

Abstract Abstract
Pulmonary hypertension of the newborn is caused by a spectrum of functional and structural abnormalities of the cardiopulmonary circuit. The existence of multiple etiologies and an incomplete understanding of the mechanisms of disease progression have hindered the development of effective therapies. Animal models offer a means of gaining a better understanding of the fundamental basis of the disease. To that effect, a number of experimental animal models are being used to generate pulmonary hypertension in the fetus and newborn. In this review, we compare the mechanisms associated with pulmonary hypertension caused by two such models: in utero ligation of the ductus arteriosus and chronic perinatal hypoxia in sheep fetuses and newborns. In this manner, we make direct comparisons between ductal ligation and chronic hypoxia with respect to the associated mechanisms of disease, since multiple studies have been performed with both models in a single species. We present evidence that the mechanisms associated with pulmonary hypertension are dependent on the type of stress to which the fetus is subjected. Such an analysis allows for a more thorough evaluation of the disease etiology, which can help focus clinical treatments. The final part of the review provides a clinical appraisal of current treatment strategies and lays the foundation for developing individualized therapies that depend on the causative factors.
Keywords: pulmonary hypertension, chronic hypoxia, ductal ligation, sheep prenatal programming
Overview/Introduction
Pulmonary hypertension of the newborn (PHN) is a clinical disease usually characterized by dysregulation or developmental abnormalities of the pulmonary microvasculature. Afflicted infants can have aberrantly reactive or overly muscular vessels, and newborns with either acute or chronic disease have difficulty adapting to breathing during the birth transition and early postnatal period. There are a number of risk factors that may predispose newborns to the development of this disease, including high-altitude living, maternal malnutrition, placental insufficiency due to environmental factors or diseases such as preeclampsia, and other pregnancy complications such as staphylococcus infection or meconium aspiration. Because of the morbidity associated with this group of maladies and the large number of people predisposed to developing disease because of environmental factors or other causes, it is imperative to further understand the etiology of PHN in the hopes of finding effective treatments. Most research related to understanding the development and treatment of this disease is now carried out on experimental models.
There are a number of techniques for causing pulmonary hypertension (PH) in animal fetuses and newborns for research purposes. Some of the most commonly used experimental models of pulmonary vascular dysfunction are in fetal lambs, including chronic ligation of the ductus arteriosus to directly increase pulmonary pressure,1-3 surgical left-to-right shunt to increase pulmonary blood flow,4,5 and long-term maternal hypoxemia to reduce fetal oxygenation.6-8 Ligation of the ductus arteriosus significantly reduces the amount of blood that bypasses the lung, forcing a larger portion of cardiac output into an underdeveloped pulmonary vascular bed. The increased blood flow through the lungs and its vasculature, which would normally occur only after birth, coupled with the high intrinsic tone of the fetal pulmonary vessels, leads to grossly elevated pulmonary vascular pressures. On the other hand, our laboratory and others find that placing pregnant sheep in a rarefied environment, resulting in long-term maternal and fetal hypoxia, leads to moderately elevated pulmonary vascular pressures in the offspring.6-8 This methodology used to induce PH in newborn animals has been used with large- as well as small-animal models, including pigs,9,10 calves,11 mice,12,13 and rats.14-16 Dr. Llanos’s research group from the Centro Internacional de Estudios Andinos (INCAS) and Universidad de Chile in Santiago6,7,17-19 and our group from Loma Linda University in California, in collaboration with the White Mountain Research Station (WMRS),8 both use maternal and postnatal high-altitude hypoxia to cause PH in sheep. In both the ductal-ligation and high-altitude experimental models, the resultant high pulmonary vascular resistance (PVR) causes back pressure on the right heart, leading to cardiac hypertrophy and insufficiency.
In this review, we compare the mechanisms associated with PH due to ductal ligation and those associated with PH due to chronic antenatal and postnatal hypoxia in sheep fetuses and newborns. By making comparisons between studies using these two models, we show that the etiology of disease is dependent on the type of stress to which the fetus is subjected. Such an analysis has the potential of providing a deeper understanding of the possible causes of PHN, which in turn can help focus future research and clinical treatments. Toward this end, we provide a brief description of both of these animal models, followed by a summary of what is known regarding their effects on regulation of pulmonary gene expression as well as pathways of vasoconstriction and vasodilation. The final part of the review provides a clinical appraisal of current and forthcoming treatment strategies with respect to what is known about the etiology of disease in these experimental models.
Impact of ductal ligation on pulmonary vascular function
Ligation of the fetal ductus arteriosus is a well-described experimental method1,2 that has been used for simulating primary PHN (PPHN) in an animal model.3,20,21 In brief, pregnant ewes in the late stages of gestation (126–135 days; normal full length is 147 days) are fasted for 24 hours and then anesthetized under pentobarbital or another anesthetic agent. A hysterotomy is performed through the anterior abdominal wall, and the left fetal forelimb is exposed under sterile conditions. Polyvinyl catheters are advanced into the aorta from the axillary artery and secured with sutures. Additional catheters are inserted into the fetal left atrium and main pulmonary artery via a left-sided thoracotomy and secured with sutures. The exposed ductus arteriosus is partially or completely occluded via ligation with umbilical tape; the fetus is then returned to the uterus, and the uterine incision is sutured. The ewes are allowed to recover while pressure and flow are monitored from the inserted catheters. The control animals used with this method include fetuses subjected to thoracotomies and vascular instrumentation without undergoing ductus ligation.
Ligation of the ductus arteriosus causes a decrease in oxygen and nutrient delivery to the growing fetus while simultaneously increasing pressures in the pulmonary vasculature and right ventricle (RV). Animals with chronic ductal ligation manifest elevated PVR and abnormal pulmonary vascular reactivity to elevated levels of inspired oxygen.3 More specifically, pulmonary artery (PA) pressures in fetuses that undergo ductal ligation rise from approximately 54 to 72 mmHg.22 Ductal ligation also results in substantial right ventricular hypertrophy, as evidenced by higher RV free-wall weights (12.5 vs. 6.8 g) due to elevated PVR and RV afterload.22 Moreover, there are changes in contractile-protein expression that contribute to vessel dysfunction. Specifically, actin and myosin expression increases, with a disproportionate elevation in actin levels and a depression in myosin ATPase levels. Taken together, these findings suggest that animals with a ligated ductus have stiffer pulmonary vascular myocytes and that these cells cannot generate as much force.22
One of the key strengths of the ductal-ligation technique is its utility for understanding the mechanisms of disease due to high intrinsic pulmonary vascular pressures before birth, as well as for evaluating novel treatments. Physiological data can be collected from the fetus while the disease develops, something not readily achieved in rodent models. The fetus can then be delivered and its lungs and pulmonary vessels harvested for various physiological and analytical studies, such as wire myography, bioimaging, mRNA quantification, Western blot analysis, and immunohistochemistry. Table 1 summarizes hemodynamic, right ventricular, and vascular changes noted in this model by previously published studies.
Table 1.
Mean pulmonary artery pressures, right ventricular hypertrophy, and pulmonary artery anatomic changes in ductus arteriosus ligation (DAL) sheep model
| Reference | Mean pulmonary artery pressure changes, mmHg | Right ventricular changes (RVH) | Pulmonary arterial anatomic changes | |||
|---|---|---|---|---|---|---|
| DAL | Control | DAL | Control | DAL | Control | |
| Grover et al.23,a | NA | NA | 0.71 ± 0.02 | 0.55 ± 0.02 | 65.4 ± 4 | 48.1 ± 3 |
| Gien et al.24,b | 77 ± 5, 79 ± 5 | 42 ± 2, 41 ± 2 | 0.82 ± 0.05 | 0.53 ± 0.03 | NA | NA |
| Belik et al.22,c | 72.3 ± 3.8 | 54.1 ± 2 | 12.5 ± 0.7 g | 6.8 ± 0.3 g | 2.2 ± 0.19 | 0.85 ± 0.06 |
| Abman et al.2,d | 62 ± 3 | 48 ± 1 | 7.4 ± 0.5 g, 0.60 ± 0.02 | 5.8 ± 0.3 g, 0.49 ± 0.01 | 76.7 ± 7.6 | 53.3 ± 2.8 |
| Jaillard et al.25,e | Day 2: 52 ± 3; day 8: 67 ± 4 | Day 2: ∼44; day 8: ∼49 | 0.75 ± 0.05 | 0.5 ± 0.04 | 17 ± 1 | 12 ± 2 |
Values are means ± SE when available. Some values are approximations of the mean based on graphs or other figures provided by the authors in the source materials, because of a lack of specific published values. RVH is expressed as 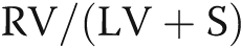 , the weight ratio of right ventricle to (left ventricle plus septum), unless noted. Pulmonary artery anatomic changes are expressed as percent of wall thickness, (2 × wall thickness/entire vessel diameter) × 100, unless otherwise noted.
, the weight ratio of right ventricle to (left ventricle plus septum), unless noted. Pulmonary artery anatomic changes are expressed as percent of wall thickness, (2 × wall thickness/entire vessel diameter) × 100, unless otherwise noted.
Ductus arteriosus constriction.
Two different groups described in the same study.
RVH expressed as absolute weight of RV free wall in grams. Pulmonary artery anatomic changes expressed as second- to fifth-generation pulmonary artery combined actin content in micrograms per milligram of wet tissue.
Partial DAL. RVH expressed as absolute weight and as RV/LV+S.
Measurements performed on day 2 and 8 after partial DAL or sham control operation.
Impact of antenatal hypoxia on pulmonary function
When a mother is at sea level and maternal alveolar partial pressure for oxygen (Po2) is roughly 100 torr, the fetal umbilical-vein Po2 is significantly lower, ∼30 torr in sheep.26 Despite this low oxygen tension, fetal oxygen delivery and whole-body growth and development requirements are facilitated, in part, by the higher oxygen affinity of fetal hemoglobin27,28 and elevations in cardiac output29 and capillary density30 that enable improved tissue oxygenation. Regardless of the facilitated oxygen delivery system, the fetus is still quite sensitive to reduced oxygen tension, which can have detrimental effects.
When a mother travels to high altitude, her alveolar Po2 falls in proportion to the altitude according to Boyle’s Law. At the highest altitudes where humans are born (La Rinconada, Peru: 5,100 m), the mother’s alveolar Po2 is reduced substantially and can be as low as 35 torr. While there are no published reports for arterial saturations of fetal humans at this extreme altitude, studies performed on our sheep housed at 3,801 m at the WMRS show that the alveolar Po2 of the pregnant ewe is ∼65 torr, while the postductal arterial Po2 of the fetus is 17–19 torr.26 This represents a 35% decrease in the alveolar Po2 in the mother and as much as a 35% drop in the arterial Po2 of the fetus relative to that at sea level.26 When these findings are extrapolated to humans, they represent a significant stress to both the mother and her unborn child. From a human health perspective, the WMRS and the INCAS station on the Andean altiplano (3,600 m) are important because they are at an altitude similar to that of major cities in the Andes (La Paz, Bolivia: 3,650 m) and Himalayas (Lhasa, Tibet: 3,650 m) and therefore are representative of large high-altitude population centers worldwide.
Diversity in acclimatization. Sheep are similar to human populations in that they are less tolerant of high-altitude antenatal hypoxia than more resilient species, such as llamas and yaks.19,31 The influence of antenatal hypoxia on human health still varies among different populations. Whereas newborn Tibetan and Andean children adapt well to birth and life at high altitude, children born to Han Chinese and European parents do not. Specifically, following pregnancy at high altitude, human infants from these nonadaptive populations display growth retardation and elevated pulmonary vascular pressures,32-35 although little is known regarding the extent of pulmonary vascular remodeling in human infants following antenatal hypoxia.32-37
Many newborns that suffer significant antenatal hypoxia adapt poorly to breathing air. Pulmonary vascular myocyte and fibroblast growth and function become aberrant. There is increased pulmonary vessel muscularization, as well as collagen and elastin deposition, which decreases internal vessel diameter and peripheral arterial density. Although we do not know the full extent of injury to the sheep lung, housing pregnant ewes at 3,801 m for an extended period during gestation damages the fetal lung. In fetal sheep, high-altitude gestation increased the medial wall thickness of distal but not proximal portions of the PA tree, compared to that in low-altitude controls.38-40 Functionally, newborn sheep born at INCAS on the Andean altiplano (3,600 m) and at WMRS in California (3,801 m) have moderately elevated pulmonary pressures, exacerbated hypoxia-induced pulmonary vasoconstriction, and modest right ventricular hypertrophy.6,8 Robust segmental-analysis procedures to assess changes in lung structure have not been performed on newborn sheep born at INCAS or WMRS,41,42 although these results suggest that antenatal and perinatal hypoxia remodels the pulmonary vascular structure. Table 2 summarizes hemodynamic, right ventricular, and vascular changes noted in this model by previously published studies.
Table 2.
Mean pulmonary artery pressures, right ventricular hypertrophy, and pulmonary artery anatomic changes in the high-altitude hypoxia (HAH) sheep model
| Reference | Mean pulmonary artery pressure changes (mmHg) | Right ventricular changes (RVH) | Pulmonary arterial anatomic changes | |||
|---|---|---|---|---|---|---|
| HAH | Control | HAH | Control | HAH | Control | |
| Herrera et al.7a | 20.9 ± 1.1 | 13.7 ± 0.5 | 0.37 ± 0.017 | 0.328 ± 0.009 | 29.98 ± 2.96 | 21.09 ± 1.73 |
| Blood et al.8b | 21.1 ± 1.9 | 16.1 ± 1.1 | NA | NA | NA | NA |
| Herrera et al.17c | ∼21 | ∼12 | NA | NA | 62 ± 4 | 48 ± 5 |
Values are means ± SE when available. Some values are approximations of the mean based on graphs or other figures provided by the authors in the source materials, because of a lack of specific published values. RVH is expressed as 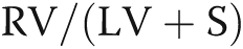 , the weight ratio of right ventricle to (left ventricle plus septum).
, the weight ratio of right ventricle to (left ventricle plus septum).
High-altitude hypoxia defined as spending 70% of gestation at altitude, followed by return to sea level. Vascular smooth muscle area of small pulmonary arteries (100–200 μm), presented as the relative difference of external and internal boundaries of the tunica media.
As measured in newborn lambs (10–14 days old) after gestation and delivery at high altitude and study at low altitude, versus low-altitude controls.
8–12-day-old newborn lambs, gestated, born, and studied at altitude versus low-altitude controls. Percent wall thickness expressed as [(external area−internal area)/external area] × 100; “areas” were defined by external and internal elastic lamina.
