI] [PMC free article] [PubMed]
- 126.Medford AR, Millar AB. Vascular endothelial growth factor (VEGF) in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): paradox or paradigm? Thorax 2006;61:621–626. [DOI] [PMC free article] [PubMed]
- 127.Lahm T, Albrecht M, Fisher AJ, et al. 17β-estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med 2012;185:965–980. [DOI] [PMC free article] [PubMed]
- 128.Lebrin F, Deckers M, Bertolino P, ten Dijke P. TGF-β receptor function in the endothelium. Cardiovasc Res 2005;65:599–608. [DOI] [PubMed]
- 129.Cross LJ, Matthay MA. Biomarkers in acute lung injury: insights into the pathogenesis of acute lung injury. Crit Care Clin 2011;27:355–377. [DOI] [PMC free article] [PubMed]
- 130.Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol 2010;298:L715–L731. [DOI] [PMC free article] [PubMed]
- 131.Medford AR, Douglas SK, Godinho SI, et al. Vascular endothelial growth factor (VEGF) isoform expression and activity in human and murine lung injury. Respir Res 2009;10:27. [DOI] [PMC free article] [PubMed]
- 132.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med 2006;27:337–349. [DOI] [PubMed]
- 133.Pucharcos C, Fuentes JJ, Casas C, et al. Alu-splice cloning of human itersectin (ITSN), a putative multivalent binding protein expressed in proliferating and differentiating neurons and overexpressed in Down syndrome. Eur J Hum Genet 1999;7:704–712. [DOI] [PubMed]
- 134.Keating DJ, Chen C, Pritchard MA. Alzheimer’s disease and endocytic dysfunction: clues from the Down syndrome-related proteins, DSCR1 and ITSN1. Ageing Res Rev 2006;5:388–401. [DOI] [PubMed]
- 135.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 2000;157:277–286. [DOI] [PMC free article] [PubMed]
- 136.Chang KT, Min KT. Upregulation of three Drosophila homologs of human chromosome 21 genes alters synaptic function: implications for Down syndrome. Proc Natl Acad Sci USA 2009;106:17117–17122. [DOI] [PMC free article] [PubMed]
- 137.Cataldo AM, Mathews PM, Boiteau AB, et al. Down syndrome fibroblast model of Alzheimer-related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am J Pathol 2008;173:370–384. [DOI] [PMC free article] [PubMed]
- 138.Hansen J, Floss T, Van Sloun P, et al. A large-scale, gene-driven mutagenesis approach for the functional analysis of the mouse genome. Proc Natl Acad Sci USA 2003;100:9918–9922. [DOI] [PMC free article] [PubMed]
- 139.Yu Y, Chu PY, Bowser DN, et al. Mice deficient for the chromosome 21 ortholog Itsn1 exhibit vesicle-trafficking abnormalities. Hum Mol Genet 2008;17:3281–3290. [DOI] [PubMed]
- 140.Marie B, Sweeney ST, Poskanzer KE, Roos J, Kelly RB, Davis GW. Dap160/intersectin scaffolds the periactive zone to achieve high-fidelity endocytosis and normal synaptic growth. Neuron 2004;43:207–219. [DOI] [PubMed]
- 141.Rose S, Malabarba MG, Krag C, et al. Caenorhabditis elegans intersectin: a synaptic protein regulating neurotransmission. Mol Biol Cell 2007;18:5091–5099. [DOI] [PMC free article] [PubMed]
- 142.Tong XK, Hussain NK, Adams AG, O’Bryan JP, McPherson PS. Intersectin can regulate the Ras/MAP kinase pathway independent of its role in endocytosis. J Biol Chem 2000;275:29894–29899. [DOI] [PubMed]
- 143.Specht K, Harbeck N, Smida J, et al. Expression profiling identifies genes that predict recurrence of breast cancer after adjuvant CMF-based chemotherapy. Breast Cancer Res Treat 2009;118:45–56. [DOI] [PubMed]
- 144.Predescu D, Zhang J, Patel M, Bardita C, Predescu S. Downregulation of intersectin-1s in human lung cancer may contribute to tumorigenesis. Cancer Res 2012;72(8, suppl 1):3264. doi:10.1158/1538-7445.AM2012-3264.