Growth factors and transcriptional regulators
Lung development is orchestrated by a wide array of growth factors and transcriptional regulators. Hypoxia inducible factor (HIF)43 and vascular endothelial growth factor (VEGF)44 are two of the best understood factors. There are other important factors that are less well studied. These include fibroblast growth factor, which is important during the pseudoglandular stage,45-47 retinoic acid, which is crucial in early lung development and likely important to alveolarization,48 and bone morphogenetic protein (BMP) and the BMP receptor 2, which have received substantial attention for their importance in familial pulmonary arterial hypertension,49 as well as transforming growth factor-β signaling, which is important to branching morphogenesis and alveolarization.50-52 Although there are some reports on the effects of ligation of the ductus arteriosus23 or antenatal hypoxia on the function of many of these growth factors, there are relatively few associated in-depth studies in the sheep models.
Hypoxia inducible factor
HIF is a transcription factor that is regulated by oxygen availability and subsequently modulates the expression of multiple genes. It is best known for its stimulation of erythropoietin transcription and consequent increase in red blood cell production.53 Still, HIF is a central regulator of the transcription of many genes. HIF levels are elevated in human fetal lung in utero,54 and its activity is central to normal lung development.43 This is exemplified in the −/− HIF-1α knockout mice that do not reach full term and have enlarged vascular structures, along with impaired lung morphogenesis.55 Further illustrating the importance of HIF to pulmonary vascular dysfunction, HIF 1α-deficient (+/−) adult mice are protected from lung malformations due to hypoxia. In these mice, PA pressure elevations and RV hypertrophy are attenuated, compared to those in wild-type mice, mainly as a result of reduced pulmonary vascular remodeling.13 Similar results were noted in HIF-2α-deficient heterozygote mice,12 suggesting that more than one isoform is important to pulmonary vascular remodeling and function.
HIF activity is certainly labile and dependent on environmental factors in the prenatal period. This idea of varied HIF activity is exemplified in a fetal-lamb model of respiratory distress, where delivery and mechanical ventilation of lambs depresses HIF expression.56 Given that HIF not only is vital to normal lung development but also responds to low oxygen tensions and plays an instigating role in vascular malformations and the development of PH, the lack of publications regarding the influence of ductal ligation or chronic hypoxia on HIF function in fetal and newborn lambs indicates that this is an important area to pursue. This is especially intriguing because fetal and newborn sheep from WMRS have an elevated hematocrit,8,57 the prototypical physiological response to tissue hypoxia that is mediated by HIF-1α activation.53 Of critical importance to the fetal lambs exposed to antenatal chronic hypoxia is that the elevated hematocrit compensates for the reduced arterial O2 saturation at high altitude, maintaining oxygen delivery to the growing tissues in the rarefied environment.
Vascular endothelial growth factor
VEGF and its receptor are critical to angiogenesis in the developing lung and are important mediators in pulmonary vascular remodeling under hypoxic conditions.58 HIF-1 is directly coupled to VEGF function, where HIF activation enhances VEGF production.59,60 The importance of VEGF to lung development is supported by studies where VEGF receptor activity was inhibited with Su-5416 in newborn rats. This novel synthetic VEGF receptor inhibitor impairs pulmonary vascular growth, reduces arterial density, causes RV hypertrophy, and increases PA wall thickness.61 These pathological changes parallel the influence of hypoxia on the cardiopulmonary circuit.
VEGF receptors are primarily responsible for endothelial cell differentiation and maintenance as well as vascular organization and permeability.62 Reduced VEGF is also known to be important to disease in humans, because infants with PHN exhibit decreased plasma levels of VEGF.63 Thus, there is an apparent association between pressurization, HIF-1, and VEGF and impaired vascular morphogenesis in response to hypoxic insults. Ductal ligation decreases VEGF production, and there appears to be a selective compensatory increase in VEGF-R2, but not VEGF-R1, receptor expression.24,64 At present, we do not know how maternal and perinatal hypoxia affects VEGF signaling in the fetal and newborn sheep lung, but since hypoxia likely increases HIF activity in these animals, as evidenced by the increased hematocrit, we suspect that there may be vast differences, as compared to the impediments in HIF-VEGF signaling due to ductal ligation, which do not have elevations in red blood cell production.65 In light of the importance of VEGF in the pathophysiology of PHN and PH, elucidating such interactions in the different models used to simulate these diseases becomes imperative.
Mechanisms of Pulmonary Vasoconstriction
PHN due to ductal ligation or antenatal hypoxia leads to aberrant vasoconstriction due to changes in varied pathways. Most of the seminal details regarding pulmonary arterial contraction are based largely on studies performed in vessels and cells from adult animals. These examinations show that there are two key mechanisms that regulate constriction of the pulmonary vasculature. Figure 1 provides an overview of these pathways, with fundamental components and the influence of ductal ligation and antenatal hypoxia described in the text. First is the distinctive process of vessel constriction in response to acute tissue hypoxia. Second is constriction to stimulatory agonists, many of which act through G protein–coupled receptors (GPCRs). There are many different agonists that cause constriction. Several of the more potent vasoconstrictors are endothelin-1 (ET-1), serotonin (5-HT), and prostaglandin F2α (PGF2α). These and other, less potent molecules, such as norepinephrine (NE)25 and platelet-activating factor (PAF), stimulate Gq-coupled receptors that cause a signaling cascade, which constricts the vessel. The signaling pathways coupled to acute tissue hypoxia and receptor stimulation overlap to a large extent and are highly organized and sophisticated.
Figure 1.

Pulmonary vasoconstriction is orchestrated. Neural, hormonal, and humoral mediators, as well as tissue hypoxia, constrict the lung vasculature in utero and maintain a high level of fetal pulmonary vascular resistance. Many of these agonists work through a Gq-coupled receptor pathway to activate a number of intracellular signaling systems that lead to pulmonary vascular smooth muscle cell contraction. These pathways are detailed in the text and ultimately cause simultaneous activation of myosin light-chain kinase (MLCK) and inhibition of myosin light-chain phosphatase (MLCP). The mechanisms associated with hypoxia-induced pulmonary vasoconstriction are controversial but overlap those of the other mediators. Adult animal studies illustrate that the hypoxia-induced pulmonary vasoconstriction response includes a combination of activation of L-type Ca2+ channels (CaL), ryanodine receptors (RyRs), Rho-kinase, nonselective cation channels, and inhibition of K+ channels. A dashed green arrow indicates an activation pathway; a dashed red line with a bar indicates an inhibition pathway; a dashed blue arrow indicates movement of calcium. CaM/CAMKII: calmodulin and Ca2+/calmodulin-dependent protein kinase II; ClCa: calcium-activated chloride channel; CPI-17: C-kinase potentiated protein phosphatase-1 inhibitor; DAG: diacylglycerol; Em: membrane potential; ET-1: endothelin-1; Gq: q alpha subunit of G protein–coupled receptor; InsP3/InsP3R: inositol 1,4,5 triphosphate and associated receptor; Kv: voltage-dependent potassium channel; MLC20/MLC20-P: 20-kDa myosin light chain and phosphorylated moiety; NE: norepinephrine; NSCC: nonselective cation channel; PAF: platelet-activating factor; PGF2α: prostaglandin F2α; PKC: protein kinase C; PLC: phospholipase C; PP1c: protein phosphatase-1c; RhoA/ROCK: Ras homolog gene family, member A, and associated kinase; SERCA: sarco/endoplasmic reticulum Ca2+-ATPase; 5-HT: serotonin.
Hypoxia-induced pulmonary vasoconstriction (HPV)
HPV is an intrinsic property of the pulmonary vasculature that serves to match ventilation to perfusion in the normal lung. In the prenatal period, the hypoxic pulmonary vasoconstrictive response helps to maintain high pulmonary vascular tone throughout the lung and diverts blood from the high-resistance pathways of the lung to the low-resistance pathway through the ductus arteriosus and foramen ovale. Chronic antenatal and perinatal hypoxia can program the lung for exaggerated hypoxia-induced pulmonary vasoconstrictive responses.6-8 Unfortunately, we did not find any reports illustrating the effects of ductal ligation on HPV responses and thus do not have a good basis for comparison.
Newborn human infants born at extreme altitudes (Morococha, Peru: 4,540 m), where the alveolar Po2 is ∼50 torr, exemplify the pulmonary vascular problems with birth at high altitude. Infants born in this rarefied environment had PA pressures of ∼60 mmHg, nearly as high as systemic pressures, for the first 72 hours after birth. This contrasts with infants born at sea level, where PA pressure falls from ∼75 to ∼20 mmHg over that time period.34 Although supplemental O2 or simply higher ambient Po2 may reduce pulmonary arterial pressures, there appears to be long-standing perinatal hypoxic programming of the cardiovascular system. Specifically, infants born at high altitude had greater RV wall thickness throughout their first year of life than sea level controls.66 This phenomenon is indicative of persistent PH and may cause lifelong complications. The elevated PA pressures in these infants can be reduced to sea level values with supplemental oxygen.34,67 This indicates that the decreased change in oxygen tension from the in utero circulation to breathing air is a critical factor. Presumably, low inspired Po2 minimizes the stimulus for vasodilation at birth. Ultimately, the reduced Po2 can promote sustained pulmonary vasoconstriction and impede vessel relaxation and the normal drop in PA pressures. The resultant elevated pressures may contribute to an increased prevalence of atrial septal defects.68 Our data and those of the Llanos group show that lambs born at high altitude in California or the Andes are similar to humans in that they have modestly elevated pulmonary pressures at sea level and exhibit highly exaggerated HPV responses when they breathe rarefied air with only 10% O2.6-8,17
Cellular mechanisms of contraction
The cellular mechanisms responsible for pulmonary arterial contraction due to acute hypoxia and GPCR activation are complex and involve modulation of ionic and nonionic signaling pathways. The mechanisms of HPV have been intensively studied for a number of decades, and multiple recent reviews and books outline these mechanisms in detail.69-71 In general, hypoxia and Gq-coupled receptors cause contraction through activation of a cascade of cellular signals. Depending on the mechanism of activation, the signal is then amplified through modulation of Ca2+-dependent and Ca2+-independent signals. Ca2+-dependent signals are highly reliant on depolarization of the plasma membrane as well as on activation of myo-inositol 1,4,5-trisphosphate (InsP3) and ryanodine receptors (RyRs). The Ca2+-independent pathways include generation of diacylglycerol and coordinated activation of protein kinase C as well as the Ras homolog gene family, member A (RhoA), and Rho-associated kinase (ROCK). Ultimately, the signals come together to enhance the phosphorylation and activity of the 20-kDa myosin light chain (MLC20). Vasoconstriction is achieved via the phosphorylating effect of myosin light-chain kinase (MLCK) on myosin and its interaction with actin. Myosin light-chain phosphatase (MLCP) acts to dephosphorylate myosin, inducing smooth muscle cell relaxation and subsequent vasodilation. The kinase and phosphatase work synergistically, where GPCR activation and acute hypoxia cause simultaneous activation of MLCK and inhibition of MLCP, resulting in much greater contraction than would occur if only the kinase were activated. Thus, the cellular and molecular mechanisms underlying vascular contraction due to hypoxia and GPCR activation provide numerous targets that hold promise for new therapeutics in the treatment of PH in the newborn. We highlight some of the cellular mechanisms that are of current interest.
Endothelin-1
ET-1 is a 21–amino acid molecule produced mostly in the vascular endothelium. It is a potent vasoconstrictor that activates 2 GPCRs in the pulmonary vasculature that are coupled to Gq signaling systems. ET-1 causes pulmonary vasoconstriction primarily through activation of the endothelin receptor type A (ETA), while activation of the endothelin receptor type B (ETB) can attenuate this effect because of its high expression in vascular endothelium.72 ET-1 signaling has received significant attention in the setting of increasing levels in the lung vasculature due to acute hypoxia and elevated PA pressures.73 ET-1 is also thought to induce pulmonary artery smooth muscle cell (PASMC) mitogenesis and is likely involved with hypoxia-induced pulmonary vascular remodeling that is critical to the development of PH.74 A recent review by Shao et al.75 provides a detailed survey of what is currently known about the contribution of endothelins to human PA hypertension. Human neonates with severe PPHN have high circulating levels of ET-1. However, use of ETA and ETB blockers in human patients is limited.75 Still, clinical studies with the nonselective ET-1 receptor blocker bosentan have showed promising results and demonstrate their therapeutic potential in adults76,77 and possibly newborns.78
Ductal ligation and chronic hypoxia both enhance ET-1-mediated pulmonary vascular contraction in sheep. Ivy et al.79 showed that in the ovine fetus, ductal ligation increased ET-1 content 3-fold, while ET-1 receptor activity was altered to promote vasoconstriction. ETB vasodilatory effects were maintained during the first few days after the onset of PH, but these eventually became suppressed. Around the same time period, ETA-mediated vasoconstriction became more pronounced, and this was concomitant with an increase in pulmonary resistance.80 In PAs of newborn sheep that were born and maintained at high altitude, chronic hypoxia increased the sensitivity of contraction to ET-1.7 Yet such programming may occur after birth because ET-1 constriction was maintained in PAs from fetal sheep exposed to maternal chronic hypoxia.81
Although no comprehensive studies have been performed in high-altitude fetal or newborn lambs, postnatal chronic hypoxia disrupts many components of ET-1 signaling in newborn piglets. More specifically, Noguchi et al.82 showed that chronic postnatal hypoxia caused persistently high circulating ET-1 levels and increased ETA density and binding throughout the entire pulmonary vasculature, without the normally seen transient postnatal ETB expression increase, a scenario that may impair vasodilation. Similarly, Schindler et al.83 showed that PAs of chronically hypoxic piglets had up to a 3-fold increase in contractile response to ET-1, a lack of ETB-NO-mediated vasodilatory response at 3 days after birth, and coconstriction with adjacent bronchi. Overall, the effects of chronic hypoxia on endothelin signaling in the pig lung are similar to those in the lamb ductal-ligation model, suggesting that endothelin signaling plays a role in PHN in both models. Specifically, the data suggest that both ligation and hypoxia enhance contraction through upregulation of ETA activity and potentially through concomitant suppression of ETB-dependent vasodilation.
Platelet-activating factor
PAF is a potent phospholipid that causes pulmonary vasoconstriction through activation of Gq-coupled receptors.84 Ishii et al.85 illustrated that in the lung, acute hypoxia upregulates PAF production; however, the effects are cell type specific. As it does for ET-1 and other signals, gestational age affects PAF in fetal sheep. Specifically, PAF levels are high in near-term lambs and fall shortly after birth.86 The high endogenous levels of PAF in near-term lambs are likely to contribute to high PVR in utero.86 Hypoxia upregulates PAF synthesis and PAF receptor function and signaling and downregulates PAF catabolism in late-stage fetal sheep, an effect that contributes to the increase in PAF function.87,88 This includes heightened PAF-dependent, InsP3-stimulated Ca2+ release from intracellular stores.87
Chronic maternal hypoxia causes an increase in fetal PAF levels through enhanced PAF synthesis and also increases PA PAF receptor expression and PAF-related cell growth.38 Although there is a substantial body of knowledge regarding PAF signaling, there are still significant knowledge gaps. Such details regarding the progression of PAF signaling in the ductal-ligation model are currently unresolved. However, the parallels with ET-1 signaling during development and with chronic maternal hypoxia indicate that it is worthwhile to understand whether ductal ligation has common effects on PAF and ET-1.