- 145.Adams A, Thorn JM, Yamabhai M, Kay BK, O’Bryan JP. Intersectin, an adaptor protein involved in clathrin-mediated endocytosis, activates mitogenic signaling pathways. J Biol Chem 2000;275:27414–27420. [DOI] [PubMed]
- 146.Wang JB, Wu WJ, Cerione RA. Cdc42 and Ras cooperate to mediate cellular transformation by intersectin-L. J Biol Chem 2005;280:22883–22891. [DOI] [PubMed]
- 147.Morissette MC, Parent J, Milot J. Alveolar epithelial and endothelial cell apoptosis in emphysema: what we know and what we need to know. Int J Chron Obstruct Pulmon Dis 2009;4:19–31. [PMC free article] [PubMed]
- 148.Bardales RH, Xie SS, Schaefer RF, Hsu SM. Apoptosis is a major pathway responsible for the resolution of type II pneumocytes in acute lung injury. Am J Pathol 1996;149:845–852. [PMC free article] [PubMed]
- 149.Guinee D Jr, Brambilla E, Fleming M, et al. The potential role of BAX and BCL-2 expression in diffuse alveolar damage. Am J Pathol 1997;151:999–1007. [PMC free article] [PubMed]
- 150.Predescu SA, Predescu DN, Knezevic I, Klein IK, Malik AB. Intersectin-1s regulates the mitochondrial apoptotic pathway in endothelial cells. J Biol Chem 2007;282:17166–17178. [DOI] [PubMed]
- 151.Tasaka S, Amaya F, Hashimoto S, Ishizaka A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid Redox Signal 2008;10:739–753. [DOI] [PubMed]
- 152.Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem 2004;279:4127–4135. [DOI] [PubMed]
- 153.Bruns RR, Palade GE. Studies on blood capillaries. I. General organization of blood capillaries in muscle. J Cell Biol 1968;37:244–276. [DOI] [PMC free article] [PubMed]
- 154.Bruns RR, Palade GE. Studies on blood capillaries. II. Transport of ferritin molecules across the wall of muscle capillaries. J Cell Biol 1968;37:277–299. [DOI] [PMC free article] [PubMed]
- 155.Popov D, Simionescu M. Structural and transport property alterations of the lung capillary endothelium in diabetes. Ital J Anat Embryol 2001;106:405–412. [PubMed]
- 156.Fra AM, Williamson E, Simons K, Parton RG. De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc Natl Acad Sci USA 1995;92:8655–8659. [DOI] [PMC free article] [PubMed]
- 157.Liu L, Pilch PF. A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J Biol Chem 2008;283:4314–4322. [DOI] [PubMed]
- 158.McMahon KA, Zajicek H, Li WP, et al. SRBC/cavin-3 is a caveolin adapter protein that regulates caveolae function. EMBO J 2009;28:1001–1015. [DOI] [PMC free article] [PubMed]
- 159.Knezevic I, Predescu D, Bardita C, et al. Regulation of dynamin-2 assembly-disassembly and function through the SH3A domain of intersectin-1s. J Cell Mol Med 2011;15:2364–2376. [DOI] [PMC free article] [PubMed]
- 160.Singla S, Predescu D, Bardita C, et al. Pro-inflammatory endothelial cell dysfunction is associated with intersectin-1s down-regulation. Respir Res 2011;12:46. [DOI] [PMC free article] [PubMed]
- 161.Wang XM, Zhang Y, Kim HP, et al. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med 2006;203:2895–2906. [DOI] [PMC free article] [PubMed]
- 162.Drab M, Verkade P, Elger M, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001;293:2449–2452. [DOI] [PubMed]
- 163.Mercier I, Lisanti MP. Caveolin-1 and breast cancer: a new clinical perspective. Adv Exp Med Biol 2012;729:83–94. [DOI] [PubMed]
- 164.Freeman MR, Yang W, Di Vizio D. Caveolin-1 and prostate cancer progression. Adv Exp Med Biol 2012;729:95–110. [DOI] [PubMed]
- 165.Wang LF, Patel M, Razavi HM, et al. Role of inducible nitric oxide synthase in pulmonary microvascular protein leak in murine sepsis. Am J Respir Crit Care Med 2002;165:1634–1639. [DOI] [PubMed]
- 166.Predescu D, Predescu S, Shimizu J, Miyawaki-Shimizu K, Malik AB. Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. Am J Physiol 2005;289:L371–L381. [DOI] [PubMed]
- 167.Sittipunt C, Steinberg KP, Ruzinski JT, et al. Nitric oxide and nitrotyrosine in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;163:503–510. [DOI] [PubMed]
- 168.Primeau M, Ben Djoudi Ouadda A, Lamarche-Vane N. Cdc42 GTPase-activating protein (CdGAP) interacts with the SH3D domain of intersectin through a novel basic-rich motif. FEBS Lett 2011;585:847–853. [DOI] [PubMed]
- 169.Jenna S, Hussain NK, Danek EI, et al. The activity of the GTPase-activating protein CdGAP is regulated by the endocytic protein intersectin. J Biol Chem 2002;277:6366–6373. [DOI] [PubMed]
- 170.Loeb CR, Harris JL, Craik CS. Granzyme B proteolyzes receptors important to proliferation and survival, tipping the balance toward apoptosis. J Biol Chem 2006;281:28326–28335. [DOI] [PubMed]
- 171.Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med 2000;161:237–243. [DOI] [PubMed]
- 172.Bem RA, van Woensel JB, Lutter R, et al. Granzyme A- and B-cluster deficiency delays acute lung injury in pneumovirus-infected mice. J Immunol 2010;184:931–938. [DOI] [PMC free article] [PubMed]
- 173.Warburton D, Tefft D, Mailleux A, et al. Do lung remodeling, repair, and regeneration recapitulate respiratory ontogeny? Am J Respir Crit Care Med 2001;164:S59–S62. [DOI] [PubMed]
- 174.Tomashefski JF Jr, Davies P, Boggis C, Greene R, Zapol WM, Reid LM. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol 1983;112:112–126. [PMC free article] [PubMed]
- 175.Martin TR, Hagimoto N, Nakamura M, Matute-Bello G. Apoptosis and epithelial injury in the lungs. Proc Am Thorac Soc 2005;2:214–220. [DOI] [PMC free article] [PubMed]
- 176.Pittet JF, Mackersie RC, Martin TR, Matthay MA. Biological markers of acute lung injury: prognostic and pathogenetic significance. Am J Respir Crit Care Med 1997;155:1187–1205. [DOI] [PubMed]
- 177.Determann RM, Royakkers A, Wolthuis EK, et al. Ventilation with lower tidal volumes as compared with conventional tidal volumes for patients without acute lung injury: a preventive randomized controlled trial. Crit Care 2010;14:R1. [DOI] [PMC free article] [PubMed]
- 178.Yilmaz M, Keegan MT, Iscimen R, et al. Toward the prevention of acute lung injury: protocol-guided limitation of large tidal volume ventilation and inappropriate transfusion. Crit Care Med 2007;35:1660–1667. [DOI] [PubMed]
- 179.Young MP, Manning HL, Wilson DL, et al. Ventilation of patients with acute lung injury and acute respiratory distress syndrome: has new evidence changed clinical practice? Crit Care Med 2004;32:1260–1265. [DOI] [PubMed]
- 180.Khan H, Belsher J, Yilmaz M, et al. Fresh-frozen plasma and platelet transfusions are associated with development of acute lung injury in critically ill medical patients. Chest 2007;131:1308–1314. [DOI] [PubMed]
- 181.Flores C, Pino-Yanes MdM, Villar J. A quality assessment of genetic association studies supporting susceptibility and outcome in acute lung injury. Crit Care 2008;12:R130. [DOI] [PMC free article] [PubMed]
- 182.Quasney MW. Genetic variation, acute lung injury, and Kipling’s six honest serving men. Crit Care Med 2008;36:2678–2680. [DOI] [PubMed]
- 183.Iles MM. What can genome-wide association studies tell us about the genetics of common disease? PLoS Genet 2008;4:e33. [DOI] [PMC free article] [PubMed]
- 184.Hattersley AT, McCarthy MI. What makes a good genetic association study? Lancet 2005;366:1315–1323. [DOI] [PubMed]
- 185.Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 2003;33:177–182. [DOI] [PubMed]
- 186.Cordell HJ, Clayton DG. Genetic association studies. Lancet 2005;366:1121–1131. [DOI] [PubMed]
- 187.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science 1996;273:1516–1517. [DOI] [PubMed]
- 188.Todd JA, Walker NM, Cooper JD, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 2007;39:857–864. [DOI] [PMC free article] [PubMed]
- 189.Zeggini E, Scott LJ, Saxena R, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 2008;40:638–645. [DOI] [PMC free article] [PubMed]
- 190.Eeles RA, Kote-Jarai Z, Giles GG, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet 2008;40:316–321. [DOI] [PubMed]
- 191.Hunter DJ, Kraft P, Jacobs KB, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 2007;39:870–874. [DOI] [PMC free article] [PubMed]
- 192.Moffatt MF, Kabesch M, Liang L, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007;448:470–473. [DOI] [PubMed]