Serotonin
Serotonin (5-HT) is one of the most potent inflammatory vasoconstrictors and is primarily released from platelets. This signaling molecule causes vasoconstriction predominantly through activation of 5-HT2 Gq-coupled receptors on smooth muscle cells. However, a small portion of their vasoconstrictor ability is through activation of Gi-coupled 5-HT1 receptors that reduce cyclic adenosine monophosphate (cAMP) vasodilatory influences.89 Serotonin activation of 5-HT2 receptors, like activation of ET-1, PAF, and other Gq-coupled receptors, causes constriction through activation of Ca2+-dependent, as well as Ca2+-independent, pathways. Recent studies indicate that 5-HT may be important to PHN, as infants born to mothers treated with selective serotonin reuptake inhibitors during pregnancy are at an increased risk of developing disease.90,91
In the fetal sheep, 5-HT exerts a constitutive vasoconstricting influence on the pulmonary vasculature via the 5-HT2A receptor, as infusion of the selective agonist ketanserin results in significant decreases in PA pressure and vascular resistance.92 Serotonin-dependent pulmonary arterial vasoconstriction is then impaired by chronic hypoxia in fetal,89 as well as newborn,8 sheep. These findings follow from studies showing development of chronic hypoxia–induced PH in rats and mice.93,94 We found that in the fetus, antenatal maternal chronic hypoxia desensitized the PAs to 5-HT at lower 5-HT concentrations. The desensitization appears to be due to loss of 5-HT1-dependent contraction, while 5-HT2 sensitivity is preserved. Moreover, the maximum contraction attained by 5-HT was maintained. In the newborn, we find that chronic hypoxia increased the maximal force due to 5-HT; however, this remains proportional to high potassium depolarization–mediated contraction, and the sensitivity of arterial contraction to 5-HT is retained. In contrast to the chronic-hypoxia model, 5-HT signaling pathways appear to be upregulated in ductal-ligated lambs. Delaney et al.92 have recently shown that ductal ligation results in an increase in 5-HT production by PA endothelial cells as well as an increase in the 5-HT2A receptor in PASMCs. These data suggest that 5-HT signaling may be differentially altered, depending on whether the fetus is exposed to chronic hypoxia or another type of stress that results in the development of PHN.
Rho-kinase
Activation of Gq-coupled receptors by a variety of vasoreactive compounds, including 5-HT, NE, and ET-1, activates RhoA signaling through ROCK, in parallel with calcium signaling pathways.95 ROCK is a protein kinase with 2 isoforms, ROCK I and II.81 Upon phosphorylation, the substrates of ROCK are activated to initiate a variety of cellular processes ranging from contraction to cellular differentiation and mitogenesis. With regard to vascular smooth muscle contraction, ROCK mediates phosphorylation of the MYPT1 subunit of MLCP. Phosphorylation at threonine 698 and 853 in the human sequence of MYPT1 enhances sensitivity to Ca2+ by inhibiting MLCP.81 Importantly, ROCK helps to maintain high PVR in the fetal lung. Thus, ROCK may contribute to the increased vascular tone in neonatal PH. Much of our understanding about ROCK is based on studies performed in rodent models, with that information being applied to the sheep models. In a chronic-hypoxia neonatal Sprague-Dawley rat model of PH, RhoA/ROCK activity and expression were increased.15 In these neonatal rats with PH, ROCK inhibition with fasudil or Y-27632 reduced the elevated PVR, and yet the animals were unresponsive to inhaled NO (iNO) or a systemic NO donor. ROCK inhibition failed to reverse RV dysfunction, although this was not necessarily unexpected, since reversal of RV remodeling may require more time once PVR is normalized.15
Examinations of ROCK function by Tourneux et al.96 in the ovine ductal-ligation model show that ROCK inhibitors reduce pulmonary pressures. Although we do not know whether ductal ligation alters the expression or function of ROCK signaling in smooth muscle, both expression and function are elevated in endothelial cells in this model.97 Studies conducted by Gao et al.81 in chronically hypoxic fetal lambs showed that ROCK II protein expression and overall ROCK activity were increased in chronically hypoxic arteries. However, in veins, ROCK II protein expression was reduced, as was ROCK activity.98 Our studies performed in PAs from these fetal sheep show that ROCK is also important to contraction due to membrane depolarization and that its function is maintained following antenatal hypoxia.99 In newborn hypoxic lambs, our recently published work shows that ROCK has great importance to 5-HT contraction and that ROCK inhibition will reduce pulmonary pressures, although it may not play a key role in the increased HPV response of the pulmonary vasculature in chronically hypoxic lambs.8
Mechanisms of Pulmonary Vasodilation
The pulmonary vasculature dilates when smooth muscle cells relax, which is primarily due to inhibition of vasoconstrictor pathways. The endothelium serves a major role in the vasodilatory process. Agonist stimulation of endothelial cells by acetylcholine, bradykinin, or other factors causes Ca2+ responses in the endothelial cells. The major pathways that have been examined in fetal and newborn sheep are depicted in Figure 2. In general, the increases in endothelial cytosolic Ca2+ stimulate a signaling cascade and activate several pathways that lead to the generation of NO100 and prostacyclins101,102 as well as to activation of gap junctions103-105 that couple endothelial cells to smooth muscle cells. This latter pathway is likely to contribute to endothelial-derived hyperpolarization factors that help induce pulmonary vasodilation.
Figure 2.

Pulmonary vasodilation is a synchronized process. A variety of substances, shear stress, and membrane stretch work together to increase prostacyclin, nitric oxide (NO) production, and other pathways defined broadly as endothelial-derived hyperpolarizing factors that collectively cause vascular dilation. The signaling molecules released from the endothelium and activated pathways work in conjunction with epinephrine and other neurohumoral substances to increase the activity of protein kinases A and G (PKA and PKG, respectively). As discussed in the text, these kinases phosphorylate many targets that reduce the level of vascular contraction, pathways that have not been fully examined in the fetus. A dashed green arrow indicates an activation pathway; a dashed red line with a bar indicates an inhibition pathway; a dashed blue arrow indicates movement of calcium. AC: adenylate cyclase; ACh: acetylcholine; AMP: adenosine monophosphate; BK: bradykinin; BKCa: calcium- and voltage-activated potassium channel; CaL: L-type Ca2+ channel; CaM/CAMKII: calmodulin and Ca2+/calmodulin-dependent protein kinase II; cAMP: cyclic adenosine monophosphate; cGMP: cyclic guanosine monophosphate; CO: carbon monoxide; CPI-17: C-kinase potentiated protein phosphatase-1 inhibitor; EDHF: endothelial-derived hyperpolarizing factors; Em: membrane potential; eNOS: endothelial nitric oxide synthase; Epi: epinephrine; Gq/Gs: q and s alpha subunits of G protein–coupled receptor; GMP: guanosine monophosphate; HO-1/2: heme oxygenase 1 and 2; MLC20/MLC20-P: 20-kDa myosin light chain and phosphorylated moiety; MLCK: myosin light-chain kinase; MLCP: myosin light-chain phosphatase; PDE3/4/5: phosphodiesterases 3, 4, and 5; PGI2: prostacyclin; PLA2: phospholipase A2; PP1c: protein phosphatase-1c; RhoA/ROCK: Ras homolog gene family, member A, and associated kinase; RyR: ryanodine receptor; SERCA: sarco/endoplasmic reticulum Ca2+-ATPase; sGC: soluble guanylate cyclase.
NO signaling
NO is a prominent vasodilatory signal for the pulmonary vasculature. The pathways associated with NO-dependent vasorelaxation have been intensively studied in the fetal and postnatal vasculature. Vasoactive NO is generated predominantly by endothelial NO synthase (eNOS), an enzyme expressed in vascular endothelium and stimulated by elevations in endothelial cytosolic Ca2+. Reduced eNOS activity is thought to be an important determinant for development of persistent PH in the newborn.106 In comparison, resident Tibetan populations have elevated NO production through eNOS upregulation, a response that may be part of the acclimatization process.107 Secondarily, decreased NO levels are associated with high-altitude pulmonary edema susceptibility in humans, which further implicates the importance of NO signaling and acclimatization to high altitude.108
With respect to the role of eNOS in the development of PH in sheep, alterations in its activity and protein expression differ, depending on which stressor lambs are exposed to. Shaul et al.109 illustrated that in the ductal-ligation model, the activity and expression of eNOS are reduced, as is the ability of exogenous NO to cause vasodilation.65 Xue et al.40 showed that chronic hypoxia, in comparison, does not alter fetal eNOS function and expression. Moreover, Herrera et al.18 showed that expression of eNOS mRNA and protein is increased in ∼2-week-old newborn lambs born at high altitude similarly to that in native Tibetans, even though their pulmonary arterial pressures are elevated. This illustrates that fetuses and newborns respond in distinct ways to high-altitude gestation, birth, and life and that there are key differences between the ductal-ligation and hypoxia models.
The cellular mechanisms that underlie the divergence in eNOS function due to ductal ligation and hypoxic stress are not completely understood. Still, studies show that ductal ligation modulates several pathways that regulate eNOS and impair its activity, in addition to reducing its expression. Specifically, Konduri et al.110 demonstrated that the ability of heat shock protein 90 (HSP90) to activate eNOS is suppressed through a decrease in the expression and association of HSP90 with eNOS. In addition, Hsu et al.111 showed that ductal ligation increases eNOS phosphorylation at threonine 495, which reduces its activity, without altering phosphorylation at the putative activation site, serine 1177. Another way that ductal ligation reduces eNOS function is by increasing NADPH oxidase–generated reactive oxygen species (ROSs). In particular, Brennan et al.112 illustrated that ductal ligation enhances expression of the P67phox subunit of NADPH oxidase, which is a critical activator of the oxidase.113 In conjunction with enhanced NADPH activation, superoxide dismutase activity, but not its expression, is depressed,112 an effect that reduces ROS degradation. This combination of increased ROS production and reduced degradation dramatically elevates plasma ROSs, including superoxide and hydrogen peroxide.112 Unfortunately, such systematic analyses of eNOS activity and ROSs have not been performed in hypoxic fetal or newborn lambs, and thus we do not know the extent to which there are alterations in eNOS functionality. The possible differences in eNOS function and NO generation due to ductal ligation and high-altitude–induced hypoxia in fetal or newborn sheep accentuate the importance of understanding the underlying disease pathophysiology. Such knowledge is vital to the development of relevant treatment strategies in patients with similar symptoms but vastly different etiologies.
Each subsequent step of the NO signaling pathway is important to vascular relaxation. The impact of imposed or environmental stress on pathway activity is inherently complex, as illustrated by the high-altitude lamb models. Changes in the sensitivity of isolated vessels to exogenous NO may compensate for or exacerbate changes in eNOS expression and activity. The pulmonary vasodilator responses to exogenous NO from sodium nitroprusside (SNP) administration were diminished slightly in PAs from fetal lambs exposed to chronic hypoxia40 as well as in newborn lambs that were born and remained at high altitude.6 In comparison, lambs born at high altitude that returned to low altitude had accentuated vasodilation to SNP.7 Moreover, ductal ligation attenuates SNP-dependent vasodilation in fetal lambs,114 even though iNO dilates pulmonary vessels in a dose-dependent fashion, with maximal effect at 100 ppm.109,115,116 Chronic antenatal hypoxia and ductal-ligation depression of NO-mediated vasodilation may account for the failure of iNO to lower pulmonary vascular pressures in some pediatric patients. From the impact of antenatal and perinatal hypoxia on NO-mediated vasodilation, it is clear that further experimentation in the WMRS and INCAS hypoxic-sheep models is needed. The NO signaling pathway should be examined in additional detail, including direct and indirect measurements of NO signaling, in order to elucidate changes in eNOS expression and function as well as downstream portions of the pathway. Decreased eNOS activity and protein expression in the ductal-ligation model suggest impairment of the eNOS signaling cascade. However, analysis could reveal additional epigenetic transcriptional or translational defects. While there may be simple alterations in mRNA or protein levels that explain the functional decrements, posttranslational modifications in eNOS would alter its ability to generate NO. Germane to this idea is the critical finding by Llanos et al.,117 mentioned above, that sheep born at INCAS have upregulated eNOS, which is distinct from the loss of eNOS in the ductal-ligation studies shown by Shaul et al.109
Soluble guanylate cyclase
NO synthesized and released by the endothelium causes vasodilation by stimulating soluble guanylate cyclase (sGC) in vascular myocytes. By producing cyclic guanosine monophosphate (cGMP) in the smooth muscle cells, sGC amplifies the NO signal;106 sGC is a heterodimeric enzyme consisting of α and β dimers. NO binding to the β subunit stimulates the conversion of guanosine triphosphate to the vasorelaxant cGMP. Using the ductal-ligation model, Chester et al.118 found increased sGC α1 and β1 protein expression in PASMCs. Still, basal cGMP content was lower in hypertensive PASMCs than in nonhypertensive PASMCs, and SNP caused equivalent cGMP production in control and hypertensive tissues. These data indicate that the increases in sGC do not fully compensate for the depression in eNOS expression shown by Shaul et al.109 and suggest that hypertension impairs sGC function.118 The effects of chronic hypoxia on sGC expression differ from those of ductal ligation and are dependent on the model examined. Herrera et al.18 showed that newborn sheep whose mothers lived at high altitude had reduced sGC protein expression. In comparison, newborn lambs born to low-altitude ewes that were brought to high altitude for gestation and birth had no change in their sGC levels,7 suggesting there may not be a change in sGC signaling.
An increase in cGMP regulates a variety of cellular functions through activation of a cGMP-dependent kinase (protein kinase G [PKG]).119 PKG causes vasodilation largely through suppression of pathways that sensitize the above-mentioned contractile proteins to calcium. In particular, PKG opposes ROCK-mediated inactivation of the MLCP by phosphorylating serine residues located adjacent to the ROCK phosphorylation site on the MYPT1 regulatory subunit. This eventually increases the rate of myosin light-chain dephosphorylation, thereby causing smooth muscle cell and vascular relaxation. Consequently, PKG reduces Ca2+ sensitization of myosin filaments by maintaining the activity of MLCP.81 Notably, Resnik et al.120 demonstrated that ductal ligation reduces mRNA and protein expression of PKG1-α in PASMCs. In contrast, Gao et al.98 showed that the expression and function of PKG are markedly increased in PAs from chronically hypoxic fetal lambs. Despite this increase, cGMP-mediated relaxation is suppressed, suggesting that there are posttranslational modifications to the kinase. Work from Negash et al.121 also indicates that acute hypoxia causes nitrosylation of PKG, which depresses pulmonary venous relaxation. This impairment can be prevented by scavenging either ROSs or reactive NO species.121 Although parallel studies have not been published for chronically hypoxic animals, the data suggest that posttranslational modifications such as nitrosylation are likely to underlie the differences in expression versus activity and function for PKG. Furthermore, there are no published reports of PKG expression or function in newborn lambs born at WMRS or INCAS. Together, the available reports indicate that changes in PKG expression and function are important to altered vasodilatory capacity in response to ductal ligation as well as to chronic hypoxia and therefore are worthwhile to pursue.