- 193.Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med 2007;357:443–453. [DOI] [PMC free article] [PubMed]
- 194.Christie JD, Wurfel MM, Feng R, et al. Genome wide association identifies PPFIA1 as a candidate gene for acute lung injury risk following major trauma. PLoS ONE 2012;7:e28268. [DOI] [PMC free article] [PubMed]
- 195.Todd JL, Goldstein DB, Ge D, Christie J, Palmer SM. The state of genome-wide association studies in pulmonary disease: a new perspective. Am J Respir Crit Care Med;184:873–880. [DOI] [PMC free article] [PubMed]
- 196.Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005;436:112–116. [DOI] [PMC free article] [PubMed]
- 197.Jerng JS, Yu CJ, Wang HC, Chen KY, Cheng SL, Yang PC. Polymorphism of the angiotensin-converting enzyme gene affects the outcome of acute respiratory distress syndrome. Crit Care Med 2006;34:1001–1006. [DOI] [PubMed]
- 198.Gao L, Flores C, Fan-Ma S, et al. Macrophage migration inhibitory factor in acute lung injury: expression, biomarker, and associations. Transl Res 2007;150:18–29. [DOI] [PMC free article] [PubMed]
- 199.Meyer NJ, Huang Y, Singleton PA, et al. GADD45a is a novel candidate gene in inflammatory lung injury via influences on Akt signaling. FASEB J 2009;23:1325–1337. [DOI] [PMC free article] [PubMed]
- 200.Flores C, Ma SF, Maresso K, Wade MS, Villar J, Garcia JG. IL6 gene-wide haplotype is associated with susceptibility to acute lung injury. Transl Res 2008;152:11–17. [DOI] [PubMed]
- 201.Gong MN, Thompson BT, Williams PL, et al. Interleukin-10 polymorphism in position −1082 and acute respiratory distress syndrome. Eur Respir J 2006;27:674–681. [DOI] [PMC free article] [PubMed]
- 202.Flores C, Perez-Mendez L, Maca-Meyer N, et al. A common haplotype of the LBP gene predisposes to severe sepsis. Crit Care Med 2009;37:2759–2766. [DOI] [PubMed]
- 203.Christie JD, Ma SF, Aplenc R, et al. Variation in the myosin light chain kinase gene is associated with development of acute lung injury after major trauma. Crit Care Med 2008;36:2794–2800. [DOI] [PubMed]
- 204.Gong MN, Zhou W, Williams PL, Thompson BT, Pothier L, Christiani DC. Polymorphisms in the mannose binding lectin-2 gene and acute respiratory distress syndrome. Crit Care Med 2007;35:48–56. [DOI] [PMC free article] [PubMed]
- 205.Zhai R, Zhou W, Gong MN, et al. Inhibitor kB-α haplotype GTC is associated with susceptibility to acute respiratory distress syndrome in Caucasians. Crit Care Med 2007;35:893–898. [DOI] [PubMed]
- 206.Marzec JM, Christie JD, Reddy SP, et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J 2007;21:2237–2246. [DOI] [PubMed]
- 207.Tsangaris I, Tsantes A, Bonovas S, et al. The impact of the PAI-1 4G/5G polymorphism on the outcome of patients with ALI/ARDS. Thromb Res 2009;123:832–836. [DOI] [PubMed]
- 208.Hong SB, Huang Y, Moreno-Vinasco L, et al. Essential role of pre-B-cell colony enhancing factor in ventilator-induced lung injury. Am J Respir Crit Care Med 2008;178:605–617. [DOI] [PMC free article] [PubMed]
- 209.Meyer NJ, Christie JD. Extracellular superoxide dismutase haplotypes and acute lung injury: reading into the genome to understand mortality? Am J Respir Crit Care Med 2009;179:89–91. [DOI] [PubMed]
- 210.Gong MN, Wei Z, Xu LL, Miller DP, Thompson BT, Christiani DC. Polymorphism in the surfactant protein-B gene, gender, and the risk of direct pulmonary injury and ARDS. Chest 2004;125:203–211. [DOI] [PubMed]
- 211.Dhainaut JF, Charpentier J, Chiche JD. Transforming growth factor-β: a mediator of cell regulation in acute respiratory distress syndrome. Crit Care Med 2003;31(suppl. 4):S258–S264. [DOI] [PubMed]
- 212.Gong MN, Zhou W, Williams PL, et al. −308GA and TNFB polymorphisms in acute respiratory distress syndrome. Eur Respir J 2005;26:382–389. [DOI] [PubMed]
- 213.Gibson G. The environmental contribution to gene expression profiles. Nat Rev Genet 2008;9:575–581. [DOI] [PubMed]
- 214.Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity 2010;105:4–13. [DOI] [PubMed]
- 215.McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008;9:356–369. [DOI] [PubMed]