Carbon monoxide (CO)
CO is another gaseous signal formed in the lung that works in a way similar to that of NO. CO is produced by endothelial, smooth muscle, and inflammatory cells, and like NO, this gaseous molecule is important for vessel dilation through activation of sGC.122 Carbon monoxide is made through the activity of heme oxygenase (HO-1 and HO-2), and acute hypoxia stimulates CO production (via upregulation of HO-1) in PASMCs. In addition to activating sGC, this gaseous mediator is also thought to modulate a heme moiety that is attached to calcium- and voltage-activated potassium (BKCa) channels.123-125 CO enhances coupling between RyR-generated Ca2+ signals and BKCa channels.126,127 The increased BKCa activity would then accentuate vasodilation by hyperpolarizing the plasma membrane and suppressing L-type Ca2+ (CaL) channel activity.
CO signaling may be an important site of acclimatization in response to chronic perinatal hypoxia. This is exemplified in studies performed on adult llamas from the Andean altiplano that are well acclimated to the rarefied environment and sheep that are not as well acclimated. Llanos’ group has shown in a series of studies18,19,117 that llamas have enhanced production of CO by inducible HO-1, while sheep have reduced HO-1 expression and CO-mediated vasodilation. Although HO-generated CO is likely to be important to pulmonary vascular tone and remodeling, its importance in fetal PAs, as well as comparisons between chronic-hypoxia and ductal-ligation models, remain unresolved.
Prostacyclin
Prostacyclin PGI2 is the predominate prostacyclin released from the endothelium and along with some other products of arachidonic acid metabolism, such as prostaglandin E2, it may provide protection from chronic hypoxic stress.128,129 These endothelial-derived substances dilate vessels and attenuate PASMC proliferation. Prostacyclins are released from the vascular endothelium and activate Gs-coupled GPCRs on smooth muscle cells. Activation of the GPCRs by prostacyclin impairs smooth muscle contraction, and these receptors are the target of iloprost, an inhaled drug used for the treatment of pulmonary arterial hypertension (PAH).130-132 Prostacyclin receptor stimulation activates adenylate cyclase (AC), which increases cAMP production, which in turn cascades to cause vasodilation through activation of protein kinase A (PKA)-dependent pathways. Activation of PKA-dependent pathways likely reduces MLCK phosphorylation and potentially enhances BKCa channel activity while impairing Ca2+ signaling as well as ROCK activation.133
The importance of prostacyclin is illustrated with prostacyclin-receptor knockout mice that exhibit a significantly greater degree of PH after exposure to hypoxia.134 This finding is also relevant to the treatment of PAH in humans. In these patients, remodeled PASMCs may exhibit decreased PGI2 synthase expression.135 This evidence suggests that chronic hypoxia can reduce prostacyclin function, at least in the adult, which can affect both pulmonary vascular remodeling and reactivity.
Ductal ligation significantly impairs PGI2 activity.136 Specifically, Lakshminrusimha et al.136 showed that lung tissue from ductal-ligation lambs has reduced PGI2-induced relaxation as a result of decreased prostacyclin receptor levels. Secondarily, they also showed decreased PGI2 synthase protein levels in hypertensive lambs, which would reduce PGI2 production. Unfortunately, neither PGI2 synthesis nor receptor function has been formally studied in the hypoxic fetal- or newborn-lamb models, so we do not know whether there are common dysfunctions. The significant progress in the use of prostacyclin analogs for treatment of PAH132,137,138 and the data from rodent and piglet studies suggesting that chronic hypoxia reduces prostacyclin signaling139,140 illustrate that it is important to further our understanding of the cellular mechanisms of vasodilation due to prostacyclins and the role they play in PHN.
Adenylate cyclase
In mammals, there are 10 Class III AC isoforms that amplify GPCR signals by producing cAMP.141 In the pulmonary vasculature, receptor signaling systems tied to cAMP generation typically cause vasodilation. These include the prostacyclin and β-adrenergic signaling systems. Thus, in many ways β-adrenergic and prostacyclin-cAMP signaling mirrors NO-cGMP-mediated vasodilation.142 For example, it has long been known that birth increases β-adrenergic-dependent signaling and dilation in the pulmonary vasculature.143 Among the various AC isoforms tied to GPCR activation, adenylate cyclase 2 (AC2) is an important enzyme for the production of cAMP in PAs.136 Using the ductal-ligation model, Lakshminrusimha et al.136 showed that AC activity and AC2 protein levels in lung tissue are maintained in hypertensive lambs. Similarly, steady-state cAMP levels were unchanged by ductal ligation. Because postreceptor coupling mechanisms that generate cAMP are intact following ductal ligation, the pathway is therapeutically relevant for inducing vasorelaxation and reducing pulmonary arterial pressures. However, besides this one study, our knowledge regarding the impact of perinatal hypoxia, as well as ductal ligation, on AC function is limited.
Phosphodiesterases (PDEs)
These are enzymes that act on vascular smooth muscle to break down cAMP and cGMP and thereby reduce vasodilation.144 There are 11 different PDE families, with PDE3, PDE5, and PDE4 being the predominate isoforms in vascular smooth muscle. In the lung, PDE5 catalyzes the breakdown of cGMP to GMP, while PDE3 and PDE4 primarily catalyze the hydrolysis of cAMP to AMP.142 Farrow et al.142 recently showed that the expression of PDE5 increases a week after birth in newborn lambs, following an initial decline observed at parturition. The depression in PDE5 expression during the transition period augments the vasodilatory activity of NO-dependent signaling pathways, potentiating vasodilatory responses. Ductal ligation was shown by Hanson et al.145 to increase PDE5 activity in hypertensive animals without enhancing levels of PDE5 mRNA expression or protein. While the total PDE5 protein content was similar in hypertensive and control lambs, phosphorylated PDE5 content was significantly higher in hypertensive animals.145 This suggests that ductal ligation causes undue activation of PDE5, restricting vasodilation due to cGMP. Herrera et al.17 showed that newborn lambs born at high altitude also did not have altered levels of PDE5 mRNA expression. However a later study by the same group of investigators similarly showed that PDE5 protein levels were significantly increased in lung tissue of hypoxic newborn lambs that were returned to sea level after birth.7 Moreover, sildenafil, a PDE5 inhibitor, suppressed the increase in PA pressure and reduced PVR in hypoxia-induced hypertensive lambs.17 Thus, hypoxic newborn lambs have a way of enhancing PDE5 function different from that of lambs with PH due to ductal ligation.17
The effects of PDE3 inhibition on vascular tone are similar to those of PDE5 inhibitors, although PDE3 preferentially degrades cAMP. Rashid et al.146 showed that inhibition of PDE3 by milrinone reduced PVR in the ductatl-ligation model. PDE3 expression and activity increase dramatically following birth, thus facilitating prostanoid and β-adrenergic-dependent vasodilation during the transition at birth.147 Moreover, this change in expression and activity follows the same trend as for modifications in PDE5 activity. However, we do not know how ductal ligation or chronic hypoxia affect PDE3 function.
Ionic regulation of pulmonary vascular tone
Regulation of ion channel activities in pulmonary arterial myocytes is a feature central to GPCR stimulation and the cellular responses to changes in oxygenation. The activities of these ion channels control membrane potential and cytosolic Ca2+ responses, effects that are critical to rapid changes in vasoreactivity as well as to longer-term changes in cell function. Coordinated changes in the membrane potential are tied to the action of Ca2+-permeable ion channels on the sarcoplasmic reticulum as well as in the plasma membrane. Such regulation causes membrane potential to be intimate to Ca2+ signals. From the standpoint of understanding fetal and perinatal physiology, it is unfortunate that a greater part of our knowledge regarding how ion channels are regulated is based on data generated in cells and tissues from adults. Thus, our understanding of ion channels in the perinatal period remains far from complete. Still, we do know that a variety of different channels are important to vascular reactivity in the fetus and newborn and that chronic hypoxia and ductal ligation affect the function of at least some of them.
Regulation of the membrane potential
Tissue hypoxia and receptor activation cause membrane depolarization and are linked to myocyte contraction. There are approximately 70 K+ channel family members,148 with voltage-gated K+ (Kv) channels and other isoforms being intensively studied for their potential involvement in the HPV response.69 Inhibition of Kv is an important component of the membrane depolarization process.149,150 These Kv channels help maintain a negative resting membrane potential and are important to the vasoconstrictive response with acute hypoxia.69 Suppressing Kv channel activity depolarizes the membrane, which elicits Ca2+ signals that cause myocyte contraction. Recent evidence from Konduri et al.151 indicates that Kv channels that are inhibited by 4-aminopyridine are reduced in the ductal-ligation model, although we do not know what impact chronic hypoxia has on these or other Kv channels in immature arterial myocytes from sheep. However, studies performed in newborn piglets may provide some insight. Extreme chronic hypoxia (ambient Po2 of 60–72 torr, ∼6,200–7,500-m equivalent altitude) caused membrane depolarization and a selective decrease in abundance of Kv1.2, but not of Kv1.5 or Kv2.1.152 Furthermore, the generalized importance of Kv channels to pulmonary arterial myocyte proliferation153 suggests that changes in Kv channel activity and expression will promote pulmonary vascular remodeling.152
Whereas membrane depolarization is important for contraction of arteries, maintaining a hyperpolarized membrane potential is important to vessel relaxation. Activation of potassium-permeable channels causes the membrane potential to hyperpolarize. However, among the numerous K+ channels that are expressed in pulmonary arterial myocytes,154 the best understood in the fetal vasculature are BKCa channels.150,155,156 Studies performed in cerebral arterial myocytes show that BKCa channels are activated by Ca2+ sparks as a result of focal activation of RyRs on the sarcoplasmic reticulum.157,158 Activation of these BKCa channels hyperpolarizes the membrane potential and causes myocyte relaxation through inhibition of voltage-dependent Ca2+ entry. One critical study by Dr. Cornfield’s group159 showed that oxygen-dependent activation of BKCa is important to vasodilation with birth. Such membrane hyperpolarization would prevent activation of voltage-dependent Ca2+ channels, reducing extracellular Ca2+ entry and thereby preventing Ca2+-dependent contraction. From a disease perspective, compression of the ductus arteriosus reduces BKCa channel expression and impairs BKCa channel activity in fetal PASMCs, effects that reduce arterial relaxation.156,160 This contrasts with newborn sheep born on the Andean altiplano at 3,600 m that have elevated BKCa expression,7 which in turn promotes vasodilator capacity. At present, we do not understand the cellular mechanisms responsible for these differences; however, one potential explanation is that BKCa expression is regulated by HIF-1, a transcriptional regulator that is depressed by ductal ligation.155
InsP3 receptor Ca2+ release pathways
InsP3 generation due to GPCR activation causes robust Ca2+ signals in PASMCs of many species, including sheep.87,161-163 While much is known regarding the activities of InsP3 signaling in myocytes from adult animals, InsP3-dependent Ca2+ responses in fetal and newborn pulmonary arterial myocytes and the impact of pulmonary vascular disease on signaling in cells from these animals are poorly understood. Our studies and those of other laboratories show that fetal-sheep pulmonary vascular myocytes are activated by various GPCR agonists. PAF causes InsP3 generation and Ca2+ responses in fetal-sheep PASMCs.87,164 We have published that ATP, phenylephrine, and 5-HT all cause Ca2+ responses in pulmonary arterial myocytes from fetal sheep.89,162 Notably, our studies indicate that antenatal hypoxia reduces the number of fetal pulmonary arterial myocytes with Ca2+ responses before and in response to 5-HT stimulation.89 On the basis of studies performed in myocytes from adult animals, we anticipate that many other agonists that stimulate GPCR, such as ET-1 and PGF2α, will elicit similar responses.165-167 Unfortunately, these influences cannot be compared to those of ductal ligation, because there are no reports regarding the influence of ductal ligation on InsP3 activity and because we do not, as yet, know whether the reduction in Ca2+ signals is specific to 5-HT or a general effect on InsP3 stimulation.
RyR Ca2+ release pathways
RyRs are important for generating highly localized Ca2+ “spark” events, and there is multifaceted regulation of their activity. In the fetus, oxygenation activates RyRs, and these localized Ca2+ events regulate vessel dilation during the fetal transition period.155,168,169 However, RyR activity is also associated with whole-cell Ca2+ responses and contraction in the setting of acute hypoxia in adults.38,170,171 Further illustrating the complexity in RyR function in pulmonary arterial myocytes is that GPCR activation with ET-1 also activates Ca2+ sparks.172 Our recent studies show that chronic prenatal hypoxia impairs Ca2+ spark activity in both the fetus and the newborn.173 These data lead to an intriguing possibility, that impaired RyR function contributes to the development of PH in lambs born at high altitude. Unfortunately, the impact of ductal ligation on RyR activity remains unknown, so we do not know whether impinging on RyR function is a common mechanism that contributes to the development of PH.
Plasmalemmal Ca2+ entry pathways
Immature pulmonary arterial myocytes have several Ca2+ entry pathways.89,99,120,162,174 L-type Ca2+ channels are the best understood and best characterized Ca2+-permeable ion channels. These channels are activated by membrane depolarization following GPCR stimulation89,175,176 or acute hypoxia.170 Our studies show that CaL channels account for about one-quarter of 5-HT89 and for potassium-dependent membrane depolarization–induced contraction in fetal sheep PAs.99 CaL channel function can be changed by chronic hypoxia. Newborn piglets exposed to severe hypoxia (Fio2 of 0.10, ∼5,800 m equivalent altitude) developed PH and had elevated CaL channel–dependent reactivity and vascular tone, compared to normoxic piglets.177 In these hypoxic animals, the decrease in transpulmonary perfusion pressure in response to nifedipine was greater than that in normoxic animals. This was also associated with an increased nifedipine-sensitive Ca2+ current density in patch voltage-clamped PASMCs of hypoxic animals, compared to controls.177 However, such enhancement in CaL expression may not occur in all species. Our studies in PAs from fetal sheep exposed to antenatal hypoxia (3,801 m) show that the role of CaL channels in potassium or 5-HT-mediated contraction is maintained, suggesting that channel expression is preserved.89,99 This preservation in function compares with ductal ligation, where Resnik et al.120 showed that there was reduced protein, but not mRNA, expression of Cav 1.2, which is the isoform that encodes for CaL currents. Nevertheless, Storme et al.178 showed that inhibition of CaL with nifedipine can reduce pulmonary pressures in ductal-ligated lambs. Still, we did not find any studies that systematically examined changes in CaL function due to ductal ligation.
Studies show that in addition to CaL channels, nonselective cation channels contribute to extracellular Ca2+ entry and are also important to fetal pulmonary arterial Ca2+ signaling and arterial reactivity.89,162,174 In particular, nonselective cation channel inhibition with SKF96365 blocked a substantial portion of the nifedipine-insensitive contraction due to 5-HT in our fetal-, as well as newborn-, sheep PAs.8,89 Data from Dr. Cornfield’s group174 shows that a number of canonical transient receptor potential channels (TRPCs)148—TRPC1, TRPC3, TRPC5, and TRPC6—are expressed in fetal pulmonary arterial myocytes. This is important because TRPCs contribute significantly to nonselective cation channel activity, which is one of the larger ion channel families, with 28 members.179 When taken together with studies regarding the role of CaL channels, the data illustrate that GPCR and hypoxia coordinate the activities of voltage-dependent and nonselective cation channels to regulate Ca2+ signaling and arterial contraction. Ductal ligation certainly impairs this coordination, because it enhances expression of TRPC6 protein while reducing the mRNA expression for TRPC1, TRPC3, and TRPC5 as well as TRPC6.174 Although the aforementioned studies illustrate that TRPC channels are important regulators of arterial contraction, more-detailed imaging and electrophysiological studies are lacking in the fetus. Moreover, we know very little regarding the effect of ductal ligation and antenatal hypoxia on the function of the aforementioned or other TRPC channels in the fetal pulmonary vasculature. Such studies are necessary to resolve the mechanistic changes that occur during fetal development, their importance during the transition period at birth, and their role in the development of PH due to antenatal hypoxia or ductal ligation.
The preceding paragraphs illustrate that fetal and newborn pulmonary vascular myocytes are dependent on highly organized ionic control systems, and yet very little is known about how antenatal hypoxia or ductal ligation influences their function. In particular, efforts have focused on just a few of the many potential players and pathways that are important to pulmonary vascular function. Systematic exploration of the various ionic signaling pathways is critical to our understanding of arterial function in the perinatal period and of the impact of disease.
Current treatment strategies
The primary goal in treating newborns with PH is to increase blood oxygenation. Multiple recent reviews outline these approaches in detail.180-183 Ductal ligation will result in a more profound increase in pulmonary pressure and hypertensive crisis emulation, requiring immediate reduction in pressures to improve survival. Antenatal hypoxia, on the other hand, causes more modest elevations in pulmonary pressure and long-lived pulmonary vascular dysfunctions that require chronic treatment to allow for adequate patient recovery and long-term health.
The therapeutic strategies that are currently used aim to simulate what should occur naturally during postnatal transition. The overarching thrust of this review is that understanding the etiology of the disease will refine the treatments for PH in the newborn. Similar strategies have led to improvements in the treatment of PH in the adult, where there have been major advances in pharmacotherapy. Unfortunately for pediatric patients, almost all of the data regarding the etiology of disease are based on adult populations, with very few parallel studies for pediatric populations. This has led clinicians to treat children on the basis of extrapolation of therapies designed for adults and expert consensus.184
Often, hypoxemia can precipitate pulmonary hypertensive crisis by causing vasoconstriction and increasing PVR, as occurs, for example, when infants are born at high altitude. Neonates with mild respiratory distress may therefore require only minimal oxygen supplementation in order to resolve the crisis by eliminating HPV. The primary rationale is that if the oxygen that is required for vasodilation is provided at birth, the arteries will dilate and PVR will fall. Although the risk of oxygen toxicity is often outweighed by the benefits of modest O2 supplementation, it should still be considered when prolonged therapy is required to maintain adequate oxygenation. Unfortunately, this type of oxygen therapy may be beneficial only to select infants with very mild disease and is of limited value in more serious cases of PH, often forcing clinicians to resort to more efficient, albeit invasive, methods of oxygenation, such as high-frequency oscillatory ventilation or extracorporeal membrane oxygenation (ECMO).180,185,186
If oxygen supplementation does not adequately reverse the process, a number of therapeutic options are now available, primarily acting as vasodilators. These include iNO, PDE5 inhibitors (PDE5is), prostacyclin analogs (PG), endothelin receptor antagonists (ERAs), and PDE3 inhibitors (PDE3is). Although these treatments mainly target the immediate issue of high PVR, some of these (e.g., bosentan and other ERAs) may also suppress vascular remodeling,187,188 which is a major component of long-term health problems.
In recent years, NO inhalation has become a widely used treatment for PPHN. This therapy improves hemodynamic and oxygenation profiles in multiple pediatric PH populations,189,190 lowers pulmonary arterial pressure, improves infant survival, and reduces the need for ECMO.191,192 However, as many as 50% of the infants do not respond to iNO, possibly because of either a dysfunction in sGC and PKG signaling or, in some cases, severe pulmonary hypoplasia with a markedly diminished pulmonary vascular bed and blood flow despite maximal vasodilation.193,194 Moreover, the high cost and technical challenges limit iNO use to mostly large, urban medical centers in developed countries. Inhibition of PDE5 with sildenafil works through the same signaling systems as iNO and has fewer practical concerns but is limited because it can also cause systemic hypotension. In addition, it may induce nonselective pulmonary vascular relaxation, increasing ventilation perfusion mismatch in the lung and worsening oxygenation. Although this could be important in newborn infants that have low systemic arterial pressures, there is now evidence in neonates with severe PPHN that PDE5is can improve oxygenation index and survival.195,196
Prostacyclin analogs, unlike the previous groups of drugs, stimulate PKA-dependent signaling, with PDE3is having similar effects by elevating cAMP-associated PKA activity. The effectiveness of these medications is based on similarities between PKA and PKG signaling, although this overlap is also responsible for their shortcomings, which is mainly a similar side-effect profile. Unlike adult PAH populations, there are fewer data on the use of prostanoids in children, with at least two trials showing improved survival in children with idiopathic PAH197 and improved hemodynamics in children with PH due to cardiac defects.198 Of note, prostanoid therapy must be administered continuously because of its short systemic half-life, resulting in increased risks associated with therapy, such as indwelling central venous catheter–related infections and thrombosis. Alternative routes of administration include continuous subcutaneous injection, with site pain and infection as common side effects, and inhaled mist treatments, which can cause bronchospasm and other upper-airway symptoms. Finally, patients may become desensitized to prostanoid therapy, especially in the setting of continuous intravenous infusions, resulting in escalating doses and a significant rebound effect upon discontinuation.
ERAs act on pathways that are unique to many of the drugs mentioned above. ET-1 binds to two distinct GPCRs (ETA and ETB) that are predominantly expressed on pulmonary vascular smooth muscle, resulting in activation of ROCK and inositol triphosphate pathways that cause vasoconstriction. Although there are some data for functional improvement and exercise capacity in children with either idiopathic or associated PAH,199,200 as well as a recent trial indicating that ERAs can be an option in newborns with PHN,78 they are still not widely used in this patient population.
As a result of the understanding that multiple pathways and various receptors are part of PH pathophysiology in the newborn, combination drug therapies are becoming more prominent. This also diminishes problems encountered with single-drug treatments, such as need for higher drug doses and increased side effects. These combination therapies, with 2 or 3 pharmacologic agents from different drug classes, have shown modest improvement in hemodynamics and/or exercise capacity in adults with PAH201-203 and are now in clinical trials on neonatal and pediatric patients.
Perspective
This review has illustrated that both ductal ligation and chronic perinatal hypoxia can cause PH in sheep, with similar vascular and cardiopulmonary malformations. The underlying causes of disease in each model are distinctive in this one species, which fuels the premise that the type of stress inducing the disease is an important consideration when treatments are being prescribed. On the one hand, there are similarities between the ligation and hypoxia models, such as functional decrements in sGC function, cGMP-dependent vasorelaxation, increased PDE5, and enhanced ET-1 contraction. On the other hand, there are marked differences between the cellular processes affected by antenatal and perinatal hypoxia and those affected by ductal ligation. In particular, studies illustrate that ductal ligation reduces eNOS as well as BKCa channels, while their expression is enhanced in chronically hypoxic newborns. More critically, however, there are many other cellular signals that still must be examined in the ductal-ligation and chronic-hypoxia models. This is illustrated by the lack of comparative data on a number of key pathways in the two models and by our lack of knowledge regarding the impact of altered Ca2+ signaling as well as transcriptional regulation due to ductal ligation or hypoxia-induced PH. Delineating the commonalities and differences in the etiology of disease due to high-altitude living, elevated pulmonary pressure or flow, diaphragmatic hernia, and other cardiopulmonary malformations is vital for our ability to properly care for afflicted infants. We already know that PHN is due to a spectrum of changes in the structure and function of the lung and cardiopulmonary circuit. Hence, treating each individual disease will require the development of refined therapies, therapies that rely on a better understanding of the fundamental basis of each disease in relevant animal models.
Source of Support: A portion of this work was performed in the Loma Linda University School of Medicine Advanced Imaging and Microscopy Core, which is supported by the National Science Foundation under Major Research Instrumentation, Division of Biological Infrastructure grant 0923559 (SMW) and the Loma Linda University School of Medicine. The work was also supported in part by US Public Health Service grants HD069746 (SMW), HL095973 (ABB), and HD/HL-03807 and HD-31226 (LD Longo).
Conflict of Interest: None declared.
References
- 1.Abman SH, Accurso FJ. Acute effects of partial compression of ductus arteriosus on fetal pulmonary circulation. Am J Physiol Heart Circ Physiol 1989;257:H626–H634. [DOI] [PubMed]
- 2.Abman SH, Shanley PF, Accurso FJ. Failure of postnatal adaptation of the pulmonary circulation after chronic intrauterine pulmonary hypertension in fetal lambs. J Clin Invest 1989;83:1849–1858. [DOI] [PMC free article] [PubMed]
- 3.Morin FC 3rd. Ligating the ductus arteriosus before birth causes persistent pulmonary hypertension in the newborn lamb. Pediatr Res 1989;25:245–250. [DOI] [PubMed]
- 4.Aggarwal S, Gross CM, Kumar S, Datar S, Oishi P, Kalkan G, Schreiber C, Fratz S, Fineman JR, Black SM. Attenuated vasodilatation in lambs with endogenous and exogenous activation of cGMP signaling: role of protein kinase G nitration. J Cell Physiol 2011;226:3104–3113. [DOI] [PMC free article] [PubMed]
- 5.Reddy VM, Meyrick B, Wong J, Khoor A, Liddicoat JR, Hanley FL, Fineman JR. In utero placement of aortopulmonary shunts: a model of postnatal pulmonary hypertension with increased pulmonary blood flow in lambs. Circulation 1995;92:606–613. [DOI] [PubMed]
- 6.Herrera EA, Pulgar VM, Riquelme RA, Sanhueza EM, Reyes RV, Ebensperger G, Parer JT, et al. High-altitude chronic hypoxia during gestation and after birth modifies cardiovascular responses in newborn sheep. Am J Physiol Regul Integr Comp Physiol 2007;292:R2234–R2240. [DOI] [PubMed]
- 7.Herrera EA, Riquelme RA, Ebensperger G, Reyes RV, Ulloa CE, Cabello G, Krause BJ, Parer JT, Giussani DA, Llanos AJ. Long-term exposure to high-altitude chronic hypoxia during gestation induces neonatal pulmonary hypertension at sea level. Am J Physiol Regul Integr Comp Physiol 2010;299:R1676–R1684. [DOI] [PMC free article] [PubMed]
- 8.Blood AB, Terry MH, Merritt TA, Papamatheakis DG, Blood Q, Ross JM, Power GG, Longo LD, Wilson SM. Effect of chronic perinatal hypoxia on the role of rho-kinase in pulmonary artery contraction in newborn lambs. Am J Physiol Regul Integr Comp Physiol 2013;304:R136–R146. [DOI] [PMC free article] [PubMed]
- 9.Fike CD, Kaplowitz MR. Nifedipine inhibits pulmonary hypertension but does not prevent decreased lung eNOS in hypoxic newborn pigs. Am J Physiol Lung Cell Mol Physiol 1999;277:L449–L456. [DOI] [PubMed]
- 10.Fike CD, Kaplowitz MR, Thomas CJ, Nelin LD. Chronic hypoxia decreases nitric oxide production and endothelial nitric oxide synthase in newborn pig lungs. Am J Physiol Lung Cell Mol Physiol 1998;274:L517–L526. [DOI] [PubMed]
- 11.Rhodes J. Comparative physiology of hypoxic pulmonary hypertension: historical clues from brisket disease. J Appl Physiol 2005;98:1092–1100. [DOI] [PubMed]
- 12.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 2003;111:1519–1527. [DOI] [PMC free article] [PubMed]
- 13.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest 1999;103:691–696. [DOI] [PMC free article] [PubMed]
- 14.Belik J, Jankov RP, Pan J, Tanswell AK. Peroxynitrite inhibits relaxation and induces pulmonary artery muscle contraction in the newborn rat. Free Radic Biol Med 2004;37:1384–1392. [DOI] [PubMed]
- 15.McNamara PJ, Murthy P, Kantores C, Teixeira L, Engelberts D, van Vliet T, Kavanagh BP, Jankov RP. Acute vasodilator effects of Rho-kinase inhibitors in neonatal rats with pulmonary hypertension unresponsive to nitric oxide. Am J Physiol Lung Cell Mol Physiol 2008;294:L205–L213. [DOI] [PubMed]
- 16.Ziino AJ, Ivanovska J, Belcastro R, Kantores C, Xu EZ, Lau M, McNamara PJ, Tanswell AK, Jankov RP. Effects of Rho-kinase inhibition on pulmonary hypertension, lung growth, and structure in neonatal rats chronically exposed to hypoxia. Pediatr Res 2010;67:177–182. [DOI] [PubMed]
- 17.Herrera EA, Ebensperger G, Krause BJ, Riquelme RA, Reyes RV, Capetillo M, Gonzalez S, Parer JT, Llanos AJ. Sildenafil reverses hypoxic pulmonary hypertension in highland and lowland newborn sheep. Pediatr Res 2008;63:169–175. [DOI] [PubMed]
- 18.Herrera EA, Reyes RV, Giussani DA, Riquelme RA, Sanhueza EM, Ebensperger G, Casanello P, et al. Carbon monoxide: a novel pulmonary artery vasodilator in neonatal llamas of the Andean altiplano. Cardiovasc Res 2008;77:197–201. [DOI] [PubMed]
- 19.Llanos AJ, Ebensperger G, Herrera EA, Reyes RV, Pulgar VM, Seron-Ferre M, Diaz M, et al. Fetal and postnatal pulmonary circulation in the Alto Andino. Placenta 2011;32(suppl 2):S100–S103. [DOI] [PubMed]
- 20.Levin DL, Mills LJ, Weinberg AG. Hemodynamic, pulmonary vascular, and myocardial abnormalities secondary to pharmacologic constriction of the fetal ductus arteriosus: a possible mechanism for persistent pulmonary hypertension and transient tricuspid insufficiency in the newborn infant. Circulation 1979;60:360–364. [DOI] [PubMed]
- 21.Morin FC 3rd, Egan EA. The effect of closing the ductus arteriosus on the pulmonary circulation of the fetal sheep. J Dev Physiol 1989;11:283–287. [PubMed]
- 22.Belik J, Halayko AJ, Rao K, Stephens NL. Fetal ductus arteriosus ligation: pulmonary vascular smooth muscle biochemical and mechanical changes. Circ Res 1993;72:588–596. [DOI] [PubMed]
- 23.Grover TR, Parker TA, Balasubramaniam V, Markham NE, Abman SH. Pulmonary hypertension impairs alveolarization and reduces lung growth in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 2005;288:L648–L654. [DOI] [PubMed]
- 24.Gien J, Seedorf GJ, Balasubramaniam V, Markham N, Abman SH. Intrauterine pulmonary hypertension impairs angiogenesis in vitro: role of vascular endothelial growth factor nitric oxide signaling. Am J Respir Crit Care Med 2007;176:1146–1153. [DOI] [PMC free article] [PubMed]
- 25.Jaillard S, Elbaz F, Bresson-Just S, Riou Y, Houfflin-Debarge V, Rakza T, Larrue B, Storme L. Pulmonary vasodilator effects of norepinephrine during the development of chronic pulmonary hypertension in neonatal lambs. Br J Anaesth 2004;93:818–824. [DOI] [PubMed]
- 26.Gilbert RD. Fetal myocardial responses to long-term hypoxemia. Comp Biochem Physiol A 1998;119:669–674. [DOI] [PubMed]
- 27.Huisman TH. The structure and function of normal and abnormal haemoglobins. Baillieres Clin Haematol 1993;6:1–30. [DOI] [PubMed]
- 28.Salhany JM, Mizukami H, Eliot RS. The deoxygenation kinetic properties of human fetal hemoglobin: effect of 2,3-diphosphoglycerate. Biochem Biophys Res Commun 1971;45:1350–1356. [DOI] [PubMed]
- 29.Rudolph AM, Heymann MA. The circulation of the fetus in utero: methods for studying distribution of blood flow, cardiac output and organ blood flow. Circ Res 1967;21:163–184. [DOI] [PubMed]
- 30.Koos BJ, Power GG. Predict fetal brain Po2 during hypoxaemia and anemia in sheep. J Dev Physiol 1987;9:517–526. [PubMed]
- 31.Durmowicz AG, Hofmeister S, Kadyraliev TK, Aldashev AA, Stenmark KR. Functional and structural adaptation of the yak pulmonary circulation to residence at high altitude. J Appl Physiol 1993;74:2276–2285. [DOI] [PubMed]
- 32.Niermeyer S, Yang P, Shanmina, Drolkar, Zhuang J, Moore LG. Arterial oxygen saturation in Tibetan and Han infants born in Lhasa, Tibet. N Engl J Med 1995;333:1248–1252. [DOI] [PubMed]
- 33.Niermeyer S. Going to high altitude with a newborn infant. High Alt Med Biol 2007;8:117–123. [DOI] [PubMed]
- 34.Niermeyer S. Cardiopulmonary transition in the high altitude infant. High Alt Med Biol 2003;4:225–239. [DOI] [PubMed]
- 35.Niermeyer S, Andrade Mollinedo P, Huicho L. Child health and living at high altitude. Arch Dis Child 2009;94:806–811. [DOI] [PubMed]
- 36.Wu T, Kayser B. High altitude adaptation in Tibetans. High Alt Med Biol 2006;7:193–208. [DOI] [PubMed]
- 37.Moore LG, Niermeyer S, Zamudio S. Human adaptation to high altitude: regional and life-cycle perspectives. Am J Phys Anthropol 1998;107(suppl 27):25–64. [DOI] [PubMed]
- 38.Bixby CE, Ibe BO, Abdallah MF, Zhou W, Hislop AA, Longo LD, Raj JU. Role of platelet-activating factor in pulmonary vascular remodeling associated with chronic high altitude hypoxia in ovine fetal lambs. Am J Physiol Lung Cell Mol Physiol 2007;293:L1475–L1482. [DOI] [PubMed]
- 39.Sheng L, Zhou W, Hislop AA, Ibe BO, Longo LD, Raj JU. Role of epidermal growth factor receptor in ovine fetal pulmonary vascular remodeling following exposure to high altitude long-term hypoxia. High Alt Med Biol 2009;10:365–372. [DOI] [PMC free article] [PubMed]
- 40.Xue Q, Ducsay CA, Longo LD, Zhang L. Effect of long-term high-altitude hypoxia on fetal pulmonary vascular contractility. J Appl Physiol 2008;104:1786–1792. [DOI] [PubMed]
- 41.Hsia CCW, Hyde DM, Ochs M, Weibel ER. How to measure lung structure—what for? on the “Standards for the Quantitative Assessment of Lung Structure.” Respir Physiol Neurobiol 2010;171:72–74. [DOI] [PubMed]
- 42.Hsia CC, Hyde DM, Ochs M, Weibel ER. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med 2010;181:394–418. [DOI] [PMC free article] [PubMed]
- 43.Shimoda LA, Semenza GL. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med 2011;183:152–156. [DOI] [PMC free article] [PubMed]
- 44.Del Moral PM, Sala FG, Tefft D, Shi W, Keshet E, Bellusci S, Warburton D. VEGF-A signaling through Flk-1 is a critical facilitator of early embryonic lung epithelial to endothelial crosstalk and branching morphogenesis. Dev Biol 2006;290:177–188. [DOI] [PubMed]
- 45.Izikki M, Guignabert C, Fadel E, Humbert M, Tu L, Zadigue P, Dartevelle P, et al. Endothelial-derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest 2009;119:512–523. [DOI] [PMC free article] [PubMed]
- 46.Yin Y, Wang F, Ornitz DM. Mesothelial- and epithelial-derived FGF9 have distinct functions in the regulation of lung development. Development 2011;138:3169–3177. [DOI] [PMC free article] [PubMed]
- 47.Yin Y, White AC, Huh SH, Hilton MJ, Kanazawa H, Long F, Ornitz DM. An FGF-WNT gene regulatory network controls lung mesenchyme development. Dev Biol 2008;319:426–436. [DOI] [PMC free article] [PubMed]
- 48.Mark M, Ghyselinck NB, Chambon P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol 2006;46:451–480. [DOI] [PubMed]
- 49.West J, Tada Y, Fagan KA, Steudel W, Fouty BW, Harral JW, Miller M, Ozimek J, Tuder RM, Rodman DM. Suppression of type II bone morphogenic protein receptor in vascular smooth muscle induces pulmonary arterial hypertension in transgenic mice. Chest 2005;128(6_suppl):553S. [DOI] [PubMed]
- 50.Jankov RP, Tanswell AK. Growth factors, postnatal lung growth and bronchopulmonary dysplasia. Paediatr Respir Rev 2004;5(suppl 1):S265–S275. [DOI] [PubMed]
- 51.Warburton D, Bellusci S, De Langhe S, Del Moral PM, Fleury V, Mailleux A, Tefft D, Unbekandt M, Wang K, Shi W. Molecular mechanisms of early lung specification and branching morphogenesis. Pediatr Res 2005;57:26R–37R. [DOI] [PubMed]
- 52.Vicencio AG, Eickelberg O, Stankewich MC, Kashgarian M, Haddad GG. Regulation of TGF-β ligand and receptor expression in neonatal rat lungs exposed to chronic hypoxia. J Appl Physiol 2002;93:1123–1130. [DOI] [PubMed]
- 53.Jelkmann W, Hellwig-Burgel T. Biology of erythropoietin. Adv Exp Med Biol 2001;502:169–187. [DOI] [PubMed]
- 54.Groenman F, Rutter M, Caniggia I, Tibboel D, Post M. Hypoxia-inducible factors in the first trimester human lung. J Histochem Cytochem 2007;55:355–363. [DOI] [PubMed]
- 55.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol 1999;15:551–578. [DOI] [PubMed]
- 56.Grover TR, Asikainen TM, Kinsella JP, Abman SH, White CW. Hypoxia-inducible factors HIF-1α and HIF-2α are decreased in an experimental model of severe respiratory distress syndrome in preterm lambs. Am J Physiol Lung Cell Mol Physiol 2007;292:L1345–L1351. [DOI] [PubMed]
- 57.Pena JP, Tomimatsu T, Hatran DP, McGill LL, Longo LD. Cerebral blood flow and oxygenation in ovine fetus: responses to superimposed hypoxia at both low and high altitude. J Physiol 2007;578:359–370. [DOI] [PMC free article] [PubMed]
- 58.Rajasekaran M, Mehta N, Baquir A, Kuntz S. Rho-kinase inhibition suppresses potassium chloride-induced bladder hyperactivity in a rat model. Urology 2007;69:791–794. [DOI] [PubMed]
- 59.Bhatt AJ, Amin SB, Chess PR, Watkins RH, Maniscalco WM. Expression of vascular endothelial growth factor and Flk-1 in developing and glucocorticoid-treated mouse lung. Pediatr Res 2000;47:606–613. [DOI] [PubMed]
- 60.Marti HH, Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci USA 1998;95:15809–15814. [DOI] [PMC free article] [PubMed]
- 61.Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol 2002;283:L555–L562. [DOI] [PubMed]
- 62.Ad Hoc Statement Committee, American Thoracic Society. Mechanisms and limits of induced postnatal lung growth. Am J Respir Crit Care Med 2004;170:319–343. [DOI] [PubMed]
- 63.Lassus P, Turanlahti M, Heikkilä P, Andersson LC, Nupponen I, Sarnesto A, Andersson S. Pulmonary vascular endothelial growth factor and Flt-1 in fetuses, in acute and chronic lung disease, and in persistent pulmonary hypertension of the newborn. Am J Respir Crit Care Med 2001;164:1981–1987. [DOI] [PubMed]
- 64.Grover TR, Parker TA, Zenge JP, Markham NE, Kinsella JP, Abman SH. Intrauterine hypertension decreases lung VEGF expression and VEGF inhibition causes pulmonary hypertension in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 2003;284:L508–L517. [DOI] [PubMed]
- 65.Deruelle P, Grover TR, Abman SH. Pulmonary vascular effects of nitric oxide-cGMP augmentation in a model of chronic pulmonary hypertension in fetal and neonatal sheep. Am J Physiol Lung Cell Mol Physiol 2005;289:L798–L806. [DOI] [PubMed]
- 66.Aparicio Otero O, Romero Gutiérrez F, Harris P, Anand I. Echocardiography shows persistent thickness of the wall of the right ventricle in infants at high altitude. Cardioscience 1991;2:63–69. [PubMed]
- 67.Gamboa R, Marticorena E. Pulmonary arterial pressure in newborn infants in high altitude. Arch Inst Biol Andina 1971;4:55–66 [in Spanish]. [PubMed]
- 68.Miao CY, Zuberbuhler JS, Zuberbuhler JR. Prevalence of congenital cardiac anomalies at high altitude. J Am Coll Cardiol 1988;12:224–228. [DOI] [PubMed]
- 69.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev 2012;92:367–520. [DOI] [PMC free article] [PubMed]
- 70.Ward JP, McMurtry IF. Mechanisms of hypoxic pulmonary vasoconstriction and their roles in pulmonary hypertension: new findings for an old problem. Curr Opin Pharmacol 2009;9:287–296. [DOI] [PMC free article] [PubMed]
- 71.Yuan JXJ. Hypoxic pulmonary vasoconstriction: cellular and molecular mechanisms. Boston: Kluwer, 2004.
- 72.Trow TK, Taichman DB. Endothelin receptor blockade in the management of pulmonary arterial hypertension: selective and dual antagonism. Respir Med 2009;103:951–962. [DOI] [PubMed]
- 73.Allen SW, Chatfield BA, Koppenhafer SA, Schaffer MS, Wolfe RR, Abman SH. Circulating immunoreactive endothelin-1 in children with pulmonary hypertension: association with acute hypoxic pulmonary vasoreactivity. Am Rev Respir Dis 1993;148:519–522. [DOI] [PubMed]
- 74.Aguirre JI, Morrell NW, Long L, Clift P, Upton PD, Polak JM, Wilkins MR. Vascular remodeling and ET-1 expression in rat strains with different responses to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 2000;278:L981–L987. [DOI] [PubMed]
- 75.Shao D, Park JE, Wort SJ. The role of endothelin-1 in the pathogenesis of pulmonary arterial hypertension. Pharmacol Res 2011;63:504–511. [DOI] [PubMed]
- 76.Rubin LJ, Badesch DB, Barst RJ, Galiè N, Black CM, Keogh A, Pulido T, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896–903. [DOI] [PubMed]
- 77.Channick RN, Sitbon O, Barst RJ, Manes A, Rubin LJ. Endothelin receptor antagonists in pulmonary arterial hypertension. J Am Coll Cardiol 2004;43:62S–67S. [DOI] [PubMed]
- 78.Mohamed WA, Ismail M. A randomized, double-blind, placebo-controlled, prospective study of bosentan for the treatment of persistent pulmonary hypertension of the newborn. J Perinatol 2012;32:608–613. [DOI] [PubMed]
- 79.Ivy DD, Ziegler JW, Dubus MF, Fox JJ, Kinsella JP, Abman SH. Chronic intrauterine pulmonary hypertension alters endothelin receptor activity in the ovine fetal lung. Pediatr Res 1996;39:435–442. [DOI] [PubMed]
- 80.Maurey C, Hislop AA, Advenier C, Vouhe PR, Israel-Biet D, Levy M. Interaction of KATP channels and endothelin-1 in lambs with persistent pulmonary hypertension of the newborn. Pediatr Res 2006;60:252–257. [DOI] [PubMed]
- 81.Gao Y, Portugal AD, Negash S, Zhou W, Longo LD, Raj JU. Role of Rho kinases in PKG-mediated relaxation of pulmonary arteries of fetal lambs exposed to chronic high altitude hypoxia. Am J Physiol Lung Cell Mol Physiol 2007;292:L678–L684. [DOI] [PubMed]
- 82.Noguchi Y, Hislop AA, Haworth SG. Influence of hypoxia on endothelin-1 binding sites in neonatal porcine pulmonary vasculature. Am J Physiol Heart Circ Physiol 1997;272:H669–H678. [DOI] [PubMed]
- 83.Schindler MB, Hislop AA, Haworth SG. Porcine pulmonary artery and bronchial responses to endothelin-1 and norepinephrine on recovery from hypoxic pulmonary hypertension. Pediatr Res 2006;60:71–76. [DOI] [PubMed]
- 84.Argiolas L, Fabi F, del Basso P. Mechanisms of pulmonary vasoconstriction and bronchoconstriction produced by PAF in the guinea-pig: role of platelets and cyclo-oxygenase metabolites. Br J Pharmacol 1995;114:203–209. [DOI] [PMC free article] [PubMed]
- 85.Ishii S, Shimizu T. Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog Lipid Res 2000;39:41–82. [DOI] [PubMed]
- 86.Ibe BO, Hibler S, Raj JU. Platelet-activating factor modulates pulmonary vasomotor tone in the perinatal lamb. J Appl Physiol 1998;85:1079–1085. [DOI] [PubMed]
- 87.Ibe BO, Portugal AM, Chaturvedi S, Raj JU. Oxygen-dependent PAF receptor binding and intracellular signaling in ovine fetal pulmonary vascular smooth muscle. Am J Physiol Lung Cell Mol Physiol 2005;288:L879–L886. [DOI] [PubMed]
- 88.Salva AM, Ibe BO, Cliborn E, Reyes G, Raj JU. Hypoxia attenuates metabolism of platelet activating factor by fetal and newborn lamb lungs. J Lipid Res 1996;37:783–789. [PubMed]
- 89.Goyal R, Papamatheakis DG, Loftin M, Vrancken K, Dawson AS, Osman NJ, Blood AB, Pearce WJ, Longo LD, Wilson SM. Long-term maternal hypoxia: the role of extracellular Ca2+ entry during serotonin-mediated contractility in fetal ovine pulmonary arteries. Reprod Sci 2011;18:948–962. [DOI] [PMC free article] [PubMed]
- 90.Chambers CD, Hernandez-Diaz S, Van Marter LJ, Werler MM, Louik C, Jones KL, Mitchell AA. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med 2006;354:579–587. [DOI] [PubMed]
- 91.Kieler H, Artama M, Engeland A, Ericsson Ö, Furu K, Gissler M, Nielsen RB, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of persistent pulmonary hypertension in the newborn: population based cohort study from the five Nordic countries. BMJ 2012;344:d8012. [DOI] [PubMed]
- 92.Delaney C, Gien J, Roe GB, Isenberg N, Kailey J, Abman SH. Serotonin contributes to high pulmonary vascular tone in a sheep model of persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol 2013;304:L894–L901. [DOI] [PMC free article] [PubMed]
- 93.Rodat L, Savineau JP, Marthan R, Guibert C. Effect of chronic hypoxia on voltage-independent calcium influx activated by 5-HT in rat intrapulmonary arteries. Pfluegers Arch 2007;454:41–51. [DOI] [PubMed]
- 94.Keegan A, Morecroft I, Smillie D, Hicks MN, MacLean MR. Contribution of the 5-HT1B receptor to hypoxia-induced pulmonary hypertension: converging evidence using 5-HT1B-receptor knockout mice and the 5-HT1B/1D-receptor antagonist GR127935. Circ Res 2001;89:1231–1239. [DOI] [PubMed]
- 95.McMurtry IF, Abe K, Ota H, Fagan KA, Oka M. Rho kinase-mediated vasoconstriction in pulmonary hypertension. Adv Exp Med Biol 2010;661:299–308. [DOI] [PubMed]
- 96.Tourneux P, Chester M, Grover T, Abman SH. Fasudil inhibits the myogenic response in the fetal pulmonary circulation. Am J Physiol Heart Circ Physiol 2008;295:H1505–H1513. [DOI] [PMC free article] [PubMed]
- 97.Gien J, Seedorf GJ, Balasubramaniam V, Tseng N, Markham N, Abman SH. Chronic intrauterine pulmonary hypertension increases endothelial cell Rho kinase activity and impairs angiogenesis in vitro. Am J Physiol Lung Cell Mol Physiol 2008;295:L680–L687. [DOI] [PMC free article] [PubMed]
- 98.Gao Y, Portugal AD, Liu J, Negash S, Zhou W, Tian J, Xiang R, Longo LD, Raj JU. Preservation of cGMP-induced relaxation of pulmonary veins of fetal lambs exposed to chronic high altitude hypoxia: role of PKG and Rho kinase. Am J Physiol Lung Cell Mol Physiol 2008;295:L889–L896. [DOI] [PMC free article] [PubMed]
- 99.Papamatheakis DG, Vemulakonda S, Patel J, Blood Q, Merritt T, Longo LD, Wilson SM. Depolarization-dependent contraction increase after birth and preservation following long-term hypoxia in sheep pulmonary arteries. Pulm Circ 2012;2:41–53. [DOI] [PMC free article] [PubMed]
- 100.Tirosh R, Resnik ER, Herron J, Sukovich DJ, Hong Z, Weir EK, Cornfield DN. Acute normoxia increases fetal pulmonary artery endothelial cell cytosolic Ca2+ via Ca2+-induced Ca2+ release. Pediatr Res 2006;60:258–263. [DOI] [PubMed]
- 101.Shaul PW, Farrar MA, Magness RR. Oxygen modulation of pulmonary arterial prostacyclin synthesis is developmentally regulated. Am J Physiol Heart Circ Physiol 1993;265:H621–H628. [DOI] [PubMed]
- 102.Watanabe K, Lam G, Jaffe EA. The correlation between rises in intracellular calcium and PGI2 production in cultured vascular endothelial cells. Prostaglandins Leukot Essent Fatty Acids 1992;46:211–214. [DOI] [PubMed]
- 103.Tran CH, Taylor MS, Plane F, Nagaraja S, Tsoukias NM, Solodushko V, Vigmond EJ, Furstenhaupt T, Brigdan M, Welsh DG. Endothelial Ca2+ wavelets and the induction of myoendothelial feedback. Am J Physiol Cell Physiol 2012;302:C1226–C1242. [DOI] [PMC free article] [PubMed]
- 104.Gairhe S, Bauer NN, Gebb SA, McMurtry IF. Serotonin passes through myoendothelial gap junctions to promote pulmonary arterial smooth muscle cell differentiation. Am J Physiol Lung Cell Mol Physiol 2012;303:L767–L777. [DOI] [PubMed]
- 105.Gairhe S, Bauer NN, Gebb SA, McMurtry IF. Myoendothelial gap junctional signaling induces differentiation of pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2011;301:L527–L535. [DOI] [PubMed]
- 106.Ho JJ, Man HS, Marsden PA. Nitric oxide signaling in hypoxia. J Mol Med (Berl) 2012;90:217–231. [DOI] [PubMed]
- 107.Beall CM, Laskowski D, Strohl KP, Soria R, Villena M, Vargas E, Alarcon AM, Gonzales C, Erzurum SC. Pulmonary nitric oxide in mountain dwellers. Nature 2001;414:411–412. [DOI] [PubMed]
- 108.Duplain H, Sartori C, Lepori M, Egli M, Allemann Y, Nicod P, Scherrer U. Exhaled nitric oxide in high-altitude pulmonary edema: role in the regulation of pulmonary vascular tone and evidence for a role against inflammation. Am J Respir Crit Care Med 2000;162:221–224. [DOI] [PubMed]
- 109.Shaul PW, Yuhanna IS, German Z, Chen Z, Steinhorn RH, Morin FC 3rd. Pulmonary endothelial NO synthase gene expression is decreased in fetal lambs with pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 1997;272:L1005–L1012. [DOI] [PubMed]
- 110.Konduri GG, Ou J, Shi Y, Pritchard KA Jr. Decreased association of HSP90 impairs endothelial nitric oxide synthase in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 2003;285:H204–H211. [DOI] [PubMed]
- 111.Hsu JH, Oishi P, Wiseman DA, Hou Y, Chikovani O, Datar S, Sajti E, et al. Nitric oxide alterations following acute ductal constriction in the fetal lamb: a role for superoxide. Am J Physiol Lung Cell Mol Physiol 2010;298:L880–L887. [DOI] [PMC free article] [PubMed]
- 112.Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Circ Res 2003;92:683–691. [DOI] [PubMed]
- 113.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245–313. [DOI] [PubMed]
- 114.Thusu KG, Morin FC 3rd, Russell JA, Steinhorn RH. The cGMP phosphodiesterase inhibitor zaprinast enhances the effect of nitric oxide. Am J Respir Crit Care Med 1995;152:1605–1610. [DOI] [PubMed]
- 115.Zayek M, Cleveland D, Morin FC 3rd. Treatment of persistent pulmonary hypertension in the newborn lamb by inhaled nitric oxide. J Pediatr 1993;122:743–750. [DOI] [PubMed]
- 116.Heidersbach RS, Johengen MJ, Bekker JM, Fineman JR. Inhaled nitric oxide, oxygen, and alkalosis: dose-response interactions in a lamb model of pulmonary hypertension. Pediatr Pulmonol 1999;28:3–11. [DOI] [PubMed]
- 117.Llanos AJ, Ebensperger G, Herrera EA, Reyes RV, Cabello G, Diaz M, Giussani DA, Parer JT. The heme oxygenase-carbon monoxide system in the regulation of cardiorespiratory function at high altitude. Respir Physiol Neurobiol 2012;184:186–191. [DOI] [PubMed]
- 118.Chester M, Seedorf G, Tourneux P, Gien J, Tseng N, Grover T, Wright J, Stasch JP, Abman SH. Cinaciguat, a soluble guanylate cyclase activator, augments cGMP after oxidative stress and causes pulmonary vasodilation in neonatal pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2011;301:L755–L764. [DOI] [PMC free article] [PubMed]
- 119.Potter LR. Guanylyl cyclase structure, function and regulation. Cell Signal 2011;23:1921–1926. [DOI] [PMC free article] [PubMed]
- 120.Resnik E, Herron J, Keck M, Sukovich D, Linden B, Cornfield DN. Chronic intrauterine pulmonary hypertension selectively modifies pulmonary artery smooth muscle cell gene expression. Am J Physiol Lung Cell Mol Physiol 2006;290:L426–L433. [DOI] [PubMed]
- 121.Negash S, Gao Y, Zhou W, Liu J, Chinta S, Raj JU. Regulation of cGMP-dependent protein kinase-mediated vasodilation by hypoxia-induced reactive species in ovine fetal pulmonary veins. Am J Physiol Lung Cell Mol Physiol 2007;293:L1012–L1020. [DOI] [PubMed]
- 122.Kourembanas S. Hypoxia and carbon monoxide in the vasculature. Antioxid Redox Signal 2002;4:291–299. [DOI] [PubMed]
- 123.Jaggar JH, Li A, Parfenova H, Liu J, Umstot ES, Dopico AM, Leffler CW. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ Res 2005;97:805–812. [DOI] [PMC free article] [PubMed]
- 124.Tang XD, Xu R, Reynolds MF, Garcia ML, Heinemann SH, Hoshi T. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature 2003;425:531–535. [DOI] [PubMed]
- 125.Horrigan FT, Heinemann SH, Hoshi T. Heme regulates allosteric activation of the Slo1 BK channel. J Gen Physiol 2005;126:7–21. [DOI] [PMC free article] [PubMed]
- 126.Leffler CW, Parfenova H, Jaggar JH. Carbon monoxide as an endogenous vascular modulator. Am J Physiol Heart Circ Physiol 2011;301:H1–H11. [DOI] [PMC free article] [PubMed]
- 127.Jaggar JH, Leffler CW, Cheranov SY, Tcheranova D, E S, Cheng X. Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+-activated K+ channels. Circ Res 2002;91:610–617. [DOI] [PubMed]
- 128.Leffler CW, Hessler JR. Pulmonary and systemic vascular effects of exogenous prostaglandin I2 in fetal lambs. Eur J Pharmacol 1979;54:37–42. [DOI] [PubMed]
- 129.Cassin S, Winikor I, Tod M, Philips J, Frisinger J, Jordan J, Gibbs C. Effects of prostacyclin on the fetal pulmonary circulation. Pediatr Pharmacol (NY) 1981;1:197–207. [PubMed]
- 130.Ceelie I, van der Starre C, Tibboel D, Stol K, Koren G, de Wildt SN. Evaluation of drug formularies for pediatric intensive care. Pediatr Crit Care Med 2011;12:e14–e19. [DOI] [PubMed]
- 131.Elliot CA, Stewart P, Webster VJ, Mills GH, Hutchinson SP, Howarth ES, Bu’Lock FA, Lawson RA, Armstrong IJ, Kiely DG. The use of iloprost in early pregnancy in patients with pulmonary arterial hypertension. Eur Respir J 2005;26:168–173. [DOI] [PubMed]
- 132.Olschewski H, Simonneau G, Galiè N, Higenbottam T, Naeije R, Rubin LJ, Nikkho S, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002;347:322–329. [DOI] [PubMed]
- 133.Barman SA, Zhu S, Han G, White RE. cAMP activates BKCa channels in pulmonary arterial smooth muscle via cGMP-dependent protein kinase. Am J Physiol Lung Cell Mol Physiol 2003;284:L1004–L1011. [DOI] [PubMed]
- 134.Hoshikawa Y, Voelkel NF, Gesell TL, Moore MD, Morris KG, Alger LA, Narumiya S, Geraci MW. Prostacyclin receptor-dependent modulation of pulmonary vascular remodeling. Am J Respir Crit Care Med 2001;164:314–318. [DOI] [PubMed]
- 135.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, Badesch D, Voelkel NF. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999;159:1925–1932. [DOI] [PubMed]
- 136.Lakshminrusimha S, Porta NF, Farrow KN, Chen B, Gugino SF, Kumar VH, Russell JA, Steinhorn RH. Milrinone enhances relaxation to prostacyclin and iloprost in pulmonary arteries isolated from lambs with persistent pulmonary hypertension of the newborn. Pediatr Crit Care Med 2009;10:106–112. [DOI] [PMC free article] [PubMed]
- 137.Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, Groves BM, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334:296–301. [DOI] [PubMed]
- 138.Simonneau G, Barst RJ, Galiè N, Naeije R, Rich S, Bourge RC, Keogh A, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002;165:800–804. [DOI] [PubMed]
- 139.Delannoy E, Courtois A, Freund-Michel V, Leblais V, Marthan R, Muller B. Hypoxia-induced hyperreactivity of pulmonary arteries: role of cyclooxygenase-2, isoprostanes, and thromboxane receptors. Cardiovasc Res 2010;85:582–592. [DOI] [PubMed]
- 140.Fike CD, Kaplowitz MR, Zhang Y, Pfister SL. Cyclooxygenase-2 and an early stage of chronic hypoxia-induced pulmonary hypertension in newborn pigs. J Appl Physiol 2005;98:1111–1118. [DOI] [PubMed]
- 141.Seifert R, Lushington GH, Mou TC, Gille A, Sprang SR. Inhibitors of membranous adenylyl cyclases. Trends Pharmacol Sci 2012;33:64–78. [DOI] [PMC free article] [PubMed]
- 142.Farrow KN, Steinhorn RH. Phosphodiesterases: emerging therapeutic targets for neonatal pulmonary hypertension. Handb Exp Pharmacol 2011;204:251–277. [DOI] [PMC free article] [PubMed]
- 143.Gao Y, Tolsa JF, Botello M, Raj JU. Developmental change in isoproterenol-mediated relaxation of pulmonary veins of fetal and newborn lambs. J Appl Physiol 1998;84:1535–1539. [DOI] [PubMed]
- 144.Manganiello V. Cyclic nucleotide phosphodiesterases: critical modulators of endocrine, metabolic, and cardiovascular function and appealing therapeutic targets. Curr Opin Pharmacol 2011;11:646–648. [DOI] [PubMed]
- 145.Hanson KA, Ziegler JW, Rybalkin SD, Miller JW, Abman SH, Clarke WR. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am J Physiol Lung Cell Mol Physiol 1998;275:L931–L941. [DOI] [PubMed]
- 146.Rashid N, Morin FC 3rd, Swartz DD, Ryan RM, Wynn KA, Wang H, Lakshminrusimha S, Kumar VH. Effects of prostacyclin and milrinone on pulmonary hemodynamics in newborn lambs with persistent pulmonary hypertension induced by ductal ligation. Pediatr Res 2006;60:624–629. [DOI] [PubMed]
- 147.Chen B, Lakshminrusimha S, Czech L, Groh BS, Gugino SF, Russell JA, Farrow KN, Steinhorn RH. Regulation of phosphodiesterase 3 in the pulmonary arteries during the perinatal period in sheep. Pediatr Res 2009;66:682–687. [DOI] [PMC free article] [PubMed]
- 148.Alexander S, Harmar A, McGrath I. New updated GRAC fifth edition with searchable online version: launch of new portal Guide to Pharmacology in association with NC-IUPHAR transporter-themed issue. Br J Pharmacol 2011;164:1749–1750. [DOI] [PMC free article] [PubMed]
- 149.Shimoda LA, Sylvester JT, Booth GM, Shimoda TH, Meeker S, Undem BJ, Sham JS. Inhibition of voltage-gated K+ currents by endothelin-1 in human pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 2001;281:L1115–L1122. [DOI] [PubMed]
- 150.Cornfield DN, Saqueton CB, Porter VA, Herron J, Resnik E, Haddad IY, Reeve HL. Voltage-gated K+-channel activity in ovine pulmonary vasculature is developmentally regulated. Am J Physiol Lung Cell Mol Physiol 2000;278:L1297–L1304. [DOI] [PubMed]
- 151.Konduri GG, Bakhutashvili I, Eis A, Gauthier KM. Impaired voltage gated potassium channel responses in a fetal lamb model of persistent pulmonary hypertension of the newborn. Pediatr Res 2009;66:289–294. [DOI] [PMC free article] [PubMed]
- 152.Fike CD, Kaplowitz MR, Zhang Y, Madden JA. Voltage-gated K+ channels at an early stage of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 2006;291:L1169–L1176. [DOI] [PubMed]
- 153.Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, Seiden JE, Rubin LJ, Yuan JX. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 2000;279:C1540–C1549. [DOI] [PubMed]
- 154.Michelakis ED, Reeve HL, Huang JM, Tolarova S, Nelson DP, Weir EK, Archer SL. Potassium channel diversity in vascular smooth muscle cells. Can J Physiol Pharmacol 1997;75:889–897. [PubMed]
- 155.Cornfield DN. Developmental regulation of oxygen sensing and ion channels in the pulmonary vasculature. Adv Exp Med Biol 2010;661:201–220. [DOI] [PubMed]
- 156.Olschewski A, Hong Z, Linden BC, Porter VA, Weir EK, Cornfield DN. Contribution of the KCa channel to membrane potential and O2 sensitivity is decreased in an ovine PPHN model. Am J Physiol Lung Cell Mol Physiol 2002;283:L1103–L1109. [DOI] [PubMed]
- 157.Jaggar JH, Wellman GC, Heppner TJ, Porter VA, Perez GJ, Gollasch M, Kleppisch T, et al. Ca2+ channels, ryanodine receptors and Ca2+-activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol Scand 1998;164:577–587. [DOI] [PubMed]
- 158.Li A, Adebiyi A, Leffler CW, Jaggar JH. KCa channel insensitivity to Ca2+ sparks underlies fractional uncoupling in newborn cerebral artery smooth muscle cells. Am J Physiol Heart Circ Physiol 2006;291:H1118–H1125. [DOI] [PMC free article] [PubMed]
- 159.Cornfield DN, Reeve HL, Tolarova S, Weir EK, Archer S. Oxygen causes fetal pulmonary vasodilation through activation of a calcium-dependent potassium channel. Proc Natl Acad Sci USA 1996;93:8089–8094. [DOI] [PMC free article] [PubMed]
- 160.Cornfield DN, Resnik ER, Herron JM, Abman SH. Chronic intrauterine pulmonary hypertension decreases calcium-sensitive potassium channel mRNA expression. Am J Physiol Lung Cell Mol Physiol 2000;279:L857–L862. [DOI] [PubMed]
- 161.Hume JR, McAllister CE, Wilson SM. Caffeine inhibits InsP3 responses and capacitative calcium entry in canine pulmonary arterial smooth muscle cells. Vasc Pharmacol 2009;50:89–97. [DOI] [PMC free article] [PubMed]
- 162.Goyal R, Creel KD, Chavis E, Smith GD, Longo LD, Wilson SM. Maturation of intracellular calcium homeostasis in sheep pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2008;295:L905–L914. [DOI] [PMC free article] [PubMed]
- 163.Zhang WM, Yip KP, Lin MJ, Shimoda LA, Li WH, Sham JS. ET-1 activates Ca2+ sparks in PASMC: local Ca2+ signaling between inositol trisphosphate and ryanodine receptors. Am J Physiol Lung Cell Mol Physiol 2003;285:L680–L690. [DOI] [PubMed]
- 164.Ibe BO, Ameer A, Portugal AM, Renteria L, Raj JU. Platelet-activating factor modulates activity of cyclic nucleotides in fetal ovine pulmonary vascular smooth muscle. J Pharmacol Exp Ther 2007;320:728–737. [DOI] [PubMed]
- 165.Shimoda LA, Sham JS, Shimoda TH, Sylvester JT. L-type Ca2+ channels, resting [Ca2+]i, and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol 2000;279:L884–L894. [DOI] [PubMed]
- 166.Cassin S. Role of prostaglandins, thromboxanes, and leukotrienes in the control of the pulmonary circulation in the fetus and newborn. Semin Perinatol 1987;11:53–63. [PubMed]
- 167.Snetkov VA, Knock GA, Baxter L, Thomas GD, Ward JP, Aaronson PI. Mechanisms of the prostaglandin F2α-induced rise in [Ca2+]i in rat intrapulmonary arteries. J Physiol 2006;571:147–163. [DOI] [PMC free article] [PubMed]
- 168.Porter VA, Reeve HL, Cornfield DN. Fetal rabbit pulmonary artery smooth muscle cell response to ryanodine is developmentally regulated. Am J Physiol Lung Cell Mol Physiol 2000;279:L751–L757. [DOI] [PubMed]
- 169.Porter VA, Rhodes MT, Reeve HL, Cornfield DN. Oxygen-induced fetal pulmonary vasodilation is mediated by intracellular calcium activation of KCa channels. Am J Physiol Lung Cell Mol Physiol 2001;281:L137–L1385. [DOI] [PubMed]
- 170.Jabr RI, Toland H, Gelband CH, Wang XX, Hume JR. Prominent role of intracellular Ca2+ release in hypoxic vasoconstriction of canine pulmonary artery. Br J Pharmacol 1997;122:21–30. [DOI] [PMC free article] [PubMed]
- 171.Ng LC, Wilson SM, Hume JR. Mobilization of sarcoplasmic reticulum stores by hypoxia leads to consequent activation of capacitative Ca2+ entry in isolated canine pulmonary arterial smooth muscle cells. J Physiol 2005;563:409–419. [DOI] [PMC free article] [PubMed]
- 172.Remillard CV, Zhang WM, Shimoda LA, Sham JS. Physiological properties and functions of Ca2+ sparks in rat intrapulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2002;283:L433–L444. [DOI] [PubMed]
- 173.Hadley SR, Blood Q, Rubalcava M, Waskel E, Lumbard B, Le P, Longo LD, Buchholz JN, Wilson SM. Maternal high-altitude hypoxia and suppression of ryanodine receptor-mediated Ca2+ sparks in fetal sheep pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 2012;303:L799–L813. [DOI] [PMC free article] [PubMed]
- 174.Resnik ER, Keck M, Sukovich DJ, Herron JM, Cornfield DN. Chronic intrauterine pulmonary hypertension increases capacitative calcium entry in fetal pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007;292:L953–L959. [DOI] [PubMed]
- 175.Papamatheakis DG, Vemulakonda S, Blood Q, Goyal R, Rubalcava M, Vrancken K, Bennett A, et al. Preservation of serotonin-mediated contractility in adult sheep pulmonary arteries following long-term high-altitude hypoxia. High Alt Med Biol 2011;12:253–264. [DOI] [PMC free article] [PubMed]
- 176.Wilson SM, Mason HS, Ng LC, Montague S, Johnston L, Nicholson N, Mansfield S, Hume JR. Role of basal extracellular Ca2+ entry during 5-HT-induced vasoconstriction of canine pulmonary arteries. Br J Pharmacol 2005;144:252–264. [DOI] [PMC free article] [PubMed]
- 177.Hirenallur SD, Haworth ST, Leming JT, Chang J, Hernandez G, Gordon JB, Rusch NJ. Upregulation of vascular calcium channels in neonatal piglets with hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2008;295:L915–L924. [DOI] [PMC free article] [PubMed]
- 178.Storme L, Parker TA, Kinsella JP, Rairigh RL, Abman SH. Chronic hypertension impairs flow-induced vasodilation and augments the myogenic response in fetal lung. Am J Physiol Lung Cell Mol Physiol 2002;282:L56–L66. [DOI] [PubMed]
- 179.Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem 2007;76:387–417. [DOI] [PMC free article] [PubMed]
- 180.Oishi P, Datar SA, Fineman JR. Advances in the management of pediatric pulmonary hypertension. Respir Care 2011;56:1314–1340. [DOI] [PubMed]
- 181.Abman SH. Recent advances in the pathogenesis and treatment of persistent pulmonary hypertension of the newborn. Neonatology 2007;91:283–290. [DOI] [PubMed]
- 182.Steinhorn RH. Pharmacotherapy for pulmonary hypertension. Pediatr Clin N Am 2012;59:1129–1146. [DOI] [PMC free article] [PubMed]
- 183.Ivy D. Advances in pediatric pulmonary arterial hypertension. Curr Opin Cardiol 2012;27:70–81. [DOI] [PMC free article] [PubMed]
- 184.Cerro MJ, Abman S, Diaz G, Freudenthal AH, Freudenthal F, Harikrishnan S, Haworth SG, et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ 2011;1:286–298. [DOI] [PMC free article] [PubMed]
- 185.Ohashi N, Matsushima M, Maeda M, Yamaki S. Advantages of oxygen inhalation therapy for postoperative pulmonary hypertension. Pediatr Cardiol 2005;26:90–92. [DOI] [PubMed]
- 186.Lazar DA, Cass DL, Olutoye OO, Welty SE, Fernandes CJ, Rycus PT, Lee TC. The use of ECMO for persistent pulmonary hypertension of the newborn: a decade of experience. J Surg Res 2012;177:263–267. [DOI] [PubMed]
- 187.Chen SJ, Chen YF, Meng QC, Durand J, DiCarlo VS, Oparil S. Endothelin-receptor antagonist bosentan prevents and reverses hypoxic pulmonary hypertension in rats. J Appl Physiol 1995;79:2122–2131. [DOI] [PubMed]
- 188.DiCarlo VS, Chen SJ, Meng QC, Durand J, Yano M, Chen YF, Oparil S. ETA-receptor antagonist prevents and reverses chronic hypoxia-induced pulmonary hypertension in rat. Am J Physiol Lung Cell Mol Physiol 1995;269:L690–L697. [DOI] [PubMed]
- 189.Goldman AP, Delius RE, Deanfield JE, Miller OI, de Leval MR, Sigston PE, Macrae DJ. Pharmacological control of pulmonary blood flow with inhaled nitric oxide after the fenestrated Fontan operation. Circulation 1996;94(9 suppl):II44–II48. [PubMed]
- 190.Bizzarro M, Gross I. Inhaled nitric oxide for the postoperative management of pulmonary hypertension in infants and children with congenital heart disease. Cochrane Database Syst Rev 2005(4):CD005055. [DOI] [PubMed]
- 191.Clark RH, Kueser TJ, Walker MW, Southgate WM, Huckaby JL, Perez JA, Roy BJ, Keszler M, Kinsella JP. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. N Engl J Med 2000;342:469–474. [DOI] [PubMed]
- 192.Schreiber MD, Gin-Mestan K, Marks JD, Huo D, Lee G, Srisuparp P. Inhaled nitric oxide in premature infants with the respiratory distress syndrome. N Engl J Med 2003;349:2099–2107. [DOI] [PubMed]
- 193.Roberts JD Jr., Fineman JR, Morin FC 3rd, Shaul PW, Rimar S, Schreiber MD, Polin RA, et al. Inhaled nitric oxide and persistent pulmonary hypertension of the newborn. N Engl J Med 1997;336:605–610. [DOI] [PubMed]
- 194.Finer NN, Barrington KJ. Nitric oxide for respiratory failure in infants born at or near term. Cochrane Database Syst Rev 2006(4):CD000399. [DOI] [PubMed]
- 195.Baquero H, Soliz A, Neira F, Venegas ME, Sola A. Oral sildenafil in infants with persistent pulmonary hypertension of the newborn: a pilot randomized blinded study. Pediatrics 2006;117:1077–1083. [DOI] [PubMed]
- 196.Steinhorn RH, Kinsella JP, Pierce C, Butrous G, Dilleen M, Oakes M, Wessel DL. Intravenous sildenafil in the treatment of neonates with persistent pulmonary hypertension. J Pediatr 2009;155:841–847. [DOI] [PubMed]
- 197.Yung D, Widlitz AC, Rosenzweig EB, Kerstein D, Maislin G, Barst RJ. Outcomes in children with idiopathic pulmonary arterial hypertension. Circulation 2004;110:660–665. [DOI] [PubMed]
- 198.Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation 1999;99:1858–1865. [DOI] [PubMed]
- 199.Maiya S, Hislop AA, Flynn Y, Haworth SG. Response to bosentan in children with pulmonary hypertension. Heart 2006;92:664–670. [DOI] [PMC free article] [PubMed]
- 200.Rosenzweig EB, Ivy DD, Widlitz A, Doran A, Claussen LR, Yung D, Abman SH, Morganti A, Nguyen N, Barst RJ. Effects of long-term bosentan in children with pulmonary arterial hypertension. J Am Coll Cardiol 2005;46:697–704. [DOI] [PubMed]
- 201.McLaughlin VV, Oudiz RJ, Frost A, Tapson VF, Murali S, Channick RN, Badesch DB, Barst RJ, Hsu HH, Rubin LJ. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med 2006;174:1257–1263. [DOI] [PubMed]
- 202.Ghofrani HA, Wiedemann R, Rose F, Olschewski H, Schermuly RT, Weissmann N, Seeger W, Grimminger F. Combination therapy with oral sildenafil and inhaled iloprost for severe pulmonary hypertension. Ann Intern Med 2002;136:515–522. [DOI] [PubMed]
- 203.Humbert M, Barst RJ, Robbins IM, Channick RN, Galiè N, Boonstra A, Rubin LJ, Horn EM, Manes A, Simonneau G. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J 2004;24:353–359. [DOI] [PubMed]